

Abstract
The effect of semaglutide, a once-weekly human glucagon-like peptide-1 (GLP-1) analog in development for type 2 diabetes (T2D), on the bioavailability of a combined oral contraceptive was investigated. Postmenopausal women with T2D (n = 43) on diet/exercise ± metformin received ethinylestradiol (0.03 mg)/levonorgestrel (0.15 mg) once daily for 8 days before (semaglutide-free) and during (steady-state 1.0 mg) semaglutide treatment (subcutaneous once weekly; dose escalation: 0.25 mg 4 weeks; 0.5 mg 4 weeks; 1.0 mg 5 weeks). Bioequivalence of oral contraceptives was established if 90%CI for the ratio of pharmacokinetic parameters during semaglutide steady-state and semaglutide-free periods was within prespecified limits (0.80–1.25). The bioequivalence criterion was met for ethinylestradiol area under the curve (AUC0–24 h) for semaglutide steady-state/semaglutide-free; 1.11 (1.06–1.15). AUC0–24 h was 20% higher for levonorgestrel at semaglutide steady-state vs. semaglutide-free (1.20 [1.15–1.26]). Cmax was within bioequivalence criterion for both contraceptives. Reductions (mean ± SD) in HbA1c (–1.1 ± 0.6%) and weight (–4.3 ± 3.1 kg) were observed. Semaglutide pharmacokinetics were compatible with once-weekly dosing; the semaglutide dose and dose-escalation regimen were well tolerated. Adverse events, mainly gastrointestinal, were mild to moderate in severity. Asymptomatic increases in mean amylase and lipase were observed. Three subjects had elevated alanine aminotransferase levels ≥3x the upper limit of normal during semaglutide/oral contraceptive coadministration, which were reported as adverse events, but resolved during follow-up. Semaglutide did not reduce the bioavailability of ethinylestradiol and levonorgestrel.
Keywords: semaglutide, GLP-1, once weekly, type 2 diabetes, ethinylestradiol, levonorgestrel
Glucagon-like peptide-1 (GLP-1) is a gut-derived incretin hormone that potentiates insulin secretion, inhibits glucagon secretion, reduces appetite, and delays the rate of gastric emptying in response to food intake.1–4 However, native GLP-1 has a very short half-life (t1/2), is rapidly degraded by dipeptidyl peptidase-4 (DPP-4),1 and is therefore unsuitable for the management of type 2 diabetes (T2D). Treatment modalities for enhancing the effect of GLP-1 receptor stimulation and action include degradation-resistant GLP-1 receptor agonists and DPP-4 inhibitors.1,5–8 GLP-1 receptor agonists have been shown to improve glycemic control by reducing fasting plasma glucose (FPG) and postprandial glucose (PPG), and to provide beneficial reductions in body weight in patients with T2D,6,8–10 and in obese patients without T2D.11
Semaglutide (Novo Nordisk A/S, Denmark), a human GLP-1 analog, is currently in phase III clinical development for the treatment of T2D. Semaglutide has 94% structural homology to native human GLP-1.12,13 Three minor but important modifications make semaglutide suitable for clinical use: amino acid substitutions at position 8 (alanine to alpha-aminoisobutyric acid, a synthetic amino acid) and position 34 (lysine to arginine), and acylation of the peptide backbone with a spacer and C-18 fatty di-acid chain to lysine at position 26.12 The fatty di-acid side chain and the spacer mediate strong binding to albumin, which is believed to result in reduced renal clearance. The amino acid substitution at position 8 makes semaglutide less susceptible to degradation by DPP-4. The reported t1/2 of semaglutide is 155–184 hours.12,14
Oral contraceptive medications, a common method of birth control, are mostly metabolized by cytochrome-P450 (CYP450).15 As semaglutide is not thought to rely on this metabolic pathway, it is not expected to inhibit or induce CYP450 enzymes or interact with the metabolism of CYP450-metabolized drugs. However, similar to native GLP-1, semaglutide may delay the rate of gastric emptying. Changes in the rate of gastric emptying could potentially delay the absorption of concomitantly administered oral therapies.16–18 In the case of oral contraceptive medications, this could result in failure to provide effective birth control.
The primary objective of this study was to investigate if semaglutide altered the pharmacokinetics of components of a commonly used combined oral contraceptive, ethinylestradiol and levonorgestrel, in postmenopausal women with T2D. Secondary objectives included evaluating semaglutide pharmacokinetics, safety, tolerability, and pharmacodynamics. Finally, this is the first study reporting the anticipated clinical dose and dose-escalation regimen of semaglutide.
Materials and Methods
Study Design and Population
This was a single-center, open-label, one-sequence crossover study. It was conducted in accordance with Good Clinical Practice19 and the Declaration of Helsinki,20 and followed the accepted rules for interaction studies according to the US Food and Drug Administration (FDA) Guidance for Industry21 and the European Medicines Agency (EMA) guidelines.22 The study was registered at http://ClinicalTrials.gov with the identifier NCT01324505.
A total of 43 postmenopausal women participated in the study. Informed consent was obtained before any study-related activities commenced. Postmenopausal women who had undergone bilateral oophorectomy or had at least 1 year of spontaneous amenorrhea, with serum follicle stimulating hormone >40 mIU/mL and estrogen deficiency (estradiol levels <30 pg/mL or a negative gestagen test), were selected for the study, with the aim of eliminating any hormonal fluctuations that might influence the interpretation of the pharmacokinetics of the oral contraceptives. Other inclusion criteria included age ≥18 years, documented T2D treated with diet and exercise ± metformin, a body mass index (BMI) of 18.5–35.0 kg/m2 and a glycosylated haemoglobin A1c (HbA1c) of 6.5%–10%. Two subjects did not receive metformin. All other subjects received metformin at least 3 months prior to treatment. Exclusion criteria included treatment with antidiabetic drugs other than metformin in the 3 months prior to start of the trial product, and the use of hormone replacement therapy in the 4 weeks prior to start of the trial product.
Drug Administration
Figure 1 shows the study design. Semaglutide was self-administered once weekly by subcutaneous injection (NordiPen®, Novo Nordisk A/S, Denmark), at any time during the day, on the same day of the week throughout the study. Semaglutide steady-state was reached using a dose-escalation regimen: subjects initiated once-weekly treatment with 0.25 mg for 4 weeks, followed by 0.5 mg for 4 weeks and, finally, 1.0 mg for 5 weeks. The oral contraceptive (1 tablet per day; 0.03 mg ethinylestradiol and 0.15 mg levonorgestrel; Microgyn®, Bayer Pharma AG, Germany) was prescribed for 8 days before (semaglutide-free) and during the last week of dosing with semaglutide 1.0 mg (steady-state); the last dose of oral contraceptive was administered 24 hours after the last dose of semaglutide.
Figure 1.
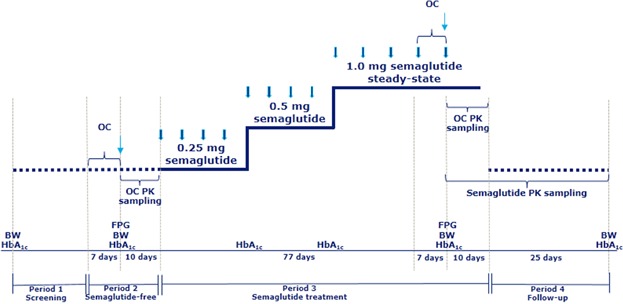
Study design. BW, body weight; FPG, fasting plasma glucose (at study site); HbA1c, glycosylated hemoglobin A1c; OC, oral contraceptive (dosing once-daily); PK, pharmacokinetic. Thick blue arrow, semaglutide (dosing once weekly); thin blue arrow, last dose of OC.
Study Endpoints and Assessments
The primary endpoint was area under the curve (AUC0–24 h) for ethinylestradiol and levonorgestrel at steady-state. Secondary endpoints included other pharmacokinetic parameters for the oral contraceptive: Cmax (maximum concentration), Ctrough (trough concentration), tmax (time to reach Cmax), t1/2, CL/F (apparent total plasma clearance), and Vz/F (apparent volume of distribution). Secondary endpoints for semaglutide included pharmacokinetics during dose escalation (Ctrough [1 week after the fourth dose at each dose level]) and at steady state (1.0 mg) (AUC0–168 h, Cmax, t1/2, CL/F, Vz/F), safety, tolerability, and pharmacodynamics.
Blood sampling for determination of the pharmacokinetics of both components of the oral contraceptive was performed after the last dose of oral contraceptive, for a total of 10 days, during the semaglutide-free and steady-state periods. For determination of plasma concentrations of ethinylestradiol and levonorgestrel, 18 serial blood samples were drawn predose (–15 minutes) and 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 30, 36, 48, 72, 96, 144, and 216 hours postdose on 2 occasions, before semaglutide treatment (semaglutide-free) and during semaglutide 1.0 mg (steady-state) (Figure 1). The 216 hour postdose sampling period corresponded to ∼5x the t1/2 of levonorgestrel—the oral contraceptive component with the longest t1/2. Ethinylestradiol and levonorgestrel were extracted by liquid/liquid extraction (LLE) and analyzed by gas chromatography-mass spectrometry (GC-MS), as described elsewhere.16
Blood sampling for determination of semaglutide pharmacokinetics was performed 1 week after the fourth dose at each dose level and for 5 weeks after the last dose of semaglutide. For determination of plasma concentrations of semaglutide, 22 serial blood samples were drawn predose (–15 minutes) and 4, 8, 12, 16, 24, 28, 32, 36, 40, 48, 54, 60, 72, 96, 120, 168, 240, 336, 504, 672, and 840 hours after the last dose of semaglutide 1.0 mg (Figure 1). In addition, a sample was taken before the first dose and trough samples were drawn after the fourth dose of semaglutide at each dose level (0.25, 0.5, and 1.0 mg). Blood samples were drawn in K3EDTA tubes and stored at –20 °C until analyzed. A liquid chromatography-tandem mass spectroscopy/mass spectroscopy (LC-MS/MS) assay was used following precipitation of the plasma proteins (Celerion Switzerland AG, Fehraltorf, Switzerland).
The LC-MS/MS assay was validated according to current guidelines for analyzing plasma samples in the concentration range 0.729–60.8 nM (3–250 ng/mL). A 5-fold dilution of each sample was validated to extend the assay range above 60.8 nM. An analog of semaglutide was used as an internal standard (IS). The analysis was carried out using an AB Sciex API QTrap® 5500 mass spectrometer monitoring positive ions in the MRM mode with mass transitions m/z 1029.1 ≥ 136.0 Da (semaglutide) and m/z 1106.8 ≥ 123.0 Da (IS), respectively. The LC system was a Waters Acquity™ UPLC® system and the LC column an Acquity UPLC® BEH300 C18, 2.1 × 50 mm. Quantification was performed by peak areas and weighted linear regression (1/x2). The lower limit of quantification (LLOQ) for semaglutide was 1.94 nmol/L.
Pharmacodynamic endpoints for semaglutide included HbA1c, FPG, and body weight. Body weight and HbA1c were measured at baseline, just before initiation of semaglutide treatment, at the end of semaglutide 1.0 mg treatment, and at follow-up. In addition, HbA1c was measured at the first- and second-dose escalation. FPG was measured at the study site before initiation of treatment and at the end of treatment.
Safety Assessments
Safety assessments during the trial included the recording of adverse events, vital signs, electrocardiogram (ECG), physical examination, antisemaglutide antibodies, hypoglycemic episodes, self-measured FPG (every second day at home), and laboratory tests (including hematology, biochemistry, calcitonin, and urinalysis). Biochemistry parameters were measured at screening and on days 7, 100, and 135. Results for self-measured FPG at home are presented with those for FPG measured at the study site.
Treatment-emergent adverse events (TEAEs) were defined as events that had an onset date on or after the first day of exposure to study drug and no later than the follow-up visit, or those that were present before randomization and increased in severity during the treatment period and no later than the follow-up visit (≤35 days after the last dose of semaglutide). All adverse events (AEs) either observed by the investigator or reported spontaneously by subjects were recorded by the investigator and evaluated. AEs were categorized according to their severity as follows: mild (no or transient symptoms, no interference with the subject's daily activities); moderate (marked symptoms, moderate interference with the subject's daily activities); severe (considerable interference with the subject's daily activities, unacceptable). All AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) coding, version 14.0.
Statistical Analysis
The full analysis set (FAS) included all 43 subjects who were exposed to at least 1 dose of trial drug. Four subjects withdrew during semaglutide treatment, 3 before entering the second period of oral contraceptive treatment. Furthermore, an additional 3 subjects did not have a valid ethinylestradiol pharmacokinetic profile during the semaglutide-free period. The statistical analysis included all subjects in the FAS, but only 40 subjects for levonorgestrel and 37 for ethinylestradiol were included in the analysis, in accordance with the prespecified statistical model. Ethinylestradiol and levonorgestrel exposure at semaglutide-free and semaglutide steady-state were established as bioequivalent if the 90%CI for the ratio of the AUC0–24 h was within the prespecified 0.80–1.25 limits (both inclusive; the power was calculated using 2 one-sided t-tests of equivalence in means on a 5% significance level). It was assumed that the true ratio between the AUC at semaglutide-free and semaglutide steady-state was 1. In order to obtain a power of at least 80%, and assuming a dropout rate of 30% and intrasubject variations of 0.22 and 0.28 for ethinylestradiol and levonorgestrel, respectively, it was recommended to include 43 subjects in the study. The analysis of AUC was performed using a linear model on log-transformed values and included semaglutide exposure and subject as fixed effects. Cmax for ethinylestradiol and levonorgestrel was analyzed in a similar way. All other pharmacokinetic and pharmacodynamic endpoints were summarized using descriptive statistics.
Safety endpoints, ECG, vital signs, physical examination, safety laboratory parameters, and antisemaglutide antibodies were summarized by descriptive statistics. All TEAEs were summarized by number, frequency, MedDRA system organ class, MedDRA preferred term, severity, and relation to study product.
Results
In total, 43 postmenopausal women were enrolled and 39 completed the study. Of the 4 subjects who withdrew, 1 was due to adverse events (intermittent nausea, vomiting, and diarrhea) and 3 because they met withdrawal criteria (withdrawal of consent, usage of other prescription drugs, and illness). Supplemental Table 1 shows the baseline demographics for the FAS population. Mean BMI was 29.4 ± 3.4 kg/m2. Mean baseline HbA1c was 7.3%, indicating that T2D was well controlled in the study population. All subjects were Caucasian. The mean age was 62.2 (±6.0) years.
Pharmacokinetics
Ethinylestradiol
Figure 2 shows the mean plasma concentration-time profile and Table 1 shows the pharmacokinetic results for ethinylestradiol at semaglutide-free and semaglutide steady-state. The bioequivalence criterion was met for both AUC0–24 h (semaglutide steady-state/semaglutide-free; 1.11 [90%CI: 1.06–1.15]) and Cmax (1.04 [90%CI: 0.98–1.10]). There were no apparent differences in any of the other pharmacokinetic parameters between the 2 treatment periods. Geometric means (coefficient of variation in percentage [CV%]) of Ctrough values for ethinylestradiol at semaglutide-free and semaglutide steady-state were 16.0 (37.7) pg/mL and 16.6 (41.6) pg/mL, respectively.
Figure 2.
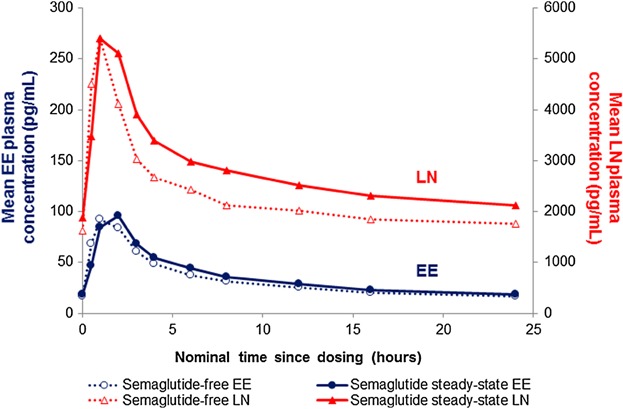
Mean concentration-time profile: 0–24 hours of levonorgestrel (n = 40) and ethinylestradiol (n = 37) during semaglutide-free and semaglutide steady-state periods. EE, ethinylestradiol; LN, levonorgestrel.
Table 1.
Pharmacokinetic Endpoints for Ethinylestradiol and Levonorgestrel During Semaglutide-Free and Semaglutide Steady-State Periods (0–24 hours)a
| Ethinylestradiol (n = 37) | Levonorgestrel (n = 40) | |||
|---|---|---|---|---|
| Parameter | Semaglutide-Free | Semaglutide Steady-State | Semaglutide-Free | Semaglutide Steady-State |
| AUC, pg·h/mL | 748.7 (28.2) | 828.9 (30.2) | 52,780 (29.3) | 63,516 (32.2) |
| Cmax, pg/mL | 93.8 (26.9) | 97.6 (30.6) | 5374 (23.9) | 5642 (31.8) |
| tmax, hours | 1 (0.5, 2) | 2 (0.5, 8) | 1 (0.5, 6) | 1 (1, 8) |
| Ctrough, pg/mL | 16.0 (37.7) | 16.6 (41.6) | 1524 (33.9) | 1745 (37.0) |
| t1/2, hours | 23.8 (41.8)b | 27.4 (83.9) c | 35.1 (20.1) | 33.5 (35.7) |
| CL/F, L/h | 40.1 (36.6) | 36.2 (37.9) | 2.8 (35.2) | 2.4 (38.9) |
| VZ/F, L | 1300 (53.7)b | 1416 (114.9)c | 144.0 (35.1) | 114.0 (58.1) |
AUC, area under the curve; CL/F, apparent total plasma clearance; Cmax, maximum concentration; Ctrough, trough concentration; t1/2, half-life; tmax, time to reach Cmax; VZ/F, apparent volume of distribution.
aData are geometric mean (coefficient of variation in percentage), except median (min, max) for tmax. For ethinylestradiol, due to no clear terminal phase for some pharmacokinetic profiles, the corresponding parameters could only be estimated for: bn = 33; cn = 36.
Levonorgestrel
Figure 2 shows the mean plasma concentration-time profile and Table 1 shows the pharmacokinetic results for levonorgestrel at semaglutide-free and steady-state. The estimated mean AUC0–24 h for levonorgestrel was 20% higher during the semaglutide steady-state than the semaglutide-free period (1.20 [90%CI: 1.15–1.26]). The bioequivalence criterion was met for Cmax for levonorgestrel (1.05 [90%CI: 0.99–1.12]). The median tmax was 1 hour for both treatment periods. However, during the semaglutide steady-state period, individual tmax values were right-shifted, indicating a slight delay of tmax (data not shown). There were no apparent differences in any of the other pharmacokinetic parameters between the 2 periods. Geometric means (CV%) of Ctrough values for levonorgestrel at semaglutide-free and steady-state were 1524 (33.9) pg/mL and 1745 (37.0) pg/mL, respectively.
Semaglutide
Figure 3 shows the mean plasma concentration-time profile for semaglutide at steady-state (1.0 mg). Geometric means (CV%) of AUC0–168 h, Cmax, and t1/2 were 4602 (16.8) nmol h/L, 33.8 (15.5) nmol/L, and 165 (14.1) hours, respectively, and median tmax was 36 (min, max: 12.0, 167.2) hours. Although some variation was observed in the profiles of individual subjects, the overall pattern was similar. Geometric mean (CV%) Ctrough values at 0.25 mg, 0.5 mg, and 1.0 mg semaglutide were 4.4 (31.5) nmol/L, 11.7 (20.2) nmol/L, and 21.2 (19.7) nmol/L, respectively.
Figure 3.
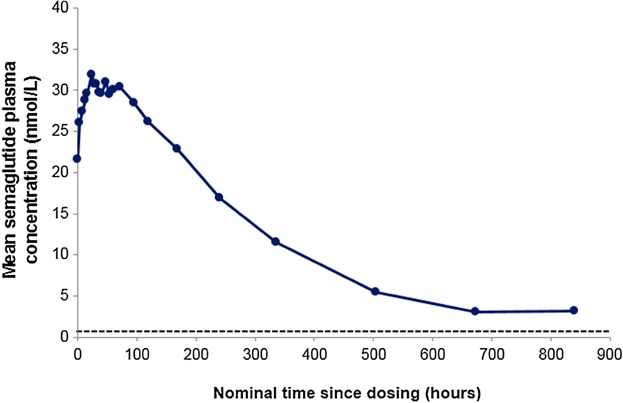
Mean concentration-time profile: 0–840 hours of semaglutide 1.0 mg at steady-state (n = 40). Dashed line represents the lower limit of quantification (LLOQ).
Pharmacodynamics
Baseline HbA1c and body weight (mean ± SD) were reduced by 1.1 ± 0.6% and 4.3 ± 3.1 kg, respectively, at the end of treatment (Supplemental Table 1). FPG measured at the study site (mean ± SD) decreased from 8.4 ± 1.8 mmol/L before initiation of semaglutide treatment to 6.5 ± 1.2 mmol/L at the end of treatment. Self-measured FPG at home (mean ± SD) recorded before initiation of semaglutide treatment and at the end of treatment were 8.8 ± 1.8 mmol/L, and 7.0 ± 1.3 mmol/L, respectively (Figure 4).
Figure 4.
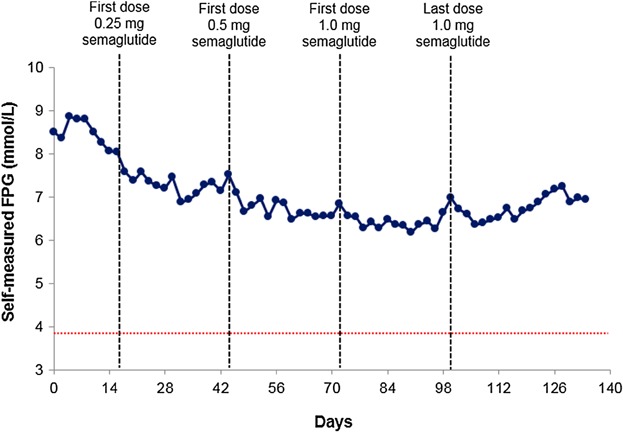
Mean self-measured fasting plasma glucose (FPG; measured every second day at home): self-measured FPG full analysis set (FAS; n = 43). Red dotted line represents the American Diabetes Association definition of documented symptomatic hypoglycemia.23 Typical symptoms of hypoglycemia are accompanied by a measured plasma glucose concentration ≤3.9 mmol/L.
Safety and Tolerability
A total of 199 TEAEs (164 events during semaglutide treatment), all with mild-to-moderate intensity, were reported by 38 of 43 (88%) subjects. Gastrointestinal TEAEs were the most common (73 events in 27 subjects [63% of subjects]); most (≥75%) were mild. Gastrointestinal TEAEs reported with semaglutide treatment included nausea, diarrhea, dyspepsia, constipation, vomiting, and abdominal distension. No serious AEs were reported in the study.
Most nausea events were intermittent. Subjects reported episodes of nausea ranging from short (minutes) to long (full days) duration (median: 3.9 days [min, max: 0.01, 95.6]). All subjects recovered before follow-up, most during semaglutide treatment and some after treatment. The proportion of subjects with nausea appeared to increase over time with increasing semaglutide dose (0.25 and 0.5 mg) up to the initiation of semaglutide 1.0 mg. Despite a further increase in mean plasma exposure toward semaglutide steady-state (1.0 mg), the incidence of nausea decreased gradually with each subsequent dose, possibly indicating tolerance development. Nausea returned to semaglutide-free levels at follow-up (Figure 5). There was a low incidence of vomiting (8 events in 4 subjects [9% of subjects] all mild or moderate) and the duration did not follow an obvious pattern. In total, 6 subjects (14%) reported diarrhea: all cases were mild or moderate. One subject withdrew due to gastrointestinal TEAEs of nausea, vomiting, and diarrhea; however, these events were intermittent and the subject recovered fully.
Figure 5.
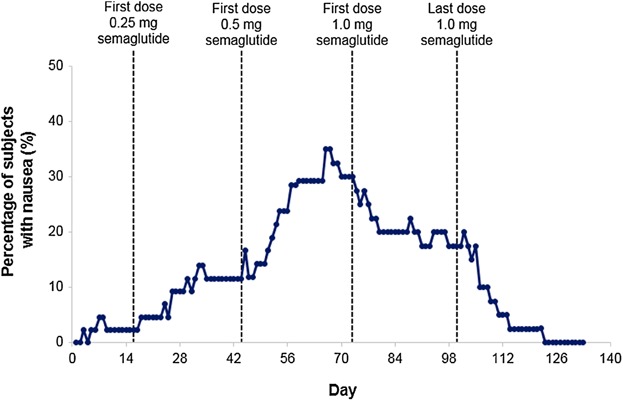
Percentage of patients with nausea by day: full analysis set (FAS; n = 43).
Based on FPG measured at the study site and self-measured FPG at home, according to the American Diabetes Association (ADA) Classification,23 no cases of severe hypoglycemia were reported, 1 subject had documented symptomatic hypoglycemia (plasma glucose 3.8 mmol/L), and 1 subject had asymptomatic hypoglycemia (plasma glucose 3.8–3.9 mmol/L).
The mean change in systolic blood pressure from baseline to just before initiation of semaglutide treatment was –8.1 mm Hg and from baseline to the end of treatment (1.0 mg) was –10.9 mm Hg. Over the same periods, the mean changes in diastolic blood pressure were –6.1 mm Hg and –5.8 mm Hg, respectively, and pulse rates were –2.3 and 4.0 bpm.
On the last day of oral contraceptive and semaglutide coadministration, 4 subjects had elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations that were reported as AEs, including 3 subjects with ALT ≥3x the upper limit of normal (ULN; 106 U/L, 119 U/L and 141 U/L, respectively, normal laboratory range 0–34 U/L). None of the cases met the criteria for Hy's law,24 and all had normal or slightly elevated bilirubin and normal alkaline phosphatase levels. Elevated ALT and AST concentrations had resolved prior to the protocol-scheduled follow-up visit (5 weeks after the last semaglutide dose).
There was a treatment-related increase in serum amylase concentration (mean ± SD) from baseline to the end of semaglutide treatment of 1.5 ± 15.3 U/L and to follow-up of 7.0 ± 14.1 U/L. No value was ≥3x ULN. Over the same periods, there was an increase in serum lipase concentration (mean ± SD) of 9.4 ± 24.3 U/L and 10.7 ± 32.6 U/L, respectively. Two subjects had values ≥3x ULN; 1 subject on the last day of semaglutide treatment and 1 subject at follow-up. Both were asymptomatic and resolved spontaneously, 3 months and 3 weeks later, respectively. No cases of pancreatitis were reported in the trial.
No antisemaglutide antibodies were detected in any subject and no injection-site reactions were reported. No clinically meaningful changes were observed in ECG or safety laboratory parameters, including calcitonin.
Discussion
Semaglutide did not reduce the bioavailability of ethinylestradiol or levonorgestrel. The prespecified bioequivalence criterion was met for ethinylestradiol but not for levonorgestrel; mean exposure was 20% higher at semaglutide steady-state. These findings are consistent with results of previous studies with GLP-1 receptor agonists, suggesting slightly higher total exposure of levonorgestrel with liraglutide16 or exenatide,18 and norethindrone with albiglutide.25 These results are not considered to be clinically relevant and are not expected to impact the efficacy or safety of these agents.16,18,25 Importantly, based on the observations in the present study, it is unlikely that coadministration with semaglutide would reduce the effectiveness of these combined oral contraceptive medications. This is an important result, in regard to low-dose oral contraceptive medications that are dependent on threshold concentrations for efficacy. Other pharmacokinetic parameters for ethinylestradiol and levonorgestrel, including Cmax, were similar before and during semaglutide treatment. The slight delay of tmax for both ethinylestradiol and levonorgestrel during the semaglutide steady-state period is consistent with the minor delay in the rate of gastric emptying reported with other GLP-1 receptor agonists.16,18,25,26
In this study, drug-drug interactions were performed at steady state for both semaglutide (at the highest intended clinical dose level) and the oral contraceptive, according to EMA and FDA guidelines.22,23 Postmenopausal women were selected for this study to avoid the potential effect of physiological hormonal changes during the menstrual cycle that may influence the pharmacokinetics of the drugs. Results from other studies suggest that the pharmacokinetics of this type of combined oral contraceptive are likely to be similar in postmenopausal women and women of childbearing age.27
Furthermore, this study confirmed the pharmacokinetic compatibility of semaglutide for once-weekly administration—ie, a long t1/2 and a low rate of apparent total plasma clearance. Long-acting GLP-1 receptor agonists, such as semaglutide, offer subjects with T2D greater convenience, together with the potential to improve treatment compliance and hence outcomes.
In this study, once-weekly semaglutide was also shown to provide clinically meaningful reductions in HbA1c, FPG, and body weight in postmenopausal women with T2D. Although the absence of a control arm means that the pharmacodynamic data should be interpreted with caution, the results are in line with a previously reported phase II semaglutide trial of the same duration.28 The rate of hypoglycemic events was low; only 1 subject had documented symptomatic hypoglycemia, according to ADA criteria.23 These findings indicate that semaglutide is highly effective at controlling blood glucose levels in combination with a low risk of hypoglycemia.
The safety results are as expected for a once-weekly GLP-1 receptor agonist; however, this study lacked a control arm.17 The most frequently reported TEAEs were gastrointestinal, which were mild or moderate. The incidence of nausea increased during the dose-escalation period, and then gradually decreased over time, despite the increase in semaglutide plasma concentration to steady-state, indicating tolerance development. Other long-acting GLP-1 receptor agonists have also been reported to exhibit improved tolerability to nausea over time, although the mechanism is unknown.29 No serious AEs were reported.
As described for other GLP-1 agonists and DPP-4 inhibitor-based therapies, some subjects experienced an asymptomatic increase in amylase and lipase levels.30 The clinical significance of this observation is unknown. Some subjects experienced elevated levels of hepatic enzymes during coadministration of the oral contraceptive and semaglutide. None of the cases met the criteria for Hy's law for drug-induced liver injury,24 and, based on normal bilirubin and alkaline phosphatase levels, cholestasis was not indicated. Abnormal liver function tests had resolved prior to follow-up. It is unclear if these effects were associated with the oral contraceptive, semaglutide, or with coadministration of semaglutide and the oral contraceptive. Although an effect on liver parameters is noted in the Microgyn® Summary of Product Characteristics,31 elevated liver enzymes were not observed following administration of oral contraceptive during the semaglutide-free period. In a phase II trial of semaglutide, no treatment effects on hepatic enzymes or transaminases were observed (data on file). Further investigation is warranted.
There were no effects on calcitonin levels and no antisemaglutide antibodies or injection-site reactions were observed. There were no cases of pancreatitis reported in this study.
In conclusion, this study—the first to investigate the intended clinical dose and the anticipated dose-escalation regimen of semaglutide—shows that treatment with semaglutide does not decrease the bioavailability of ethinylestradiol or levonorgestrel in postmenopausal women with T2D. Therefore, the coadministration of semaglutide is not expected to affect the efficacy of oral contraceptive medications. Furthermore, the study confirms the pharmacokinetic compatibility of semaglutide for once-weekly dosing and corroborates recent phase II study results, with regard to improved glycemic control and reduced body weight, in combination with a low risk of hypoglycemia.28 With dose escalation, the incidence of nausea declined, indicating tolerance development. The elevated liver enzyme results will be further investigated in the ongoing semaglutide phase III clinical program. The semaglutide dose and dose-escalation regimen used in this study have been selected for the further development of semaglutide.
Acknowledgments
CK acts as a guarantor and takes responsibility for the content of the paper. The authors thank all subjects who took part in this study. The authors also thank Novo Nordisk A/S for their contribution in the study design, data management, and data analysis. Editorial assistance was provided by Dr Ian Seymour from AXON Communications, and funded by Novo Nordisk A/S.
Declaration of Conflicting Interests
The study was sponsored by Novo Nordisk A/S, the manufacturer of semaglutide. CK is a shareholder of Profil, which has received research grants from Novo Nordisk. LN did not have a conflict of interest. AF, HH, CJ, and LJ are employees in and own shares in Novo Nordisk A/S.
Author Contributions
CJ and AF designed the study. CJ, AF, LJ, CK, and LN conducted the study and collected the data. CJ, AF, LJ, HH, CK, and LN performed the data analysis, wrote, and revised the manuscript. All authors gave final proof of the manuscript.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Table S1. Baseline characteristics (mean ± SD).
References
- 1.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 2.Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101(3):515–520. doi: 10.1172/JCI990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naslund E, Barkeling B, King N, et al. Energy intake and appetite are suppressed by glucagon-like peptide-1 (GLP-1) in obese men. Int J Obes Relat Metab Disord. 1999;23(3):304–311. doi: 10.1038/sj.ijo.0800818. [DOI] [PubMed] [Google Scholar]
- 4.Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7–36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab. 1996;81(1):327–332. doi: 10.1210/jcem.81.1.8550773. [DOI] [PubMed] [Google Scholar]
- 5.Pratley RE, Gilbert M. Targeting incretins in Type 2 diabetes: role of GLP-1 receptor agonists and DPP-4 inhibitors. Rev Diabet Stud. 2008;5(2):73–94. doi: 10.1900/RDS.2008.5.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garber AJ. Novel incretin-based agents and practical regimens to meet needs and treatment goals of patients with type 2 diabetes mellitus. J Am Osteopath Assoc. 2011;111(7 Suppl 5):S20–S30. [PubMed] [Google Scholar]
- 7.Martin JH, Deacon CF, Gorrell MD, Prins JB. Incretin-based therapies—review of the physiology, pharmacology and emerging clinical experience. Intern Med J. 2011;41(4):299–307. doi: 10.1111/j.1445-5994.2011.02439.x. [DOI] [PubMed] [Google Scholar]
- 8.Koliaki C, Doupis J. Incretin-based therapy: a powerful and promising weapon in the treatment of type 2 diabetes mellitus. Diabetes Ther. 2011;2(2):101–121. doi: 10.1007/s13300-011-0002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deacon CF. Potential of liraglutide in the treatment of patients with type 2 diabetes. Vasc Health Risk Manag. 2009;5(1):199–211. doi: 10.2147/vhrm.s4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drucker DJ, Dritselis A, Kirkpatrick P. Liraglutide. Nat Rev Drug Discov. 2010;9(4):267–268. doi: 10.1038/nrd3148. [DOI] [PubMed] [Google Scholar]
- 11.Wadden T, Hollander P, Klein S, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond) 2013;37(11):1443–1451. doi: 10.1038/ijo.2013.120. [DOI] [PubMed] [Google Scholar]
- 12.Nauck MA, Petrie JR, Sesti G. The once-weekly human GLP-1 analogue semaglutide provides significant reductions in HbA1c and body weight in patients with type 2 diabetes. Diabetologia. 2012;55(Suppl 1):S7(2-OP). [Google Scholar]
- 13.Gotfredsen CF, Molck AM, Thorup I, et al. The human GLP-1 analogs liraglutide and semaglutide: Absence of histopathological effects on the pancreas in nonhuman primates. Diabetes. 2014;63(7):2486–2497. doi: 10.2337/db13-1087. [DOI] [PubMed] [Google Scholar]
- 14. Novo Nordisk.;1; Data on file, 2013.
- 15.Rhoda Lee C. Drug interactions and hormonal contraception. Trends Urol. Gynecol Sexual Health. 2009;14(3):23–26. [Google Scholar]
- 16.Jacobsen LV, Vouis J, Hindsberger C, Zdravkovic M. Treatment with liraglutide—a once-daily GLP-1 analog—does not reduce the bioavailability of ethinyl estradiol/levonorgestrel taken as an oral combination contraceptive drug. J Clin Pharmacol. 2011;51(12):1696–1703. doi: 10.1177/0091270010389471. [DOI] [PubMed] [Google Scholar]
- 17.Tzefos M, Harris K, Brackett A. Clinical efficacy and safety of once-weekly glucagon-like peptide-1 agonists in development for treatment of type 2 diabetes mellitus in adults. Ann Pharmacother. 2012;46(1):68–78. doi: 10.1345/aph.1Q379. [DOI] [PubMed] [Google Scholar]
- 18.Kothare PA, Seger ME, Northrup J, Mace K, Mitchell MI, Linnebjerg H. Effect of exenatide on the pharmacokinetics of a combination oral contraceptive in healthy women: an open-label, randomised, crossover trial. BMC Clin Pharmacol. 2012;12:8. doi: 10.1186/1472-6904-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Harmonised Tripartite Guideline for Good Clinical Practice E6 (R1). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf.
- 20.World Medical Association. Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. 52nd WMA General Assembly, Edinburgh, Scotland, October 2000. Last amended with Note of Clarification on Paragraph 29 by the WMA General Assembly, Washington 2002; and Note of Clarification on Paragraph 30 by the WMA General assembly, Tokyo 2004. http://www.wma.net/en/30publications/10policies/b3/index.html.
- 21.US Food and Drug Administration (FDA) Guidance for Industry. Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf.
- 22.European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions. CPMP/EWP/560/95/Rev. 1 Corr, June 21, 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf.
- 23.American Diabetes Association. Defining and reporting hypoglycemia in diabetes: a report from the American Diabetes Association Workgroup on Hypoglycemia. Diabetes Care. 2005;28(5):1245–1249. doi: 10.2337/diacare.28.5.1245. [DOI] [PubMed] [Google Scholar]
- 24.Temple R. Hy's law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241–243. doi: 10.1002/pds.1211. [DOI] [PubMed] [Google Scholar]
- 25.Bush M, Scott R, Watanalumlerd P, Zhi H, Lewis E. Effects of multiple doses of albiglutide on the pharmacokinetics, pharmacodynamics, and safety of digoxin, warfarin, or a low-dose oral contraceptive. Postgrad Med. 2012;124(6):55–72. doi: 10.3810/pgm.2012.11.2613. [DOI] [PubMed] [Google Scholar]
- 26.Kapitza C, Flint A, Hindsberger C, Zdravkovic M. The effect of the once-daily human GLP-1 analogue liraglutide on the pharmacokinetics of paracetamol. Diabetes Care. 2008;57(suppl 1):A593. [Google Scholar]
- 27.Blode H, Schurmann R, Benda N. Novel ethinyl estradiol-beta-cyclodextrin clathrate formulation does not influence the relative bioavailability of ethinyl estradiol or coadministered drospirenone. Contraception. 2008;77(3):171–176. doi: 10.1016/j.contraception.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Nauck MA, Petrie JR, Sesti G, et al. The once-weekly human GLP-1 analogue semaglutide provides significant reductions in HbA1c and body weight in patients with type 2 diabetes. EASD. 2012 Oral presentation 2 (Trial: NN9535–1821) [Google Scholar]
- 29.Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- 30.Lando HM, Alattar M, Dua AP. Elevated amylase and lipase levels in patients using glucagonlike peptide-1 receptor agonists or dipeptidyl-peptidase-4 inhibitors in the outpatient setting. Endocr Pract. 2012;18(4):472–477. doi: 10.4158/EP11290.OR. [DOI] [PubMed] [Google Scholar]
- 31. Summary of Product Characteristics. Microgynon® 30. http://www.medicines.org.uk/emc/medicine/1827/spc.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Baseline characteristics (mean ± SD).