

Abstract
The aim of this work was to prepare a novel class of 64Cu(II) labeled complexes with the new macrocyclic ligands 1,10-dithia-4,7-diazacyclododecane-3,8-dicarboxylic acid (NEC-SE, 1), 1,10-dithia-4,7-diazacyclotridecane-3,8-dicarboxylic acid (NEC-SP, 2) and 1,10-dithia-4,7-diazacyclotetradecane-3,8-dicarboxylic acid, (NEC-SB, 3) to evaluate the usefulness of these macrocycles for potential utility as 64Cu(II) chelators. The corresponding non-radioactive complexes [Cu(NEC-SE)]·3H2O (4), [Cu(NEC-SP)]·3H2O (5) and [Cu(NEC-SB)] (6) were prepared and their 64Cu-analogs, [64Cu(NEC-SE)] (7) and [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9) were produced in >98% radiochemical purity. Rats were injected with complex 7, 8 or 9 and were euthanized at 1, 4 and 24 h. All three complexes are cleared from the blood over the first hour following injection but there is poor clearance of this activity over 24 h. A similar pattern of retention was noted in the liver where the levels of activity in this tissue at 1 h are not statistically different from those at 24 h. Molecular mechanics and DFT studies were performed on the complexes in order to gain insight into the lower stability.
Introduction
Copper radionuclides offer an attractive foundation for the development of disease-targeting radiopharmaceuticals; with a wide range of half-lives and decay schemes they have utility in both Positron Emission Tomography (PET) (60Cu, 61Cu, 62Cu and 64Cu) and targeted radiotherapy (64Cu and 67Cu).1,2 Attachment of these nuclides to biomolecules, such as peptides and antibodies, require conjugation of the nuclide to the biological entity via a stable bifunctional ligand that can withstand metabolism and demetalation in vivo; the in vivo stability of the metal chelate is perhaps one of the most important considerations in the design of a disease-targeting copper radiopharmaceutical. The identification of kinetically inert radiocopper complexes, rather than copper complexes with high thermodynamic stability, is the first stage in identifying a suitable bifunctional chelator for copper nuclides.3,4 Macrocyclic ligands form metal complexes which in general are thermodynamically more stable than complexes with analogous open-chain ligands and this has been termed the “macrocyclic effect”.3,5–12 In addition macrocyclic metal complexes are often inert towards acid dissociation.13 Due to the interest in macrocycles for use as bifunctional chelators, a large number of comparative studies on macrocycles with different donors and ring sizes have been recently published.14–16 The majority of bifunctional ligands used for complexing copper have centered around the tetraaza-based macrocycles.16 In general, studies have involved conjugation of 1,4,7,10-tetraazacyclododecane-N,N′,N″N‴-tetraacetic acid (DOTA) or 1,5,8,11-tetraazacyclotetradecane-N,N′,N″,N‴-tetraacetic acid (TETA) to the terminal amine of tumor-targeting peptides for subsequent labeling with 64Cu (t1/2 = 12.7 h, 17.4% β+, 41% EC, 40% β−, Eavg = 278 keV) for PET imaging applications.17–21 However, image quality is often less than optimal due to the slow release of uncoordinated 64Cu by decomposition of the complex in the blood or transchelation in the liver, causing elevated uptake in non-target tissues such as the liver.4,22 New cross-bridged cyclam chelates (e.g., CB-TE2A)4,23 have recently been developed for 64Cu and have shown significant metal-chelate stability and as a consequence a lowering in non-target tissue accumulation.22,24
As mentioned, the dominant moieties for copper chelates have been the tetraaza-based (N4) macrocyclic ligands, with little or no studies done with N2S2-based macrocycles. Thiaazacycloalkanes exhibit interesting properties intermediate between those of cyclic polyamines and polythioethers.25 The discovery of (N2S2)4− donor sites in biology based on cysteine-X-cysteine tripeptide motifs has prompted the examination of small molecule models that build on a well known literature that is based on (N2S2)2− ligands, which evolved over several decades.26,27 Although N,N-ethylene-di-L-cysteine (EC) has been known for a long time,28 it is surprising that macrocyclic ligands based on the EC skeletons have not been extensively investigated. EC copper(II),29 nickel(II),30 cobalt(III),31 [Tc(V)O]3+,32,33 [Re(V)O]3+,34–36 molybdenum(VI),37 indium(III) and gallium(III)38 complexes have been synthesized and characterized. EC can be labeled with 99mTc easily and efficiently with high radiochemical purity and stability.39–43 Compared to metals with an oxidation state of +3 or higher, in the metal(II) complexes (copper(II) and nickel(II)) the EC acts as tetradentate N2S2 ligand to form complexes with nearly planar cis-NiN2S230 and cis-CuN2S2 units.29 The bulky carboxylic or ester groups are equatorially oriented on the λ conformation NS chelate rings.44 The non-macrocyclic N2S2 moiety has been used as a means to attach radiocopper to biomolecules.45,46 Siegfried and Kaden measured the rate of formation and dissociation of a series of N2S2 macrocycles with Cu2+ and Ni2+.15 The ring size (from 12- to 16-membered cycles) and the geometry (cis or trans) of the arrangement of the donor groups was systematically varied in order to investigate how these two parameters affect the mechanism of complex formation and dissociation. In particular this study showed that the 14-membered macrocycle (cis-arrangment) forms complexes in which both Cu2+ and Ni2+ are ideally coordinated so that the dissociation becomes extremely slow.47–49 The marked dependence of the nature of the backbone on the specificity of copper complexes of EC led us to investigate further EC-proligands with extended 12-, 13- and 14-member backbones as ligands for the development of new copper radiopharmaceuticals. It was hoped that these ligands would better accommodate both Cu(I) and Cu(II). A series of 12-, 14- and 16-membered N2S2-macrocycles and the related Cu(I) and Cu(II) derivatives were synthesized and their stabilities, redox and spectroscopic properties were explored.15,50–52 In general it was shown that complexes with the smaller rings react as flexible open-chain ligands directly with H+, with the larger rings reacting in a similar manner as rigid ligands.15 It was shown that the 14-membered macrocycle forms complexes in which the metal ions are ideally coordinated so that their dissociation becomes extremely slow. In view of a radiochemical application, N2S2-macrocycles with ring sizes varying between 12 and 16, as well as two 12-membered N2S2-rings with a pendant carboxylic and amino group linked to the N atoms, respectively, were also synthesized, and related Ag(I) complexes were tested in blood serum for their stability.53
In this current study we aimed to prepare three novel N2S2 macrocyclic ligands based on the L,L-ethylenedicysteine skeletons. We present the synthesis and characterization of these novel ligands, the non-radioactive copper complexes and the corresponding 64Cu-analogs. We further report the in vivo characteristics of these 64Cu complexes in an animal model using acute biodistribution and we discuss the usefulness of these agents as bifunctional chelates for copper nuclides.
Results
Synthesis and characterization of the macrocyclic ligands
The reaction scheme of the syntheses of 1,10-dithia-4,7-diazacyclododecane-3,8-dicarboxylic acid (NEC-SE, 1), 1,10-dithia-4,7-diazacyclotridecane-3,8-dicarboxylic acid (NEC-SP, 2) and 1,10-dithia-4,7-diazacyclotetradecane-3,8-dicarboxylic acid, (NEC-SB, 3) is shown in Fig. 1. The starting material ethylenedicysteine was prepared by the sodium reduction of the precursor L-thiazolidine-4-carboxylic acid in liquid ammonia according to methods described by Blondeau et al.32 The ligands (1, 2 or 3) were purified by washing with water and ethanol and were obtained in high yield. Their authenticity was confirmed by 1H-NMR, 13C-NMR, elemental analysis, IR and ESI-MS spectroscopy. The novel ligands 1 and 2 are white, the ligand 3 is yellow. They are microcrystalline solids, soluble in water solutions at pH > 7 and insoluble in common organic solvents; the ligand 3 is soluble in methanol solution. The infrared spectra of 1–3 showed broad absorptions in the range 3390–3412 cm−1 due to the NH stretching vibrations. The presence of the COO moiety is detected by intense, broad absorptions in the range 1586–1633 cm−1, due to the ionized COO− stretching bands, and by medium or weak absorptions at 1726–1732 cm−1, attributable to the COOH un-ionized stretching frequencies. This behavior is in accordance with the coexistence of the predominant zwitterion together with a minority of the un-ionized carboxylic groups.54 The formation of the macrocycle is confirmed by the presence of the S(CH2)nS protons in the 1H-NMR at δ 2.63–2.70 for 1, at δ 1.72 and 2.49–2.77 for 2 and at δ 1.50 and 2.40–2.60 for 3. In the 13C{1H}NMR spectrum of 1 the chemical shifts of the S–CH2 carbons are observed at δ 34.74 and 37.21; for 2 the corresponding 13C chemical shifts of the S–CH2 carbons are at δ 33.56 and 37.07, while for 3 the 13C chemical shifts of the S–CH2 carbons are at δ 30.52 and 33.44. In addition, in 2, the chemical shift of the C–CH2–C carbon is at δ 31.48, while for 3 the corresponding 13C chemical shift of the C–CH2CH2–C carbons are at δ 27.11. The ESI-MS negative-ion spectrum of 1 in H2O/NH4OH is dominated by the deprotonated species [M – H]− and [2M – H]− at m/z 293 (100%) and 588 (80%), respectively. The ESI-MS negative-ion spectrum of 2 in H2O/NH4OH is dominated by the deprotonated ligands [M – H]− and [M – 2H]2− at m/z 307 (100%) and 153 (30%), respectively. The ESI-MS negative-ion spectrum of 3 in H2O/NH4OH is dominated by the deprotonated species [M – 2H]2− at m/z 160 (100%).
Fig. 1.
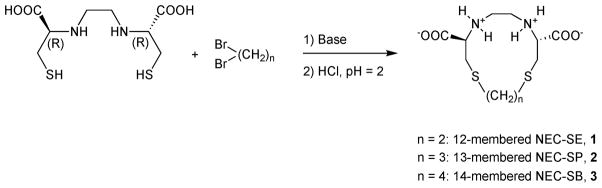
The reaction schemes of the syntheses of 1,10-dithia-4,7-diazacyclododecane-3,8-dicarboxylic acid (NEC-SE, 1), 1,10-dithia-4,7-diazacyclotridecane-3,8-dicarboxylic acid (NEC-SP, 2) and 1,10-dithia-4,7-diazacyclotetradecane-3,8-dicarboxylic acid, (NEC-SB, 3).
Synthesis and characterization of the non-radioactive Cu(II) complexes
Ligands 1–3 were used in the preparation of the copper(II) complexes [Cu(NEC-SE)]·3H2O (4), [Cu(NEC-SP)]·3H2O (5), and [Cu(NEC-SB)] (6); the chemical scheme of the syntheses of the compounds is given in Fig. 2. The stoichiometry of the compounds was not affected by either reducing or increasing the amount of the corresponding ligand. The blue copper complexes are insoluble in common organic solvents and not very soluble in water solutions at pH >7. The authenticity of 4–6 was confirmed by elemental analysis and IR spectroscopy. The infrared spectra of the derivatives 4–6 showed broad absorptions in the range 3397–3412 cm−1 due to the NH stretching vibrations. The presence of the COO moiety is detected by intense broad absorptions in the ranges 1601–1617 cm−1 and 1358–1384 cm−1, due to the asymmetric and symmetric stretching modes, respectively; the shift to blue of the coordinated COO asymmetric stretchings with respect to the free ligands being observed upon complex formation. The observed range is in accordance with the values reported for coordinated COO stretching bands in amino-carboxylate metal complexes.54 The magnitude of νasymCOO – νsymCOO (Δν) separation can be used to explain the type of carboxylate structure present in the solid state. Δν values for 4–6 are of about 250 cm−1, characteristic of monodentate coordination compounds. In the low-frequency region the copper–nitrogen stretching frequencies fall in the range 440–470 cm−1, while the copper–oxygen vibrations are detected in the range 300–380 cm−1.55
Fig. 2.
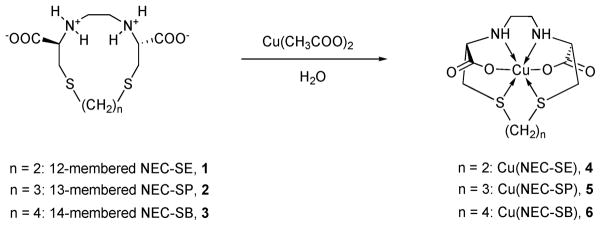
The reaction schemes of the syntheses of Cu(NEC-SE) (4), Cu(NEC-SP) (5) and Cu(NEC-SB) (6).
Synthesis and characterization of the 64Cu(II) complexes
The corresponding 64Cu-analogs, [64Cu(NEC-SE)] (7) and [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9) were produced from reacting 64CuCl2 and either 1, 2 or 3 in 0.1 M ammonium acetate buffer (pH 8.0) at 90 °C for 1 h. It was necessary to dissolve 3 in 20 μL methanol prior to diluting with the 0.1 M ammonium acetate buffer. The presence in solution of the 64Cu-complexes was confirmed by radio-TLC by comparison with the TLC profile of an authentic sample of non-radioactive complex (4, 5 or 6). The complexes were produced in a radiochemical purity of >98%. Compound 9 was additionally analyzed by radio-HPLC and was shown to have a radioactive peak at tR = 12.61 min, which corresponded to the UV peak for the analogous cold complex.
In vivo data
The biodistribution data (percent injected dose per gram, %ID/g) of [64Cu(NEC-SE)] (7) and [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9) in male Lewis rats is summarized in Table 1. Fig. 3 presents a comparison between the biodistribution data (%ID/organ) of 7, 8 and 9 and selected 64Cu–tetraazamacrocycles at 24 h post injection.56 The data obtained from complexes 7, 8 and 9 are indistinguishable. Although blood clearance of these complexes is observed over the first hour post injection (7, 0.850 ± 0.051 %ID/g; 8, 0.820 ± 0.046 %ID/g; 9, 0.874 ± 0.140 %ID/g) the clearance from the blood was poor over 24 h (7, 0.768 ± 0.040 %ID/g; 8, 0.774 ± 0.031 %ID/g; 9, 0.692 ± 0.151 %ID/g). The kidney did demonstrate partial clearance of the radioactivity over 24 h but still remained at relatively high levels. The liver and kidney uptakes of the complexes were high at 1 h post injection (liver: 7, 3.919 ± 0.363 %ID/g; 8, 3.151 ± 0.286 %ID/g; 9, 4.105 ± 0.465 %ID/g; kidney: 7, 8.662 ± 0.551 %ID/g; 8, 9.438 ± 1.019 %ID/g; 9, 8.895 ± 0.612 %ID/g), with little or no clearance observed for either complex from the liver over 24 h (liver: 7, 3.314 ± 0.476 %ID/g; 8, 3.182 ± 0.188 %ID/g; 9, 3.296 ± 0.728 %ID/g; kidney: 7, 4.906 ± 0.211 %ID/g; 8, 5.070 ± 0.518 %ID/g; 9, 4.738 ± 0.617 %ID/g). The poor clearance of these complexes suggests that the complexes are dissociating in tissues and 64Cu remains trapped in these tissues hindering clearance.57
Table 1.
Biodistribution (percent dose per gram, %ID/g) of [64Cu(NEC-SE)] (7), [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9) in male Lewis rats. Rats were injected with ~40 μCi of complex and were euthanized at 3 time points (n = 4 per group at 1, 4 and 24 h). Following euthanasia, 19 tissues and organs of interest were removed and weighed and the radioactivity present was measured in a γ-counter. Given are the %ID/g values for selected organs. The complete biodistribution data for all 19 tissues, including percent dose per organ (%ID/organ) values, can be found in the ESI†
| [64Cu(NEC-SE)] (7), %ID/g
|
[64Cu(NEC-SP)] (8), %ID/g
|
[64Cu(NEC-SB)] (9), %ID/g
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Tissue | 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | 1 h | 4 h | 24 h |
| Blood | 0.850 ± 0.051 | 0.897 ± 0.080 | 0.768 ± 0.040 | 0.820 ± 0.046 | 0.835 ± 0.021 | 0.774 ± 0.031 | 0.874 ± 0.140 | 0.879 ± 0.112 | 0.692 ± 0.151 |
| Lung | 0.838 ± 0.065 | 0.768 ± 0.045 | 0.723 ± 0.036 | 0.776 ± 0.089 | 0.737 ± 0.009 | 0.771 ± 0.061 | 1.673 ± 0.225 | 0.949 ± 0.109 | 0.746 ± 0.143 |
| Liver | 3.919 ± 0.363 | 3.905 ± 0.540 | 3.314 ± 0.476 | 3.151 ± 0.286 | 3.418 ± 0.371 | 3.182 ± 0.188 | 4.105 ± 0.465 | 3.866 ± 0.635 | 3.296 ± 0.728 |
| Spleen | 0.560 ± 0.025 | 0.731 ± 0.174 | 1.046 ± 0.106 | 0.483 ± 0.054 | 0.704 ± 0.014 | 1.078 ± 0.099 | 0.698 ± 0.168 | 0.815 ± 0.137 | 0.924 ± 0.207 |
| Kidney | 8.662 ± 0.551 | 7.849 ± 1.099 | 4.906 ± 0.211 | 9.438 ± 1.019 | 7.818 ± 0.663 | 5.070 ± 0.518 | 8.895 ± 0.612 | 7.096 ± 1.059 | 4.738 ± 0.617 |
| Muscle | 0.380 ± 0.032 | 0.344 ± 0.012 | 0.309 ± 0.026 | 0.393 ± 0.055 | 0.352 ± 0.017 | 0.319 ± 0.031 | 0.411 ± 0.056 | 0.330 ± 0.050 | 0.302 ± 0.033 |
| Fat | 0.334 ± 0.054 | 0.266 ± 0.068 | 0.163 ± 0.026 | 0.318 ± 0.074 | 0.214 ± 0.024 | 0.153 ± 0.019 | 0.371 ± 0.073 | 0.218 ± 0.037 | 0.136 ± 0.035 |
| Heart | 0.672 ± 0.086 | 0.774 ± 0.096 | 1.041 ± 0.070 | 0.657 ± 0.025 | 0.736 ± 0.048 | 1.013 ± 0.078 | 0.839 ± 0.228 | 0.752 ± 0.059 | 0.923 ± 0.189 |
| Bone | 1.131 ± 0.072 | 1.274 ± 0.082 | 0.984 ± 0.076 | 1.099 ± 0.098 | 1.255 ± 0.040 | 0.999 ± 0.065 | 1.143 ± 0.195 | 1.155 ± 0.161 | 0.855 ± 0.188 |
Fig. 3.
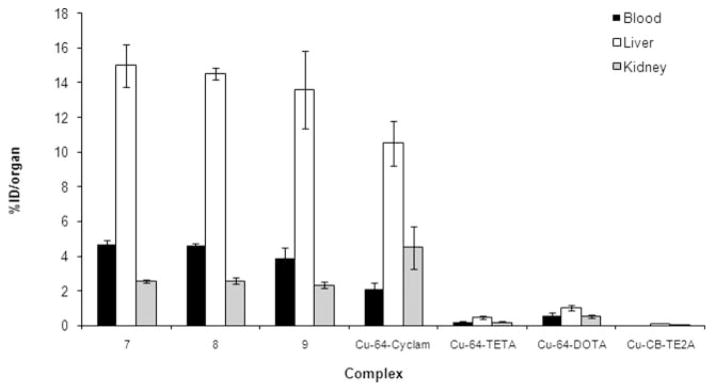
Comparison between the biodistribution data (%ID/organ) of [64Cu(NEC-SE)] (7), [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9) and selected 64Cu-tetraazamacrocycles at 24 h post injection. Data for 7, 8 and 9 obtained in male Lewis Rats. 64Cu-Cyclam, 64Cu-TETA, 64Cu-DOTA and 64Cu-CB-TE2A obtained in female Sprague-Dawley rats.4,56
Discussion
In this current study we prepared three novel N2S2 macrocyclic ligands 1,10-dithia-4,7-diazacyclododecane-3,8-dicarboxylic acid (NEC-SE, 1), 1,10-dithia-4,7-diazacyclotridecane-3,8-dicarboxylic acid (NEC-SP, 2), and 1,10-dithia-4,7-diazacyclotetradecane-3,8-dicarboxylic acid (NEC-SB, 3) based on the L,L-ethylenedicysteine skeleton. It is anticipated that the use of macrocyclic N2S2 ligands would result in an increase in in vivo metal–chelate stability and therefore lead to an improvement in target to non-target tissue ratios. This has been demonstrated before and has been termed to “macrocycle-effect”.3,5–12 We synthesized and characterized all the non-radioactive (4–6) and the 64Cu complexes (7–9) of these three ligands (Fig. 1 and 2). We further studied the in vivo biodistribution of complexes 7, 8 and 9 in healthy rats to compare the biokinetics of these three compounds with previously reported data obtained with selected 64Cu-tetrazamacrocycles.
The biodistribution of complexes 7, 8 and 9 were very similar to each other and very few statistical differences between the data exist. All three complexes were cleared from the blood over the first hour following injection but there was poor clearance of this activity over 24 h (Table 1). A similar pattern of retention is noted in the liver where the levels of activity in this tissue at 1 h are not statistically different from those at 24 h. The kidney did demonstrate partial clearance of the complexes over 24 h but still remained at relatively high levels. The retention of activity in tissues was similar to that observed with 64Cu-cyclam29 (Fig. 3) and 64Cu-monooxo-tetrazamacrocyclic complexes.4 However, on comparison with 64Cu-TETA and 64Cu-DOTA, the uptake and retention of 7, 8 and 9 are orders-of-magnitude higher (Fig. 3). This could in part be attributed to the kinetics of these hydrophilic complexes, but is more likely due to very rapid dissociation of the 64Cu from the chelates in vivo. This results in a biodistribution being more similar to “free” 64Cu than the biokinetics of intact N2S2 complexes. This same dissociation phenomenon is observed (although slower) with N4-macrocyclic chelates such as DOTA. 64Cu has been shown to dissociate in vivo from DOTA and DOTA-conjugates16 and then undergoes metabolism and transchelation to superoxide dismutase and other proteins causing increased blood and liver accumulation.22,24 The poor clearance of 7, 8 and 9 suggests that the 64Cu complexes are rapidly degraded in blood and tissues and the 64Cu is sequestered by proteins and thereby remaining trapped in these tissues hindering clearance. The cross-bridged cyclam chelator, CBTE2A, has demonstrated improved in vivo stability and consequently a reduction in transchelation when compared to the non-bridged analog, TETA.4,16,22,24 Conjugation of this chelate to tumor-targeting peptides has caused an improvement in non-target organ clearance and higher tumor ratios compared with either the 64Cu-TETA and 64Cu-DOTA analogs.16,24
In a recent report by Siegfried and Kaden, the kinetics of the formation and dissociation of Cu2+ from a series N2S2 macrocycles was studied.15 The ring size and the geometry of the arrangement of the donor groups were varied and the kinetic characteristics of the complexes were compared. Since all attempts to obtain crystal structures of the copper complexes were unsuccessful, molecular modeling was performed to gain insight into the geometries of the complexes that may be contributing to in vivo instability. The molecular modeling studies provide support for the role of the carboxylic acid groups in coordinating the copper atoms. The preferred structure for 7 is a square pyramidal complex with the base formed from one of the N atoms, two O atoms each donated by one of the carboxylate groups, and one of the S atoms. The apical position is filled by the remaining ring nitrogen, and the remaining ring S is 2.957 Å from the copper. The complex retains an essentially planar arrangement of the N and S atoms, planes defined by N–Cu–N and S–Cu–S intersect with a 27° angle comparable to the 21° found by Schugar.40 Complex 8 prefers an octahedral coordination arrangement with the equatorial positions filled by the ring nitrogens and sulfurs, and the axial positions filled by carboxylate oxygens. The N and S atoms are even more planar than in 7 with the N–Cu–N and S–Cu–S planes separated by only 13.2°. Complex 9 was found to favor a distorted tetrahedral arrangement with the copper being coordinated by the carboxylates and both ring sulfurs. As with 8, the N–Cu–N and S–Cu–S planes are separated by only 13.4°. The structures of these models are shown in Fig. 4, and a structural summary in Table 2.
Fig. 4.
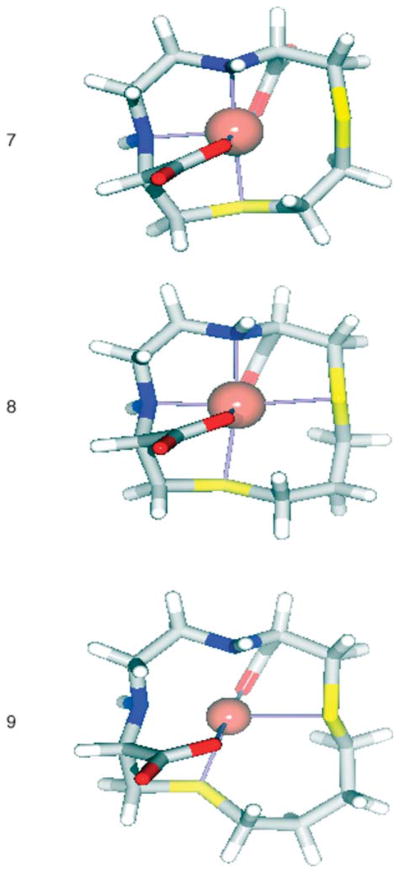
The DFT (B3LYP/LACVP*) optimized structures of Cu(NEC-SE) (7), Cu(NEC-SP) (8) and Cu(NEC-SB) (9).
Table 2.
Structural features of B3LYP optimized complexes
| Complex 7 | Complex 8 | Complex 9 | |||
|---|---|---|---|---|---|
| Cu–L (Å) | Cu–L (Å) | Cu–L (Å) | |||
| N9 | 2.193 | N10 | 2.154 | N11 | 2.213 |
| N12 | 1.997 | N13 | 2.150 | N14 | 2.188 |
| S3 | 2.957 | S3 | 2.553 | S3 | 2.565 |
| S6 | 2.341 | S7 | 2.550 | S8 | 2.540 |
| O17 | 1.948 | O18 | 1.967 | O19 | 1.953 |
| O19 | 2.053 | O20 | 1.968 | O21 | 1.955 |
| Angle (°) | Angle (°) | Angle (°) | |||
| N–Cu–N | 91.94 | N–Cu–N | 88.21 | N–Cu–N | 84.66 |
| S–Cu–S | 98.77 | S–Cu–S | 103.55 | S–Cu–S | 112.21 |
| O–Cu–N | 86.02, 79.03 | O–Cu–N | 81.73, 81.49 | O–Cu–N | 81.62, 81.05 |
The NBO analysis provides a method for analyzing the strength of the bonding interactions present in the complexes; an examination of the relative strengths for each donor–copper interaction provides a means to determine the predominate interactions for each complex. The copper center in complex 7 is strongly coordinated by one of the nitrogens (N12 242.62 kcal mol−1) and sulfur (S6 679.65 kcal mol−1) and oxygen (O17 246.44 kcal mol−1), while the other nitrogen (N9 161.36 kcal mol−1) and oxygen (O19 173.08 kcal mol−1) display weaker interactions with the copper. Complex 8 is more balanced in terms of its bonding interactions, with both nitrogens (N10 and N14, 138.7 and 154.61 kcal mol−1) and sulfurs (S3 and S7, 370.61 and 375.46 kcal mol−1) having relatively equal interactions. In a similar trend both oxygen donors (O18 and O20) have respective bond strengths of 183.27 and 183.04 kcal mol−1. Complex 9 also shows relatively equivalent interaction strengths between pairs of donor atoms and the copper center, the two nitrogens (N11 and N14) have energies of 113.58 and 130.29 kcal mol−1, while the two sulfurs (S3 and S8) have energies of 351.91 and 376.49 kcal mol−1. The two oxygen donors also interact in an equivalent fashion (O19 and O21) 175.58 and 174.6 kcal mol−1.
The stability of these complexes therefore appear to be a balance of favorable donor–copper interactions and steric strain caused by their macrocyclic nature. In all cases the macrocycle is able to adopt a conformation in which the copper can sit in the plane of the ring, however the interactions between the donor atoms and copper vary greatly. All three systems contain three types of donor groups: nitrogen, sulfur, and oxygen; no one type of metal donor interaction seems strong enough to override the others. In complex 7 it appears that macrocyclic constraints prevent optimal overlap of the donor atoms with the copper center, most likely lowering the in vivo stability of the complex. Complexes 8 and 9 appear to be equally well coordinated with the available donor atoms, although it does appear that there may be less than maximal coordination with the sulfur donors when compared with the strength of the Cu–S6 bond found in complex 7. This would suggest that the lower stability may arise from weaker Cu–S interactions than would be found in a non-macrocyclic ligand.
Conclusions
We prepared three novel N2S2 macrocyclic ligands NEC-SE (1), NEC-SP (2) and NEC-SB (3) based on the L,L-ethylenedicysteine skeleton. From ligands 1, 2 and 3 we have synthesized and characterized the non-radioactive copper complexes (4–6) and the analogous 64Cu complexes (7–9). To the best of our knowledge, this is the first report of 64Cu labeled to this form of (N2S2) macrocyclic ligand. It is however evident that the biological behavior of these complexes is less than ideal and additional study is required to improve the in vivo stability of these agents. In fact, the new ligands described in this manuscript are susceptible to further functionalization, for example by modification of the ring dimensions, and/or, by change of the donor atoms set.
Experimental
Methods and materials
Chemicals and analysis
All syntheses and handling were carried out under an atmosphere of dry oxygen-free dinitrogen, using standard Schlenk techniques or a glove box. All solvents were dried, degassed and distilled prior to use. Elemental analyses (C, H, N, S) were performed in house with a Fisons Instruments 1108 CHNS-O Elemental Analyser. Melting points were taken on an SMP3 Stuart Scientific Instrument. IR spectra were recorded as neat from 4000 to 600 cm−1 with a Perkin-Elmer Spectrum One System and as Nujol mulls from 600 to 100 cm−1 with a Perkin-Elmer System 2000 FT-IR instrument. IR annotations used: m = medium, mbr = medium broad, s = strong, sbr = strong broad, sh = shoulder, w = weak. 1H- and 13C-NMR spectra were recorded on a Oxford-400 Varian spectrometer (400.4 MHz for 1H, 100.1 MHz for 13C). Chemical shifts (δ) are quoted relative to internal SiMe4. NMR annotations used: br = broad, m = multiplet, qn = quintet, t = triplet. Electrospray mass spectra (ESI-MS) were obtained in positive- or negative-ion mode on a Series 1100 MSD detector HP spectrometer, using an acetone mobile phase. The compounds were added to reagent grade methanol to give solutions of approximate concentration 0.1 mM. These solutions were injected (1 μL) into the spectrometer via a HPLC HP 1090 Series II fitted with an autosampler. The pump delivered the solutions to the mass spectrometer source at a flow rate of 300 μL min−1, and nitrogen was employed both as a drying and nebulizing gas. Capillary voltages were typically 4000 V and 3500 V for the positive- and negative-ion mode, respectively. Confirmation of all major species in this ESI-MS study was aided by comparison of the observed and predicted isotope distribution patterns, the latter calculated using the IsoPro 3.0 computer program.
Radiochemistry
All chemicals unless otherwise stated were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Water was distilled and then deionized (18 MΩ cm−2) by passing through a Milli-Q water filtration system (Millipore Corp., Milford, MA). Radioactivity was counted with a Beckman Gamma 8000 counter containing a NaI crystal (Beckman Instruments, Inc., Irvine, CA). Care was taken to ensure all buffers and vials were as free of metal contaminants as possible. The non-radioactive copper complexes (4, 5 and 6) were used as fully characterised non-radioactive standards to confirm the identity of the 64Cu analogs (7, 8 and 9). Identification of the 64Cu complexes was performed using radio thin-layer chromatography (radio-TLC). Radio-TLC detection was accomplished using a BIOSCAN AR2000 Imaging Scanner (Washington, DC). TLC was performed using glass-backed silica gel and a 10% aqueous (CH3COO)NH4 solution : methanol (1 : 1) as mobile phase. Reaction progress and purity were also monitored by analytical reversed-phase HPLC, which was performed on a Waters 600E (Milford, MA) chromatography system with a Waters 991 photodiode array detector and an Ortec Model 661 radioactivity detector (EG & G Instruments, Oak Ridge, TN). A Vydak C-18 column was employed with a gradient that changes from 0.1% TFA in water to 5 : 95 0.1%TFA in Water : 0.1 %TFA in CH3CN over the course of 25 min. Unreacted 64Cu was eluted in the void volume, tR < 1.0 min.
Ligand synthesis and characterization
Synthesis of 1,10-dithia-4,7-diazacyclododecane-3,8-dicarboxylic acid (NEC-SE, 1)
Ethylenedicysteine was synthesised according to literature methods.26 Ethylenedicysteine (5.50 g, 20.50 mmol) and 1,2-dibromoethane (3.85 g, 20.50 mmol) were added to 100 mL of a NaHCO3 solution (6.89 g, 82.01 mmol). The mixture was gradually heated up to 100 °C and was stirred for 24 h to obtain a clear pale yellow solution. The solution was cooled to room temperature and acidified to pH 2. The resulting precipitate which separated out was filtered and washed with water (3 × 50 mL) and ethanol (3 × 15 mL) and dried under reduced pressure to yield derivative 1 in 65% yield. Melting point, 225–228 °C. 1H NMR (D2O/NaOD, 293 K): δ 2.49 (br, 4H, CH 2N), 2.63–2.70 (m, 8H, CH 2S), 3.10 (t, 2H, CH ). 13C{1H}NMR (D2O/NaOD, 293 K): δ 34.74, 37.21 (CH2S), 49.26 (CH2N), 65.75 (CH), 182.81 (CO2H). IR (cm−1): 3390br (νNH), 1730sh (νasymCOOH), 1586s (νasymCOO−), 610br, 532s, 459m, 399w, 376sbr, 267w, 255w. ESI-MS (major negative-ions, H2O/NH4OH), m/z (%): 293 (100) [M – H]−, 588 (60) [2M – H]−. Anal. Calcd. for C10H18N2O4S2: C, 40.80; H, 6.16; N, 9.52; S, 21.78%. Found: C, 41.05; H, 6.26; N, 9.37; S, 21.53%.
Synthesis of 1,10-dithia-4,7-diazacyclotridecane-3,8-dicarboxylic acid (NEC-SP, 2)
The ligand NEC-SP, 2, was prepared analogously to 1. Ethylenedicysteine (5.50 g, 20.50 mmol) and 1,3-dibromopropane (4.14 g, 20.50 mmol) were added to a NaHCO3 (6.89 g, 82.01 mmol) solution (100 mL). The mixture was gradually heated up to 100 °C and stirred for 24 h to obtain a clear yellow solution. The solution was cooled to room temperature and acidified to pH 2. The precipitate which separated out was filtered, washed with water (3 × 50 mL) and ethanol (3 × 25 mL) and dried under reduced pressure to yield derivative 2 in 72% yield. Melting point, 212–215 °C. 1H NMR (D2O/NaOD, 293 K): δ 1.72 (qn, 2H, CH2). 2.54 (br, 4H, CH2N), 2.61–2.77 (m, 8H, CH2S), 3.22 (t, 2H, CH), 13C{1H}NMR (D2O/NaOD, 293 K): δ 31.48 (CH2), 33.56 (CH2S), 37.07 (CH2S), 48.57 (CH2N), 65.55 (CH), 181.68 (CO2H). IR (cm−1): 3412br (νNH), 1732m (νasymCOOH), 1588s (νasymCOO−), 633w, 573w, 536sbr, 439m, 373s, 279w. ESI-MS (major negative-ions, H2O/NH4OH), m/z (%): 153 (30) [M – 2 H]2−, 307 (100) [M – H]−. Anal. Calcd. for C11H20N2O4S2: C, 42.84; H, 6.54; N, 9.08; S, 20.79%. Found: C, 42.75; H, 6.61; N, 8.97; S, 20.33%.
Synthesis of 1,10-dithia-4,7-diazacyclotetradecane-3,8-dicarboxylic acid, (NEC-SB, 3)
Ethylendicysteine (5.50 g, 20.50 mmol) was added to a methanol/water (4 : 1) solution (100 mL) of NaOH (3.28 g, 82.01 mmol) and the solution was stirred at room temperature for 2 h. 1,4-dibromobutane (4.43 g, 20.50 mmol) were added and the mixture was stirred for 48 h to obtain a clear yellow solution. The solution was acidified to pH 2. The precipitate which separated out was filtered, washed with water (3 × 50 mL) and ethanol (2 × 25 mL) and dried under reduced pressure to yield derivative 3 in 70% yield. Melting point, 202 °C (decomposition). 1H NMR (D2O/NaOD, 293 K): δ 1.50 (br, 4H, CH2), 2.54 (br, 4H, CH2N), 2.64 (m, 8H, CH2S), 3.02 (t, 2H, CH). 13C{1H}NMR (D2O/NaOD, 293 K): δ 27.11 (CH2), 30.52 (CH2S), 33.44 (CH2S), 45.73 (CH2N), 61.90 (CH), 179.46 (CO2H). IR (cm−1): 3397br (νNH), 1726m (νasymCOOH), 1633s (νasym COO−), 634w, 616w, 569m, 552m, 536m, 457w, 376w, 280m. ESI-MS (major negative-ions, H2O/NH4OH), m/z (%): 160 (100) [M – 2 H]2−. Anal. Calcd. for C12H22N2O4S2: C, 44.70; H, 6.88; N, 8.69; S, 19.89%. Found: C, 44.55; H, 6.72; N, 8.38; S, 19.44%.
Complex synthesis and characterization
Synthesis of [Cu(NEC-SE)]·3H2 O (4)
Complex 4 was prepared by stirring 1 (0.293 g, 1.0 mmol) and (CH3COO)2Cu (0.200 g, 1.0 mmol) in 30 mL of H2O for 6 h at room temperature. The resulting blue solid was filtered off, washed with water (3 × 50 mL) and ethanol (3 × 25 mL) and dried under reduced pressure to yield 4 in 85% yield. Melting point, 179–182 °C (decomposition). IR (cm−1): 3409br (νNH), 3210w (νOH), 1610 s (νasymCOO), 1362 s (νsymCOO), 644mbr, 556br, 528br, 467br, 430w, 400w, 377m, 337m, 310br. Anal. Calcd. for C10H22CuN2O7S2 : C, 29.30; H, 5.41; N, 6.83; S, 15.64%. Found: C, 29.41; H, 5.48; N, 6.60; S, 15.46%.
Synthesis of [Cu(NEC-SP)]·3H2 O (5)
Complex 5 was prepared by stirring 2 (0.308 g, 1.0 mmol) and (CH3COO)2Cu (0.200 g, 1.0 mmol) in 30 mL of H2O at room temperature for 6 h. The resulting blue precipitate was filtered off, washed with water (3 × 50 mL) and ethanol (3 × 25 mL) and dried under reduced pressure to yield 5 in 77% yield. Melting point, 173–176 °C (decomposition). IR (cm−1): 3412br (νNH), 3162wbr (νOH), 1617s (νasymCOO), 1384s (νsymCOO), 640br, 460 m, 445sh, 377w, 350br, 268w, 255w. Anal. Calcd. for C11H24CuN2O7S2 : C, 31.16; H, 5.71; N, 6.61; S, 15.12%. Found: C, 31.11; H, 5.58; N, 6.41; S, 14.97%.
Synthesis of [Cu(NEC-SB)] (6)
Complex 6 was prepared by stirring 3 (0.322 g, 1.0 mmol) and (CH3COO)2Cu (0.200 g, 1.0 mmol) in 30 mL of H2O at room temperature for 6 h. The resulting blue precipitate was filtered off, washed with water (3 × 50 mL) and ethanol (3 × 25 mL) and dried under reduced pressure to yield 6 in 67% yield. Melting point, 187 °C (decomposition). IR (cm−1): 3397br (νNH), 3209br (νOH), 1601s (νasymCOO), 1358s (νsymCOO), 617m, 592w, 568w, 553br, 400br, 385br, 330br, 301w, 280m. Anal. Calcd. for C12H20CuN2O4S2: C, 37.54; H, 5.25; N, 7.30; S, 16.70%. Found: C, 37.35; H, 5.07; N, 7.19; S, 16.53%.
Synthesis of radiocomplexes [64Cu(NEC-SE)] (7), [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9)
Copper-64 was produced and purified as previously described.58 64CuCl2 (200 μCi) was diluted in 100 μL of 0.1 M ammonium acetate buffer (pH 7.5). 0.25 mg of 1, 2, or 3 was dissolved in 100 μL of the same buffer, and then added to the 64Cu solution. It was necessary to dissolve 3 in 20 μL methanol prior to diluting with the 0.1 M ammonium acetate buffer. After vigorous stirring of the resulting mixture at 90 °C for 1 h, deprotonation and coordination of the enantiomerically pure macrocycle to the metal center occurred. The presence in solution of the labeled complexes was established by TLC (7, Rf = 0.32; 8, Rf = 0.35; 9, Rf = 0.55) by comparison with the TLC profile of an authentic sample of the corresponding non-radioactive complex. The complexes were produced in a radiochemical purity of >98%. For the biodistribution studies the radioactive complexes were diluted with 0.9% saline.
Biodistribution studies
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals established by Washington University’s Animal Studies Committee. Male Lewis rats (30-day old, Charles River Laboratories, Inc., Wilmington, MA) were injected with either complex 7, 8 or 9 (~40 μCi in 150 μL). Rats were examined at 3 time points (n = 4 per group at 1, 4 and 24 h). In all studies following euthanasia, 19 tissues and organs of interest were removed and weighed, and the radioactivity present was measured in a γ-counter. The percent dose per gram (%ID/g) and percent dose per organ (%ID/organ) were then calculated on comparison to known standards.
Molecular modeling
All three copper complexes were studied utilizing a three-step approach. First each copper complex was built by hand and geometry optimized in the program PCModel59, using the GMX force field.60 This is a MM2-like valence force field and uses a Urey-Bradley force field about metal centers. Monte Carlo based conformational searches, GMMX, similar to that developed by Saunders et al61 were performed on each complex in order to optimize the geometry about the copper atoms. The lowest energy conformation of each complex was then exported to the program AMPAC62 and optimized using the SAM/1D Hamiltonian. The optimized structures were then exported into JAGUAR and DFT calculations (B3LYP/LACVP*) were then performed on each complex followed by a NBO analysis.63
Supplementary Material
Acknowledgments
We are very grateful for the technical assistance of Christopher Sherman. Thanks also to Tom Voller and the WUSTL cyclotron facility staff for radionuclide production. This work was partially supported by grants from the National Institutes of Health/National Cancer Institute (NIH/NCI) (CA86307 and CA93375), by University of Camerino (FAR 2007), and by Ministero dell’Istruzione dell’Università e della Ricerca (PRIN 2007).
Abbreviations
- DOTA
1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid
- EC
N,N-Ethylene-di-L-cysteine
- NMR
Nuclear magnetic resonance
- ESI-MS
Electrospray ionization mass spectroscopy
- IR
Infra-red
- NEC-SE
1,10-Dithia-4,7-diazacyclododecane-3,8-dicarboxylic acid
- NEC-SP
1,10-Dithia-4,7-diazacyclotridecane-3,8-dicarboxylic acid
- NEC-SB
1,10-Dithia-4,7-diazacyclotetradecane-3,8-dicarboxylic acid
- PET
Positron emission tomography
- TETA
1,5,8,11-Tetraazacyclotetradecane-N,N′,N″,N‴-tetraacetic acid
- TLC
Thin layer chromatography
Footnotes
Electronic supplementary information (ESI) available: Complete biodistribution data ( %ID/g and %ID/organ) for 19 harvested tissues at 1, 4 and 24 h for [64Cu(NEC-SE)] (7), [64Cu(NEC-SP)] (8) and [64Cu(NEC-SB)] (9) in male, Lewis rats. See DOI: 10.1039/b808831d
Contributor Information
Jason S. Lewis, Email: lewisj2@mskcc.org.
Carlo Santini, Email: carlo.santini@unicam.it.
Notes and references
- 1.Blower PJ, Lewis JS, Zweit J. Nucl Med Biol. 1996;23:957–980. doi: 10.1016/s0969-8051(96)00130-8. [DOI] [PubMed] [Google Scholar]
- 2.McQuade P, Rowland DJ, Lewis JS, Welch MJ. Curr Med Chem. 2005;12:807–818. doi: 10.2174/0929867053507397. [DOI] [PubMed] [Google Scholar]
- 3.Moi MK, Meares CF, McCall MJ, Cole WC, DeNardo SJ. Anal Biochem. 1985;148:249–253. doi: 10.1016/0003-2697(85)90653-0. [DOI] [PubMed] [Google Scholar]
- 4.Sun X, Wuest M, Weisman GR, Wong EH, Reed DP, Boswell CA, Motekaitis R, Martell AE, Welch MJ, Anderson CJ. J Med Chem. 2002;45:469–477. doi: 10.1021/jm0103817. [DOI] [PubMed] [Google Scholar]
- 5.Anderson CJ, Welch MJ. Chem Rev. 1999;99:2219–2234. doi: 10.1021/cr980451q. [DOI] [PubMed] [Google Scholar]
- 6.Cabbiness DK, Margerum DW. J Am Chem Soc. 1969;91:6540–6541. [Google Scholar]
- 7.Heeg MJ, Jurisson SS. Acc Chem Res. 1999;32:1053–1060. [Google Scholar]
- 8.Hubin TJ. Coord Chem Rev. 2003;241:27–46. [Google Scholar]
- 9.Kaden TA. Dalton Trans. 2006:3617–3623. doi: 10.1039/b606410h. [DOI] [PubMed] [Google Scholar]
- 10.Li M, Meares CF, Zhong GR, Miers L, Xiong CY, DeNardo SJ. Bioconjugate Chem. 1994;5:101–104. doi: 10.1021/bc00026a001. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Edwards DS. Bioconjugate Chem. 2001;12:7–34. doi: 10.1021/bc000070v. [DOI] [PubMed] [Google Scholar]
- 12.Meyer M, Dahaoui-Gindrey V, Lecomte C, Guilard RC. Coord Chem Rev. 1998;178–180:1313–1405. [Google Scholar]
- 13.Izatt RM, Bradshaw JS, Nielsen SA, Lamb JD, Christensen JJ, Sen D. Chem Rev. 1985;85:271–339. [Google Scholar]
- 14.Delgado R, Félix V, Lima LMP, Price DW. Dalton Trans. 2007:2734–2745. doi: 10.1039/b704360k. [DOI] [PubMed] [Google Scholar]
- 15.Siegfried L, Kaden TA. Dalton Trans. 2005:1136–1140. doi: 10.1039/b417405b. [DOI] [PubMed] [Google Scholar]
- 16.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Curr Pharm Des. 2007;13:3–16. doi: 10.2174/138161207779313768. [DOI] [PubMed] [Google Scholar]
- 17.Anderson CJ, Dehdashti F, Cutler PD, Schwarz SW, Laforest R, Bass LA, Lewis JS, McCarthy DW. J Nucl Med. 2001;42:213–221. [PubMed] [Google Scholar]
- 18.Anderson CJ, Jones LA, Bass LA, Sherman ELC, McCarthy DW, Cutler PD, Lanahan MV, Cristel ME, Lewis JS, Schwarz SW. J Nucl Med. 1998;39:1944–1951. [PubMed] [Google Scholar]
- 19.Lewis JS, Lewis MR, Cutler PD, Srinivasan A, Schmidt MA, Schwarz SW, Morris MM, Miller JP, Anderson CJ. Clin Cancer Res. 1999;5:3608–3616. [PubMed] [Google Scholar]
- 20.Lewis JS, Lewis MR, Srinivasan A, Schmidt MA, Wang J, Anderson CJ. J Med Chem. 1999;42:1341–1347. doi: 10.1021/jm980602h. [DOI] [PubMed] [Google Scholar]
- 21.McQuade P, Miao Y, Yoo J, Quinn TP, Welch MJ, Lewis JS. J Med Chem. 2005;48:2985–2992. doi: 10.1021/jm0490282. [DOI] [PubMed] [Google Scholar]
- 22.Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL, Anderson CJ. J Med Chem. 2004;47:1465–1474. doi: 10.1021/jm030383m. [DOI] [PubMed] [Google Scholar]
- 23.Wong EH, Weisman GR, Hill DC, Reed DP, Rogers ME, Condon JS, Fagan MA, Calabrese JC, Lam KC, Guzei IA, Rheingold AL. J Am Chem Soc. 2000;122:10561–10572. [Google Scholar]
- 24.Spraugue JE, Peng Y, Sun X, Weisman GR, Wong EH, Achilefu S, Anderson CJ. Clin Cancer Res. 2004;10:8674–8682. doi: 10.1158/1078-0432.CCR-04-1084. [DOI] [PubMed] [Google Scholar]
- 25.Westerby BC, Juntunen KL, Leggett GH, Pett VB, Koenigbauer MJ, Purgett MD, Taschner MJ, Ochrymowycz LA, Rorabacher DB. Inorg Chem. 1991;30:2109–2120. [Google Scholar]
- 26.Bouwman E, Driessen WL, Reedijk J. Coord Chem Rev. 1990;104:143–172. [Google Scholar]
- 27.Busch DH, Jicha DC. Inorg Chem. 1:884–887. [Google Scholar]
- 28.Crooks HM. The Chemistry of Penicillin. Princeton University Press; Princeton, NJ: 1949. [Google Scholar]
- 29.Bharadwaj PK, Potenza JA, Schugar HJ. J Am Chem Soc. 1986;108:1351–1352. [Google Scholar]
- 30.Stibrany RT, Fox S, Bharadwaj PK, Schugar HJ, Potenza JA. Inorg Chem. 2005;44:8234–8242. doi: 10.1021/ic050114h. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto K, Fushimi N, Konno T, Hidaka J. Bull Chem Soc Jpn. 1991;64:2635–2643. [Google Scholar]
- 32.Blondeau P, Berse C, Gravel D. Can J Chem. 1967;45:49–52. [Google Scholar]
- 33.Davison A, Jones AG, Orvig C, Sohn M. Inorg Chem. 1981;20:1629–1632. [Google Scholar]
- 34.Hansen L, Lipowska M, Taylor A, Marzilli LG. Inorg Chem. 1995;34:3579–3580. [Google Scholar]
- 35.Hansen L, Hirota S, Xu X, Taylor AT, Marzilli LG. Inorg Chem. 2000;39:5731–5740. doi: 10.1021/ic000183q. [DOI] [PubMed] [Google Scholar]
- 36.Hansen L, To Yue K, Xu X, Lipowska M, Taylor A, Marzilli LG. J Am Chem Soc. 1997;119:8965–8972. [Google Scholar]
- 37.Berg JM, Spira DJ, Hodgson KO, Bruce AE, Miller KF, Corbin JL, Stiefel EI. Inorg Chem. 1984:23. [Google Scholar]
- 38.Anderson CJ, John CS, Li YJ, Hancock RD, McCarthy TJ, Martell AE, Welch MJ. Nucl Med Biol. 1995;22:165–173. doi: 10.1016/0969-8051(94)00106-t. [DOI] [PubMed] [Google Scholar]
- 39.Van Nerom CG, Bormans GM, De Roo MJ, Verbruggen AM. Eur J Nucl Med. 1993;20:738–746. doi: 10.1007/BF00180902. [DOI] [PubMed] [Google Scholar]
- 40.Verbruggen AM, Nosco DL, Van Nerom CG, Bormans GM, Adriaens PJ, De Roo MJ. J Nucl Med. 1992;33:551–557. [PubMed] [Google Scholar]
- 41.Yang DJ, Azhdarinia A, Wu P, Yu DF, Tansey W, Kalimi SK, Kim EE, Podoloff DA. Cancer Biotherapy Radiopharm. 2001;16:73–83. doi: 10.1089/108497801750096087. [DOI] [PubMed] [Google Scholar]
- 42.Zareneyrizi F, Yang DJ, Oh CS, Ilgan S, Yu DF, Tansey W, Liu CW, Kim EE, Podoloff DA. Anti-Cancer Drugs. 1999;10:685–692. doi: 10.1097/00001813-199908000-00009. [DOI] [PubMed] [Google Scholar]
- 43.Yang DJ, Ilgan S, Higuchi T, Zareneyrizi F, Oh CS, Liu CW, Kim EE, Podoloff DA. Pharm Res. 1999;16:743–750. doi: 10.1023/a:1018836911013. [DOI] [PubMed] [Google Scholar]
- 44.Buchanan I, Minelli M, Ashby MT, King TJ, Enemark JH, GCD Inorg Chem. 1984;23:495–500. [Google Scholar]
- 45.Anderson CJ, Rocque PA, Weinheimer CJ, Welch MJ. Nucl Med Biol. 1993;20:461–467. doi: 10.1016/0969-8051(93)90077-8. [DOI] [PubMed] [Google Scholar]
- 46.Fujibayashi Y, Matsumoto K, Arano Y, Yonekura Y, Konishi J, Yokoyama A. Chem Pharm Bull Tokyo. 1990;38:1946–1948. doi: 10.1248/cpb.38.1946. [DOI] [PubMed] [Google Scholar]
- 47.Rorabacher DB. Inorg Chem. 1966;5:1891–1899. [Google Scholar]
- 48.Taylor RW, Stepien HK, Rorabacher DB. Inorg Chem. 1974;13:1282–1289. [Google Scholar]
- 49.Turan TS, Rorabacher DB. Inorg Chem. 1972;11:288–295. [Google Scholar]
- 50.Siegfried L, Kaden TA. Helv Chim Acta. 1984;67:29–38. [Google Scholar]
- 51.Balakrishnan KP, Kaden TA, Siegfried L, Züberbuhler AD. Helv Chim Acta. 1984;67:1060–1069. [Google Scholar]
- 52.Kaden TA, Kaderli S, Sager W, Siegfried-Hertli LC, Züberbuhler AD. Helv Chim Acta. 1986;69:1216–1223. [Google Scholar]
- 53.Riesen PC, Kaden TA. Helv Chim Acta. 1995;78:1325–1333. [Google Scholar]
- 54.Nakamoto K. Applications in Coordination, Organometallic, and Bioinorganic. John Wiley & Sons, Inc; New York: 1997. pp. 62–73. [Google Scholar]
- 55.Herlinger AW, Wenhold SL, Long TV., II J Am Chem Soc. 1970;4:6474–6481. [PubMed] [Google Scholar]
- 56.Jones-Wilson TM, Deal KA, Anderson CJ, McCarthy DW, Kovacs Z, Motekaitis RJ, Sherry AD, Martell AE, Welch MJ. Nucl Med Biol. 1998;25:523–530. doi: 10.1016/s0969-8051(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 57.Bass LA, Wang M, Welch MJ, Anderson CJ. Bioconjugate Chem. 2000;11:527–532. doi: 10.1021/bc990167l. [DOI] [PubMed] [Google Scholar]
- 58.McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS, Anderson CJ, Welch MJ. Nucl Med Biol. 1997;24:35–43. doi: 10.1016/s0969-8051(96)00157-6. [DOI] [PubMed] [Google Scholar]
- 59.PCModel. Serena Software; Bloomington, IN: 2003. [Google Scholar]
- 60.Gajewski JJ, Gilbert KE, Kreek TW. J Comput Chem. 1997;19:1167–1178. [Google Scholar]
- 61.Saunders M, Houk KN, Wu YD, Still WC, Lipton M, Chang G, Guida WC. J Am Chem Soc. 1990;112:1419–1427. [Google Scholar]
- 62.AMPAC. Semichem, Inc; Shawnee Mission: 2004. [Google Scholar]
- 63.Glendenning ED, Badenhoop JK, Reed AE, Carpenter JE, Bohman JA, Morales CM, Weinhold F. NBO 5.0. Theoretical Chemistry Institute, University of Wisconsin; Madison, WI: 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.