

Abstract
Aims
The SDF-1α/CXCR4 dyad was previously shown by us and others to be instrumental in intimal hyperplasia as well as early stage atherosclerosis. We here sought to investigate its impact on clinically relevant stages of atherosclerosis in mouse and man.
Methods and results
Immunohistochemical analysis of CXCR4 expression in human atherosclerotic lesions revealed a progressive accumulation of CXCR4+ cells during plaque progression. To address causal involvement of CXCR4 in advanced stages of atherosclerosis we reconstituted LDLr−/− mice with autologous bone marrow infected with lentivirus encoding SDF-1α antagonist or CXCR4 degrakine, which effects proteasomal degradation of CXCR4. Functional CXCR4 blockade led to progressive plaque expansion with disease progression, while also promoting intraplaque haemorrhage. Moreover, CXCR4 knockdown was seen to augment endothelial adhesion of neutrophils. Concordant with this finding, inhibition of CXCR4 function increased adhesive capacity and reduced apoptosis of neutrophils and resulted in hyperactivation of circulating neutrophils. Compatible with a role of the neutrophil CXCR4 in end-stage atherosclerosis, CXCR4 expression by circulating neutrophils was lowered in patients with acute cardiovascular syndromes.
Conclusion
In conclusion, CXCR4 contributes to later stages of plaque progression by perturbing neutrophil function.
Keywords: atherosclerosis, neutrophils, CXCR4, SDF-1α, senescence
1. Introduction
Leukocyte recruitment to the plaque is a crucial step in the progression of atherosclerosis [1–3]. Chemokines are chemotactic cytokines involved in the recruitment of leukocytes to the plaque [4–6] and several members of the CC- and CXC-chemokine family and their receptors have been implicated in the progression of cardiovascular disease. One of these CXC chemokines, CXCL12 (SDF-1α), was demonstrated to be expressed by vascular smooth muscle cells (vSMCs), endothelial cells, and macrophages in atherosclerotic but not healthy arteries [7,9]. Although these findings may suggest a potential role in atherosclerosis, the exact role of the SDF-1α/CXCR4 axis in atherosclerosis has long been unclear. A number of reports pointed to a pro-atherosclerotic role for SDF-1α due to its participation in platelet activation and leukocyte chemotaxis [7–9], while blockade of the SDF-1α/CXCR4 axis was demonstrated to inhibit neointima formation [10]. Conversely, Damås and colleagues demonstrated reduced SDF-1α levels in patients with unstable angina pectoris, and a causal role in this disease was inferred from the anti-inflammatory and matrix-stabilizing effects of SDF-1α [8]. This notion was corroborated by recent study by us showing that systemic blockade of CXCR4 by the CXCR4 antagonist AMD3465 enhanced plaque initiation due to increased neutrophil recruitment [11].
In this study, we mapped the actual expression levels of SDF-1α and its receptor CXCR4 at different stages of human atherosclerosis. The observed differential expression of SDF-1α/CXCR4 expression in human atherosclerosis led us to investigate effects of CXCR4 blockade on plaque formation and progression in mice by a loss-of-function approach using lentivirus encoding SDF-1α antagonist P2G [12] (LV.SDF-1α(P2G)) or CXCR4 degrakine (LV.CXCR4deg), which effects proteasomal CXCR4 degradation [13]. Our human and experimental data support a role of the CXCR4 axis in advanced atherosclerosis, potentially by regulating neutrophil senescence, and neutrophils adhesive and functional capacity.
2. Materials and Methods
For the detailed Methods section, see Supplementary material online.
2.1. Human cohort studies
2.1.1. Plaque analysis
Carotid artery samples were obtained either from patients undergoing carotid endarterectomy (Academic Hospital Maastricht or Maasland Hospital, Sittard, The Netherlands) or from autopsies performed at Department of Pathology, Academic Hospital Maastricht (The Netherlands). Carotid plaque tissue was classified for progression stage according to the criteria designed by Virmani and coworkers [14]. All human work was approved by the Ethical Committee of the University Hospital Sittard/Maastricht and was performed in accordance with the “Code for Proper Secondary Use of Human Tissue”.
Two further transcriptomics studies have been interrogated to corroborate the results from this study: (1) advanced versus early plaques from the GSE28829 study, and (2) advanced stable versus unstable plaques from the human plaque transcriptomics study [15].
2.1.2. Blood leukocyte analysis
To measure CXCR4 protein expression on human circulating granulocytes, granulocytes were purified from freshly isolated blood specimen from a cohort of healthy controls (n = 13), carotid endarterectomy (CEA, n = 5) and unstable angina pectoris patients (UAP, n = 19) using Lymphoprep™ according to the manufacturer’s protocol. This human work was approved by the Ethical Committee of the University Hospital Oslo. Signed informed consent for participation in the study was obtained from all individuals. Granulocytes were lysed and CXCR4 protein expression was determined by western blot analysis using a CXCR4 specific antibody (Abcam, Cambridge, USA). Detailed methodology of these studies is provided in the Supplemental Method section.
2.2. Animals
All animal work was approved by the regulatory authority of Leiden and Maastricht University and performed in compliance with the Dutch government guidelines. Female LDLr−/− mice received a bone marrow transplantation with bone marrow cells infected with lentivirus (LV). Empty, LV.SDF-1α(P2G) or LV.CXCR4deg (m.o.i. = 15) [16]. After 6 weeks of recovery, mice were placed on a Western type diet for another 6 (n = 8 per group) and 10 weeks (n = 8–11 per group). Mice were anaesthetized by subcutaneous injection of ketamine (60 mg/kg, Eurovet Animal Health, Bladel, the Netherlands), fentanyl citrate and fluanisone (1.26 mg/kg and 2 mg/kg respectively, Janssen Animal Health, Sauderton, UK).
2.3. Neutrophil differentiation in vitro
HL60 cells were differentiated into neutrophils by incubating them for 5–7 days in medium containing 2.5 μM Retinoic Acid (RA) either in the presence or absence of the CXCR4 antagonist AMD3100 (500 ng/mL) [17,18]. To measure proliferation, the differentiated cells were incubated with [3H]thymidine (Amersham, Uppsala, Sweden). To measure cell survival, HL60 cells were stained with Annexin V and propidium iodide (Sigma-Aldrich); cell survival rate (defined by non-apoptotic, non-necrotic cells) was determined by flow cytometry analysis. To determine neutrophil myeloperoxidase activity in the presence or absence of CXCR4 antagonist AMD3100, cells were lysed in Triton X-100 (Fluka, Zwijndrecht, the Netherlands). Cell lysate was diluted with 10 mM citrate (pH 5) after which substrate (100 μg/mL TMB, 0.003% H2O2, 13 μg/mL recorsinol in 10 mM citrate (pH5)) was added. The reaction was stopped with 2 M H2SO4 and absorbance was read at 450 nm. Total RNA was extracted, reverse transcribed using M-MuLV reverse transcriptase (RevertAid, MBI Fermentas, Leon-Roth, Germany) and gene expression (Table 1) was measured by qPCR as described previously [19].
Table 1.
Primer sequences used for RT-PCR analysis (m = murine, h = human).
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| mFAK | GAGAATCCAGCTTTGGCTGTT | GGCTTCTTGAAGGAACTTCT |
| mAKT | GGTATTTTGATGAGGAGTTCACG | ACACACTCCATGCTGTCATCTT |
| mCXCR4 | GCCTTATCCTGCCTGGTATTGTC | GCGAAGAAAGCCAGGATGAGGAT |
| mSDF-1α | CTGTGCCCTTCAGATTGTTG | TAATTTCGGGTCAATGCACA |
| hCXCR4 | CTGCTGACTATTCCCGACTT | TGAAACTGGAACACAACCAC |
| hSDF-1α | GATTCTTCGAAAGCCATGTT | CACTTTAGCTTCGGGTCAAT |
2.4. Neutrophil senescence
LDLr−/− mice fed a Western type diet received daily an intraperitoneal injection of AMD3100 (2 μg/mouse) or PBS. Neutrophil senescence was assessed by flow cytometry analysis (CD11b+Ly6GhighDHR123+) on blood leukocytes, after incubation at 37 °C with 1 μM dihydrorhodamine 123 (DHR123) and subsequent stimulation for 30 min with 20 μg/ml phorbol myristate acetate (PMA) to allow generation of intracellular H2O2.
2.5. Statistical analysis
Data are expressed as mean ± SEM. A 2-tailed Student’s t-test was used to compare individual groups. To determine the significance of the relative mRNA expression levels, statistical analysis was performed on Ct values. Non-parametric data were analyzed using a Mann–Whitney U test. Microarray data were processed as described in the supplemental methods section, and presented significances were adjusted for multiple testing. Multiple groups were analyzed using ANOVA with a subsequent Newman–Keuls Multiple comparisons post test. Contingency data were analyzed by means of the Fisher’s exact test. Data were considered statistically significant when P < 0.05.
3. Results
3.1. Differential expression of CXCR4 during human and mouse atherosclerotic lesion progression
First we have mapped CXCR4 and SDF-1α expression in atherosclerotic human carotid artery tissue obtained by endarterectomy (advanced stable versus advanced unstable plaque study; cohort 1) or after autopsy (early versus advanced stable plaque study; cohort 1) (for definitions of plaque stage see methods and supplemental methods sections). For all microarray studies tissues were sectioned and used for histology or transcriptomic analysis. The progression stage of transcriptomics sections was considered early (intimal thickening, IT), advanced stable (thick fibrous cap atheroma, TfcA) or advanced unstable (thin capped fibroatheroma with intraplaque hemorrharge (IPH), cap break or dissection, or intra/extramural thrombi) if both of the adjacent sections were graded as such according to the morphological criteria of Virmani and coworkers [14]. Microarray (Fig. 1A) showed that CXCR4 expression was progressively up-regulated throughout disease progression, being significantly higher in advanced stable versus early plaques and most pronounced in advanced unstable lesions. This pattern could be confirmed in independent validation cohorts (cohorts 2), representing microarray studies of advanced versus early stable plaques (GSE28829; autopsy material) and of advanced unstable versus advanced stable plaques (endarterectomy specimen [15]). Increased SDF-1α mRNA expression was shown in human advanced stable versus early atherosclerotic lesions, whereas SDF-1α expression was significantly reduced in advanced unstable versus advanced stable lesions (Fig. 1A). Of note, both SDF-1α and CXCR4 mRNA expression levels were increased in atherosclerotic plaques of LDLr−/− in mice as compared to a non-diseased artery (Supplemental Fig. 1). Next, SDF-1α and CXCR4 mRNA expression data were verified at protein level by immunohistochemistry. Both proteins could be detected in normal arteries (Fig. 1B). SDF-1α expression was primarily localized in intimal leukocytes and medial vSMCs, and its expression decreased during plaque progression (Figs. 1B and C). These findings were confirmed by subsequent co-staining showing moderate expression of SDF-1α in plaque macrophages (Supplemental Fig. 2A), vSMC (Supplemental Fig. 2B) and CD3+ T cells (Supplemental Fig. 2C). CXCR4 protein expression in advanced lesions was higher than in early lesions. However ruptured lesions showed an altered CXCR4 expression pattern, with hotspots at sites of neovascularization, but a reduction in overall staining intensity, which contrasts with the observed increment in mRNA expression (Figs. 1B and C). Co-staining showed moderate expression of CXCR4 by foam cells (Supplemental Fig. 3A) and by vSMC (Supplemental Fig. 3B). CXCR4 was only expressed by a fraction of plaque macrophages and vSMC, and we hypothesize that its expression may be linked to the differentiation stage of these cell subsets. In addition, almost all CD3+ T cells in the plaque were seen to express CXCR4 (Supplemental Fig. 3C). Plaque neutrophils were also seen to express CXCR4 (Supplemental Fig. 3D). A mouse IgG2b isotype control did not show any staining (Fig. 1D).
Fig. 1. Analysis of CXCR4 and SDF-1α expression in atherosclerotic human carotid artery plaques.

A, Microarray analyses of CXCR4 and SDF-1α mRNA expression in human atherosclerosis. Four two-armed plaque cohorts were analyzed for mRNA expression patterns: advanced versus early stable carotid artery plaque cohorts obtained at autopsy (advanced:early cohort 1; n = 9 and 8, resp), advanced versus early stable carotid artery plaque cohort from GSE28829 which served as validation cohort (early/advanced cohort 2); advanced unstable versus stable plaque cohort (n = 12 and 3, respectively, unstable:stable cohort 1); a second advanced unstable versus stable plaque cohort from the human plaque transcriptomics study [16] (n = 22 and 22, stable/unstable cohort 2), both obtained by endarterectomy surgery. Values represent relative CXCR4 expression intensity in the first versus the second tissue component; all expression values were false-discovery-rate (FDR) corrected (*P < 0.05, **P < 0.01) B, CXCR4 and SDF-1α protein expression in non-diseased arteries and early lesions, as well as in advanced and ruptured plaques (magnification: 100×). Histological classification is described in the supplemental methods (* = lumen, I = intima and # = media). C, CXCR4 and SDF-1α expression by intimal cells (magnification: 400×). D, Mouse isotype IgG2b control (magnification: 100×).
3.2. Effective impairment of CXCR4 function by SDF-1α antagonist and CXCR4 degrakine lentivirus in mice
The progressively altered SDF-1α/CXCR4 expression in human atherosclerosis led us to investigate effects of CXCR4 blockade at different stages of plaque formation by a lentivirus loss-of-function approach. Lentiviruses were generated encoding CXCR4 degrakine (LV.CXCR4deg) or SDF-1α antagonist (LV.SDF1α(P2G)) under the CMV promotor. An empty lentivector (LV.Empty) was used as control. To establish functionality of the lentivector, basal and SDF-1α induced motility of FDCP-MixA4 cells in the absence or presence of LV.SDF-1α(P2G) was measured in a slope-well assay (Supplemental Fig. 4). SDF-1α induced cell migration was completely blocked upon LV.SDF-1α (P2G) or LV.CXCR4deg infection (multiplicity of infection, MOI: 15) (Fig. 2A), comparable to infection with a lentivirus containing a shRNA sequence targeting CXCR4 (shX4HM), while SDF-1α was able to induce LV.Empty infected cell migration. Neutrophils isolated from mice transplanted with either LV.Empty or LV.CXCR4deg infected bone marrow displayed similarly reduced migration in response to SDF-1α (Fig. 2B).
Fig. 2. In vitro and in vivo analysis of functionality of lentiviral CXCR4 degrakine and SDF-1α antagonist.
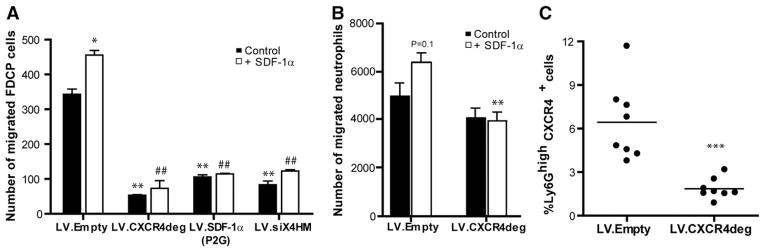
A, Quantification of migrated FDCP-MixA4 cells infected with LV.Empty, LV.SDF1-α(P2G), LV.CXCR4deg or a shRNA targeting CXCR4 (shX4HM) without stimulation (black bars) or in response to SDF-1α (white bars) in a slope well assay. (*P < 0.05 and **P < 0.01 compared to LV.Empty control, ##P < 0.01 compared to LV.Empty + SDF-1α). B, Quantification of migrated neutrophils, that were isolated from either LV.Empty control transplanted mice, or LV.CXCR4deg chimeras in a transwell migration assay. LV.CXCR4 deg expressing neutrophils migrated significantly less towards SDF-1α (**P < 0.01 compared to LV.Empty + SDF-1α). C, Flow cytometry analysis of relative circulating CXCR4+ neutrophils in LV.CXCR4deg versus LV.Empty chimeras (***P < 0.005).
Next, we tested LV.CXCR4deg functionality in vivo by reconstituting irradiated LDLr−/− mice with LV.CXCR4deg infected bone marrow. The total number of circulating CXCR4+ neutrophils (defined as CD11b+Ly6Ghigh cells as illustrated in Supplemental Fig. 5) was significantly reduced compared to that in control mice (Fig. 2C). When normalized to total neutrophil numbers, the percentage of CXCR4+ neutrophils was reduced from 41% ± 5% in the LV.Empty mice to 17 %± 3% in the LV.CXCR4deg mice (P < 0.001). Furthermore, CXCR4 expression (mean fluorescence intensity, MFI) per neutrophil was reduced from 43.4 ± 2.1 in LV.Empty to 34.9 ± 1.6 in LV.CXCR4deg mice (P < 0.01). CXCR4 expression on CD3+ T cells was reduced (−38%: 74.2 ± 12.8 in controls versus 45.5 ± 5.8 in LV.CXCR4deg mice), as well as on CD19+ B cells (−30%, MFI: 65.5 ± 9.0 versus 46.3 ± 5.6) and F4/80+ cells (−30%, MFI: 55.9 ± 11.7 versus 31.8 ± 1.6) in LV.CXCR4deg bone marrow transplanted mice compared to LV.Empty controls at 16 weeks after bone marrow transplantation, demonstrating that LV.CXCR4deg is an efficient tool to reduce CXCR4 protein levels on leukocytes.
3.3. Hematopoietic CXCR4 deficiency aggravates atherosclerotic lesion progression and induces intraplaque hemorrhages in LDLr−/− mice
To address the role of CXCR4 blockade on atherosclerosis, we examined lesion development and progression in LDLr−/− mice reconstituted with LV.CXCR4deg and LV.SDF-1α(P2G) infected bone marrow and fed a Western type diet. For plaque initiation and progression, aortic root lesions were examined after 6 and 10 weeks of Western type diet feeding, respectively. CXCR4 blockade increased plaque progression in LV.CXCR4deg treated mice compared to control mice (10 weeks after Western type diet; Fig. 3A, left panel and Supplemental Figs. 6A,B). Lesion progression also tended to be increased in LV.SDF-1α (P2G) bone marrow reconstituted mice, however this did not reach significance (P = 0.06). Atherosclerotic lesion development was not notably affected in the plaque initiation study (6 weeks of Western type diet feeding, Fig. 3A, right panel and Supplemental Figs. 6A,B). Interestingly, more lesions (5/8) of LV.CXCR4deg chimeras in the plaque initiation study displayed intraplaque hemorrhages (IPH) compared to LV.Empty controls (1/8) (Fig. 3B), and the area of extravasated intraplaque erythrocytes was enlarged in LV.CXCR4deg lesions (Fig. 3C). Analysis of the iron content of advanced lesions showed somewhat increased Perl’s Iron staining from 0.12% ± 0.05% in the LV.Empty group to 1.1% ± 0.8% in the LV.CXCR4deg mice, which may be illustrative of previous hemorrhage. There was no difference in plaque collagen and vSMC content (data not shown), while plaque macrophage content was unaltered as well (Fig. 3D). In addition, plaque T cell content did not differ between groups (Supplemental Fig. 6C). While as expected B cells were completely absent within the lesions, in the adventitia a few scattered B cells could be detected (data not shown). During the experiments, no differences were noticed between treatment groups in total body weight, plasma total cholesterol levels and lipid distribution (Fig. 3E).
Fig. 3. CXCR4 and SDF-1α lentiviral blockade deteriorates atherosclerotic plaque progression.

LDLr−/− mice were transplanted with LV.empty, LV.CXCR4deg or LV.SDF1(P2G) bone marrow and placed on Western type diet for 6 (plaque initiation study, n = 8 per group) and 10 weeks (plaque progression study, n = 8–11 per group). A, Oil-Red-O staining of the aortic root. Blockade of CXCR4 by LV.CXCR4deg aggravated lesion progression after 10 weeks of diet feeding in LDLr−/− mice (**P < 0.01 compared to LV.Empty, left graph). Similarly, hematopoietic overexpression of the SDF-1α(P2G) antagonist tended to induce lesion progression after 10 weeks of diet feeding (P = 0.06 compared to LV.Empty, left graph). Right panel: representative lesions of each group (50× magnification). Initial plaque development as measured after 6 weeks of western-type diet feeding was not affected by CXCR4 blockade (P = 0.25 compared to LV.Empty, right graph). B, Intraplaque hemorrhage in a LV.CXCR4deg plaque indicated by the arrow (upper left: 50× magnification, lower right: 200× magnification). C, Increased intraplaque hemorrhage was observed as measured by the relative erythrocyte surface area in plaques of LV.CXCR4deg chimeras (*P < 0.01). Data are represented for the 6 weeks group. D, Plaque macrophage (MOMA-2) content, represented as the percentage of MOMA2+ cells per plaque area. Data are represented for the 10 weeks group. E, Plasma cholesterol level distribution did not differ between the groups.
3.4. Neutrophils show increased plaque adherence in the absence of CXCR4
In mouse plaques, neutrophils were seen to accumulate during lesion progression (Supplemental Fig. 7). As the SDF-1/CXCR4 dyad is important for neutrophil homeostasis, we analyzed effects on neutrophils in our study. The total number of CD11b+Ly6GhighCD71− blood neutrophils remained unaffected in LV.CXCR4deg versus LV.Empty transplanted chimeras (Fig. 4A). The number of intraplaque neutrophils was somewhat increased in the plaque initiation study (32 ± 10 neutrophils versus 21 ± 6 in controls), while neutrophil numbers decreased during plaque progression in both groups (data not shown). Interestingly, neutrophil adhesion to the plaque was highly increased in the LV.CXCR4deg transplanted mice compared to LV.Empty control mice both at 6 and 10 weeks of Western type diet feeding (Fig. 4B). Next, we addressed effects of CXCR4 blockage on neutrophil adherence in vitro. HL60 cells were differentiated into neutrophils with retinoic acid (Fig. 4C) [17,18]. Flow cytometry analysis confirmed that retinoic acid induced neutrophil differentiation as demonstrated by increased CD11b+Ly6GhighCD71−neutrophil numbers (data not shown). The adhesive capacity of HL60 cells differentiated in the presence of AMD3100 and retinoic acid was increased compared to HL60 cells differentiated in the sole presence of retinoic acid (26 ± 2 versus 18 ± 2 cells/microscopic field, Fig. 4D). HL60 cell adherence to fibronectin coated wells was increased by 55%, however that of HL60 cells differentiated in the presence of AMD3100 was even 2-fold higher (202%) compared to uncoated wells. Adherence of differentiated HL60 cells to gelatin remained unaltered by AMD3100 treatment. The augmented adhesive capacity was associated with altered outside in signaling as witness the 3-fold increased relative focal adhesion kinase (FAK) mRNA expression in HL60 derived neutrophils differentiated in the presence of AMD3100 (Fig. 4E). To confirm these in vitro findings, we next isolated bone marrow neutrophils from LV.Empty and LV.CXCR4deg transplanted mice and tested them for adhesive capacity to matrix and endothelial cells. Similarly as when using differentiated HL60 myeloblasts, adhesion of LV.CXCR4deg neutrophils to fibronectin coated wells was significantly increased as compared to LV.Empty control neutrophils (Fig. 4F). Furthermore, relative FAK mRNA expression was increased almost 2-fold from 1.0 ± 0.4 *10−4 in LV.Empty control neutrophils to 1.9 ± 0.6*10−4 in LV.CXCR4deg neutrophils. In addition, neutrophil adhesion from LV.SDF-1α(P2G) expressing neutrophils to murine endothelial cells (H5V cells) was significantly increased as compared to LV.Empty neutrophils, while also the adhesion of LV.CXCR4deg expressing neutrophils tended to be increased (Fig. 4G).
Fig. 4. CXCR4 blockage induces neutrophil adhesion.

A, Flow cytometry analysis of the percentage of Ly6GhighCD11b+(CD71−) neutrophils in blood from LV.Empty (n = 4) versus LV.CXCR4deg (n = 4) bone marrow transplanted mice. Data are represented for the 6 weeks group. B, Napthol CAE staining showing adhering neutrophils to the plaque endothelium (left panel, upper picture: 400× magnification, lower picture: 1000× magnification). Represented in the graphs are the number of adhering neutrophils per endothelial cell length in LV.Empty versus LV.CXCR4deg transplanted LDLr−/− bone marrow chimeras 6 and 10 weeks after Western type diet feeding (**P < 0.01 compared to LV.Empty). C, HL60 cells were treated with retinoic acid to induce neutrophil development. D, Adhesion of non-treated versus AMD3100 treated neutrophils in control, fibronectin (FN) and gelatin coated wells. *P < 0.05 compared to HL60 + RA, ##P < 0.01 compared to uncoated controls, **P < 0.01 compared to HL60 + RA (FN). E, Relative gene expression of FAK in neutrophils differentiated in the presence or absence of AMD3100 (*P < 0.05). F, Adhesion of neutrophils isolated from bone marrow of LV.Empty versus LV.CXCR4deg transplanted mice to either control, fibronectin (FN) and gelatin coated wells. **P < 0.05 compared to LV.control, ###P < 0.001 compared to uncoated controls. G, Adhesion of neutrophils isolated from bone marrow of LV.Empty, LV.SDF-1α(P2G), LV.CXCR4deg transplanted mice to mouse endothelial cells. *P < 0.05 compared to LV.Empty control.
We thus hypothesize that in our in vivo model, as the percentage of circulating CXCR4+ neutrophils is reduced by over 70%, the neutrophils adhering to the plaque are predominantly CXCR4low or even negative. Taken together, these findings indicate that disruption of the CXCR4/SDF-1α axis affects neutrophil adhesion in vitro as well as in vivo.
3.5. Impaired in vivo homing of neutrophils in response to SDF-1α but not KC and thioglycollate in LV.CXCR4deg transplanted mice
The increased adherent plaque neutrophils in LV.CXCR4deg mice led us to investigate effects of CXCR4 blockade on neutrophil migration in vivo. Leukocyte recruitment towards the peritoneum was measured in LDLr−/− mice, transplanted with either bone marrow infected with LV.Empty or LV.CXCR4deg, in response to PBS (control), KC or SDF-1α. Peritoneal homing of CD11b+Ly6Ghigh neutrophils in response to KC, which induces CXCR2 dependent neutrophil chemotaxis, did not differ between LV.Empty and LV.CXCR4deg chimeras (Fig. 5A). In contrast, the neutrophil migratory response to SDF-1α was decreased in LV.CXCR4deg transplanted mice (Fig. 5B). Importantly, CXCR4 blockade on leukocytes did not affect thioglycollate elicited peritoneal neutrophil recruitment in AMD3100 treated LDLr−/− mice (Fig. 5C), demonstrating that decreased CXCR4 expression on leukocytes per se does not affect neutrophil migration in vivo. Apparently, while SDF-1α induced neutrophil migration was impaired in LV.CXCR4 transplanted chimeras, the general migratory capacity of neutrophils remained unaltered.
Fig. 5. CXCR4 blockage does not alter neutrophil recruitment.
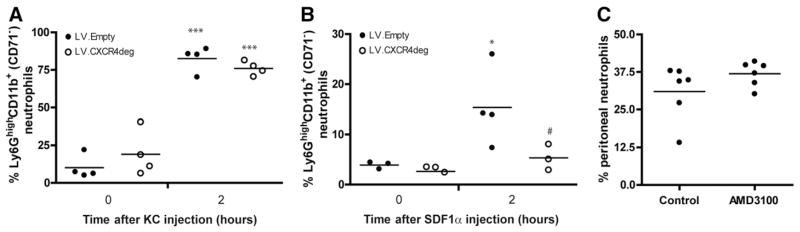
In vivo chemokinesis assay in LDLr−/− mice, transplanted with either bone marrow infected with LV.Empty or LV.CXCR4deg A, Percentage of peritoneal Ly6GhighCD11b+(CD71−) neutrophils at 2 h after KC injection. ***P < 0.001 compared to T = 0. B, Percentage of recruited neutrophils in response to SDF-1α. *P < 0.05 compared to T = 0, #P < 0.05 compared to LV.Empty. C, Percentage of peritoneal neutrophils in response to 3% Brewer’s Thioglycollate in control versus AMD3100 treated mice.
3.6. CXCR4 regulates neutrophil homeostasis
Focal adhesion kinase (FAK) expression is critical for cell survival. Therefore, we next studied the impact of CXCR4 dysfunction on neutrophil homeostasis. AMD3100 treatment throughout retinoic acid induced neutrophil differentiation was seen to inhibit the formation of Ly6GhighCXCR4+ and Ly6GhighCXCR2+ neutrophil subsets (Fig. 6A). Moreover, AMD3100 reduced HL60 cell proliferation during retinoic acid differentiation (Fig. 6B), while augmenting neutrophil myeloperoxidase (MPO) activity (Fig. 6C), illustrative of enhanced neutrophil activation. Furthermore, AMD3100 treated HL60 cells at 7 days after start of retinoic acid induced neutrophil differentiation showed increased cell survival (Fig. 6D). In line, expression of cell survival factor AKT was enhanced (Fig. 6E).
Fig. 6. Neutrophils lacking functional CXCR4 expression show increased cell survival.

A, Treatment of HL60 cells with AMD3100 inhibits differentiation of HL60 cells into neutrophils as demonstrated by a reduction in the percentage of Ly6GhighCXCR4+ (left panel) and Ly6GhighCXCR4+ cells (right panel) (**P < 0.01 compared to control HL60 cells, ##P < 0.01 compared to RA differentiated HL60 cells). B, AMD3100 reduced neutrophil precursor proliferation during differentiation (**P < 0.01). C, Myeloperoxidase activity (**P < 0.01). D, Percentage of cell survival (***P < 0.001). E, Relative gene expression of Akt in neutrophils differentiated in the presence or absence of AMD3100 (*P < 0.05).
The CXCR4 blockade associated cell survival response could well have impacted neutrophil senescence in vivo. Upon ageing neutrophils upregulate CXCR4 expression which mediate re-entry into the bone marrow, where they undergo apoptosis in a TNF-related apoptosis-inducing ligand (TRAIL) dependent manner [20]. To investigate whether defective CXCR4 expression leads to neutrophil retention in circulation, LDLr−/− mice were placed on Western type diet for 3 weeks and treated with AMD3100. AMD3100 treated mice showed higher CD11b+Ly6Ghigh granulocyte counts in blood than control mice (Fig. 7A). Flow cytometry of blood granulocytes for DHR123 revealed elevated levels of intracellular hydrogen, reflective of augmented reactive oxygen species (ROS) production and senescence, in this subset in AMD3100 treated mice (Fig. 7B). Thus, CXCR4 dysfunction leads to an increase of neutrophil survival, senescence and activation, which may be accompanied by enhanced neutrophil adhesion to vascular endothelium, and in this way to aggravated atherosclerosis. Damås and coworkers have shown that coronary artery disease (CAD) patients have lowered plasma levels of SDF-1α [8]. In a final experiment we sought to investigate the relevance of our findings for human disease. Supportive of our animal experimental findings, CXCR4 protein expression on circulating neutrophils isolated from CEA as well as from UAP patients was significantly lowered than compared to neutrophils from healthy controls (Fig. 7C). When further analyzing human atherosclerotic lesions, we observed that plaque MPO+ neutrophil content is >10 fold increased in advanced as compared to early stages of atherosclerosis (IT) (Supplemental Fig. 8), with more abundant presence in the plaque shoulder and cap regions. MPO-CXCR4 double-staining furthermore revealed that a large number of MPO+ neutrophils are CXCR4+ (thus activated) in IT and TfcA, with a trend towards increased presence of CXCR4− MPO+ cells in plaques with intraplaque hemorrhage (47% ± 12% versus 36% ± 5% for TfcA, and 25% in early lesions).
Fig. 7. Increased numbers of senescent neutrophils in the absence of CXCR4.
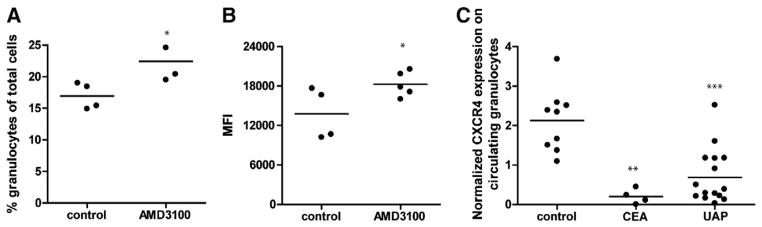
LDLr−/− mice on western type diet were treated with AMD3100 for 3 weeks. Control mice received PBS injections. A, Circulating Ly6GhighCD11b+ granulocyte numbers (*P < 0.05). B, Mean fluorescence intensity for DHR123 (*P = 0.05). C, Normalized CXCR4 protein expression on circulating granulocytes isolated from healthy (control) persons versus carotid endarterectomy (CEA) and patients with unstable angina pectoris (UAP, defined in the online methods) (**P < 0.01, ***P < 0.001 compared to controls).
4. Discussion
Previously we demonstrated that the SDF-1α/CXCR4 axis is instrumental in early stage atherosclerosis by controlling circulating neutrophil counts in mice [11]. In this study we have extended this notion, focusing on later, clinically relevant stages of plaque development in mice and man. We show that plaque SDF-1α/CXCR4 expression is progressively deregulated during disease progression. Moreover, CXCR4 blockade was seen to aggravate advanced plaque formation and stability as judged by an increased frequency of intraplaque hemorrhages. These adverse effects of CXCR4 blockade were accompanied by enhanced endothelial adhesion of neutrophils, possibly due to enhanced focal adhesion. Our studies point to CXCR4 controlled neutrophil survival, senescence, and subsequent hyperactivation as conceivable cause of CXCR4’s atheroprotective activity.
The actual contribution of neutrophils to human atherosclerotic lesion development and progression has long been elusive, due to their scarce presence in human plaques, amongst others. However, evidence is mounting that neutrophils play a role in atherosclerosis [21]. The present study identifies CXCR4 as a regulator of neutrophil function in advanced atherosclerosis in mice, and suggests that this disease axis may hold for human atherosclerosis as well.
Our histological data largely confirm previous work by Abi-Younes et al. [7], showing strong expression of SDF-1α and to a lesser extent also CXCR4 by endothelium, vSMCs and macrophages in the atherosclerotic plaque but not in healthy vessels. Moreover, we extend these findings, demonstrating that normal arteries do express SDF-1α, and that advanced unstable plaques show decreased SDF-1α expression, congruent with their decreased vSMC content. In contrast, CXCR4 expression was increased in more advanced lesions. In particular plaque leukocytes displayed high CXCR4 expression, possibly in response to the prevailing hypoxia [22,23]. Moreover, as plasma levels of this chemokine were shown to be progressively reduced in patients with stable and even more so with more unstable angina pectoris [8], the plaque CXCR4+ leukocyte accumulation may partly result from an increased SDF-1α gradient between circulation and plaque.
To delineate the contribution of leukocyte-derived SDF-1α/CXCR4 to plaque development and progression, we generated lentivirus vectors encoding an SDF-1α antagonist [12] and CXCR4 degrakine, respectively [13]. LDLr−/− mice reconstituted with LV.CXCR4deg lentivirus transduced bone marrow indeed displayed partially reduced CXCR4 protein expression by leukocytes compared to LV.Empty mice. Early plaque development was not affected by the reduced hematopoietic CXCR4 expression, however we did observe an increase in intraplaque erythrocyte accumulation, which was described to be involved in plaque destabilization [24,25]. The latter was confirmed by increased hemosider-in deposition in advanced plaques in both the LV.SDF-1α(P2G) and the LV.CXCR4degrakine group. Interestingly, at this stage lesion progression was severely increased in mice in which SDF-1α/CXCR4 signaling was inhibited, which may be explained by the increased accumulation of intraplaque erythrocytes with subsequent cholesterol deposition [26]. Previously, inhibition of CXCR4 signaling in a wire injury model of restenosis was associated with a decrease in vascular progenitor cell recruitment [10], resulting in reduced intimal thickening. However, in advanced atherosclerosis, plaque vSMC were seen to exclusively originate from arterial vSMC, and not to be of hematopoietic origin [27]. Furthermore, in this atherosclerosis model, no effects on lesional smooth muscle cell content was observed, suggesting that the relative contribution of vascular progenitor cells to the development atherosclerotic lesions in this model is not significant.
Blockage of CXCR4 has been shown to result in increased neutrophil release from bone marrow into circulation [28,29]. In ApoE−/− mice, complete and chronic blockage of CXCR4 with AMD3465, a CXCR4 antagonist, has been shown to increase blood neutrophil numbers, resulting in an increased accumulation of neutrophils within the atherosclerotic vessel wall and subsequently promoting plaque progression [11]. Partial blockage of CXCR4 in LV.CXCR4deg transfected bone marrow chimeras failed to induce neutrophil release into circulation, possibly due to compensatory effects such as reduced neutrophil production. Despite the lack of neutrophilia, CXCR4 blockade did aggravate plaque progression, possibly due to the increased adhesion of circulating neutrophils to plaque endothelium. In addition, neutrophils with impaired CXCR4 function showed increased intracellular ROS accumulation. Activated neutrophils elaborate a range of enzymes such as neutrophil elastase and cathepsin G, antibacterial proteins, myeloperoxidases and radicals, and chemokines and cytokines.
In addition to increased activity, neutrophils lacking functional CXCR4 expression showed increased cell survival, as witness the impaired apoptosis and increased expression of the cell survival factor Akt in vitro. These data suggest a role for neutrophil senescence in atherosclerosis, however this still needs to be confirmed in vivo. As neutrophils age, they upregulate the expression of CXCR4 and acquire the ability to migrate back to bone marrow where they undergo apoptosis [20,30]. However, our study indicates that if mature neutrophils fail to upregulate functional CXCR4 expression, they will be retained in circulation to migrate towards inflamed tissue, such as the atherosclerotic vessel wall, where they may further promote inflammation, possibly due to excessive activation. It is tempting to speculate that cardiovascular disease (CVD) patients have dysfunctional CXCR4 expression which in turn will lead to increased neutrophil senescence in blood and thus increased to neutrophil adherence to and activity in lesions. Indeed, in this study, we observed reduced number of CXCR4+ neutrophils in unstable human atherosclerotic plaques. It should be noted however that impaired CXCR4 function as in advanced human and mouse atherosclerosis probably acts deleterious by impeding activated neutrophil regress into bone marrow, and not only at the level of the plaque itself. Altogether, our data coincide with previous findings showing delayed neutrophil apoptosis in patients with unstable angina [29,30]. Whether neutrophil ageing is a cause or consequence of enhanced activation is still unknown.
A few limitations of this study are worth mentioning in this regard. First, while recent studies in mouse models of atherosclerosis have given new impetus to the notion that neutrophils are instrumental in the pathophysiology of disease [11,20,31], the actual impact of neutrophils in human disease is still controversial. Partly due to issues of technical (most human neutrophil antibodies lack specificity), and of intrinsic nature (neutrophils are very short-lived), only few reports have documented the presence of neutrophils in human plaques. Mostly their presence was confined to vulnerable and ruptured lesions, not coincidentally episodes of fulminant inflammation. In that respect, although our human and mouse data seem to point towards an increase in CXCR4 dependent neutrophil senescence and neutrophil adhesion, in particular in later stages of disease progression, translation of these findings to human disease should be handled with caution and yet remains to be established.
In conclusion, we provide clear indications for perturbed vascular homeostasis of SDF-1α and CXCR4 at later stages of atherogenesis in humans. Furthermore, we show that blockade of the SDF-1α/CXCR4 axis on leukocytes induces atherosclerotic plaque progression in mice, a finding that may be associated with an increased adherence to plaque endothelium and increased activity of neutrophils with impaired CXCR4 function. In addition, CXCR4 dysfunctional neutrophils also show increased cell survival, suggesting an important role for neutrophil senescence in atherosclerosis.
Supplementary Material
Acknowledgments
Funding
This work was supported by the Netherlands Organization for Scientific Research [grant 916.86.046 to I.B.], the Netherlands Heart Foundation [grant 2003 T201 to E.B.], the Deutsche Forschungsgemeinschaft [FOR809, WE1913/11-2 and ZE827/1-2 to A.Z. and C.W.] and the National Institutes of Health [AI41356 and AI048407 to L.S.].
We would like to thank Vigids Bjerkeli for excellent technical assistance.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.yjmcc.2014.04.021.
Footnotes
Disclosures
None declared.
References
- 1.Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–72. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- 2.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17:1410–22. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 3.Shah PK. Molecular mechanisms of plaque instability. Curr Opin Lipidol. 2007;18:492–9. doi: 10.1097/MOL.0b013e3282efa326. [DOI] [PubMed] [Google Scholar]
- 4.Braunersreuther V, Mach F, Steffens S. The specific role of chemokines in atherosclerosis. Thromb Haemost. 2007;97:714–21. [PubMed] [Google Scholar]
- 5.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–21. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 6.Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 7.Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P, Luster AD. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res. 2000;86:131–8. doi: 10.1161/01.res.86.2.131. [DOI] [PubMed] [Google Scholar]
- 8.Damås JK, Waehre T, Yndestad A, Ueland T, Müller F, Eiken HG, et al. Stromal cell-derived factor-1alpha in unstable angina: potential antiinflammatory and matrix-stabilizing effects. Circulation. 2002;106:36–42. doi: 10.1161/01.cir.0000020001.09990.90. [DOI] [PubMed] [Google Scholar]
- 9.Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation. 2003;10:335–50. doi: 10.1038/sj.mn.7800198. [DOI] [PubMed] [Google Scholar]
- 10.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Möpps B, et al. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. 2005;96:784–91. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- 11.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–17. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 12.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coffield VM, Jiang Q, Su L. A genetic approach to inactivating chemokine receptors using a modified viral protein. Nat Biotechnol. 2003;21:1321–7. doi: 10.1038/nbt889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–75. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 15.Goossens P, Gijbels MJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, et al. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 2010;12:142–53. doi: 10.1016/j.cmet.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Bot I, Guo J, Van Eck M, Van Santbrink PJ, Groot PH, Hildebrand RB, et al. Lentiviral shRNA silencing of murine bone marrow cell CCR2 leads to persistent knockdown of CCR2 function in vivo. Blood. 2005;106:1147–53. doi: 10.1182/blood-2004-12-4839. [DOI] [PubMed] [Google Scholar]
- 17.Spellberg BJ, Collins M, French SW, Edwards JE, Jr, Fu Y, Ibrahim AS. A phagocytic cell line markedly improves survival of infected neutropenic mice. J Leukoc Biol. 2005;78:338–44. doi: 10.1189/jlb.0205072. [DOI] [PubMed] [Google Scholar]
- 18.Wallington LA, Durham J, Bunce CM, Brown G. Growth of single HL60 cells in liquid culture: analysis of the influences of differentiative agents. Leuk Res. 1996;20:821–9. doi: 10.1016/s0145-2126(96)00040-9. [DOI] [PubMed] [Google Scholar]
- 19.‘t Hoen PA, Van der Lans CA, Van Eck M, Bijsterbosch MK, Van Berkel TJ, Twisk J. Aorta of ApoE-deficient mice responds to atherogenic stimuli by a prelesional increase and subsequent decrease in the expression of antioxidant enzymes. Circ Res. 2003;93:262–9. doi: 10.1161/01.RES.0000082978.92494.B1. [DOI] [PubMed] [Google Scholar]
- 20.Lum JJ, Bren G, McClure R, Badley AD. Elimination of senescent neutrophils by TNF-related apoptosis-inducing ligand. J Immunol. 2005;175:1232–8. doi: 10.4049/jimmunol.175.2.1232. [DOI] [PubMed] [Google Scholar]
- 21.Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–88. doi: 10.1161/CIRCRESAHA.111.257535. [DOI] [PubMed] [Google Scholar]
- 22.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karshovska E, Zernecke A, Sevilmis G, Millet A, Hristov M, Cohen CD, et al. Expression of HIF-1alpha in injured arteries controls SDF-1alpha mediated neointima formation in apolipoprotein E deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:2540–7. doi: 10.1161/ATVBAHA.107.151050. [DOI] [PubMed] [Google Scholar]
- 24.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 25.Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 26.Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–61. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 27.Bentzon JF, Sondergaard CS, Kassem M, Falk E. Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice. Circulation. 2007;116:2053–61. doi: 10.1161/CIRCULATIONAHA.107.722355. [DOI] [PubMed] [Google Scholar]
- 28.Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583–93. doi: 10.1016/s1074-7613(03)00263-2. [DOI] [PubMed] [Google Scholar]
- 29.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;1:2423–31. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biasucci LM, Liuzzo G, Giubilato S, Della Bona R, Leo M, Pinnelli M, et al. Delayed neutrophil apoptosis in patients with unstable angina: relation to C-reactive protein and recurrence of instability. Eur Heart J. 2009;30:2220–5. doi: 10.1093/eurheartj/ehp248. [DOI] [PubMed] [Google Scholar]
- 31.Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.