

Abstract
The conserved cylindromatosis (CYLD) codes for a deubiquitinating enzyme and is a crucial regulator of diverse cellular processes such as immune responses, inflammation, death, and proliferation. It directly regulates multiple key signaling cascades, such as the Nuclear Factor kappa B [NF-kB] and the Mitogen-Activated Protein Kinase (MAPK) pathways, by its catalytic activity on polyubiquitinated key intermediates. Several lines of emerging evidence have linked CYLD to the pathogenesis of various maladies, including cancer, poor infection control, lung fibrosis, neural development, and now cardiovascular dysfunction. While CYLD-mediated signaling is cell type and stimuli specific, the activity of CYLD is tightly controlled by phosphorylation and other regulators such as Snail. This review explores a broad selection of current and past literature regarding CYLD’s expression, function and regulation with emerging reports on its role in cardiovascular disease.
Keywords: Cardiovascular, CYLD, cylindromatosis, deubiquitination, K63, MAPK, NF-kB
1. INTRODUCTION
1.1. CYLD
The cylindromatosis gene CYLD is a cornerstone of the inflammatory response and is a critical regulator, via control over NF-kB-mediated inflammation, of many cellular processes. Much research into CYLD’s function and mechanics has been reported in the literature, but there is a lack of knowledge in the role of CYLD in the cardiovascular system. Since recent developments in the body of evidence for a CYLD/cardiovascular interaction have opened up a new frontier of CYLD research, this review will present a broad slice of current knowledge (form, function, regulation) and then examine the newest data that potentially links CYLD to cardiovascular disease, along with some speculation as to the mechanisms by which this link occurs.
1.2. Brief History
The cylidromatosis gene CYLD’s existence and location on chromosome 16q12-13 were first elucidated in 1995 by Biggs and colleagues, exploring CYLD’s role in skin cylidromatosis tumors [1]. In 1999, Thomson and colleagues followed with more detail on CYLD’s genetic association with the cylindromatosis tumors. A landmark report in 2000 by Bignell and colleagues established CYLD’s coding region by exploring the genomic mutations in multiple cases of the tumors [2]. This paper also reported three Cap-Gly domains in CYLD. Other studies explored cases of CYLD mutation in 21 families afflicted with cylindromatosis and found that truncation of the functional Cap-Gly domains was present [3]. After sparking interest, a spate of papers were published in rapid succession exploring the role of CYLD in the regulation of the NF-kB pathway [4–7]. Once the importance of CYLD in NF-kB signaling was confirmed, Saito and colleagues used nuclear magnetic resonance and computer fit modeling to discover that a specific Cap-Gly domain (aa470-684) in CYLD binds with the proline-enriched portion of NEMO, providing evidence of direct interaction [8]. Exploration then continued with the thought of CYLD as being a tumor suppressor and inflammatory control factor, especially in the skin [9, 10]. As more pathogen responses and developmental aspects of cellular biology began to be linked to inflammation, CYLD’s role as a master regulator of NF-kB-induced inflammation came into the forefront as a factor that could switch off NF-kB and prevent tissue damage that leads to necrosis, cancer and loss of cell proliferation [11–13]. From 2009, many reports detailing specific instances in diverse tissue types (such as lungs, immune cells, breast tissue and bone) where CYLD loss of function is deleterious have been published [14–22]. From the first discovery of NF-kB regulation by deubiquitination to key roles in immune response and cell maintenance to recent studies in cellular necrosis, CYLD is proving to be important in controlling critical cellular pathways. Accordingly, it is not surprising that CYLD has been implicated in the pathogenesis of several maladies including cardiovascular disease.
2. CYLD OVERVIEW
2.1. Structure, Catalytic Function, Location, and Regulation of CYLD Deubiquitinase
CYLD is a gene of 60 kb in length on Chromosome 16q12.1 in humans (Genecards.org). It encodes for a thioesterase enzyme that is 956 amino acids in length and contains three Cap-Gly domains for interaction with targets such as NEMO in the NF-kB pathway [8, 14]. It is capable of cleaving ester sulfhydryl groups and also contains a C-terminal USP catalytic domain that acts on specific lysine residues in ubiquitin [23]. Furthermore, a triumvirate of Cys601, His871, and Asp889 in the α1 helical subdomain of CYLD effects a nucleophilic attack on the K63 of ubiquitin [23]. This region was resolved to 2.8A.
CYLD is found primarily in the cytoplasm and per-inuclear spaces of multiple cell types. Array data based on RNA transcript abundance at Genecards.org indicates that CYLD is found in low levels in a large percentage of somatic cells, with immune cells expressing more of it [24]. Unlike other deubiquitinases such as A20, CYLD is constitutively expressed, albeit at a low basal level [25, 26]. Regulation of both CYLD transcription and translation is highly variable, with multiple mechanisms available, including direct phosphorylation by kinases, treatment with allosteric caspase inhibitors like zVAD, protealytic cleavage, external serum receptor transduction and subsequent modification via kinases [such as SRF to MAPK and CaMKII pathways], control by other transcription factors and gene interactions, and direct binding by miRNAs [27–37]. (Fig. 2A) Reactive oxygen species [ROS] caused by serum starvation or ROS-generating chemicals like meniadone were shown to negatively regulate transcription of CYLD and other cell regulatory genes in a HepG2 hepatocyte cell line while serum levels regulate CYLD through the action of MAPK [30, 38]. A review by Sun extensively details CYLD structure, function and known regulation [39].
Fig. 2.

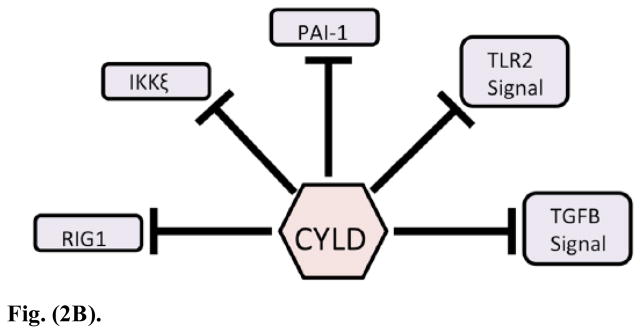
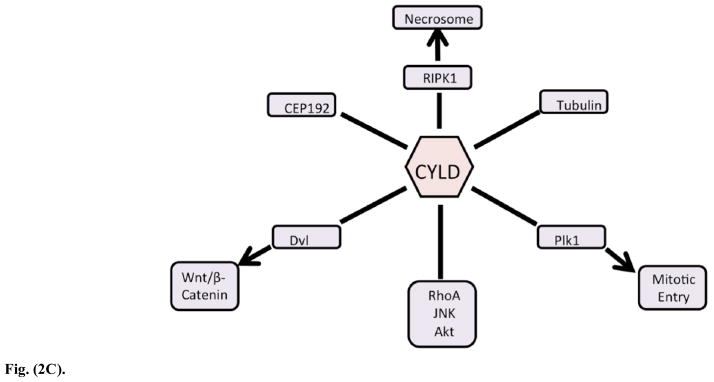
Fig. (2A) The regulation of CYLD is complex and multimodal. As the key effector of K63-linked deubiquitination, which transduces cellular signals, CYLD is tightly regulated at the transcript and protein level, as well as by phosphorylation.
Fig. (2B). CYLD’s regulation of the immune system.
Fig. (2C). CYLD’s interaction with important regulators of necrosis and development.
Evidence of regulation of CYLD expression by pharmacological agents is scarce in the literature, but it may be possible to indirectly regulate CYLD levels by agents that act on regulatory factors for CYLD, such as Serum Response Factor [SRF] or kinase inhibitors. Clinical disease due to a lack of CYLD function resulting in cylindromatosis can be modulated by agents known to downregulate inflammation, such as aspirin [4]. Levels of CYLD may be indirectly increased by allosteric inhibition of caspase 8 (which cleaves CYLD) by zVAD-FMK [40].
Multiple mutations of CYLD are possible, with mutations in the catalytic domain causing an ablation of deubiquitinating activity [14, 15]. Medical literature contains no current reports on clinical mutations that overexpress CYLD.
2.2. Ubiquitination as a Key Process in Cellular Homeostasis
Ubiquitin was first characterized by Ciehanover and colleagues in 1978, and its interaction with the proteasome was elucidated in 1990 by Driscoll and Goldberg. It is a 76-amino acid small protein that is controlled primarily by 3 classes of ubiquitin-related enzymes: E1 ubiquitin-activators, E2 ubiquitin-conjugators, and E3 ubiquitin ligases [41–44]. Ubiquitin has a C-terminal end that can attach to these enzymes and, on the N terminus, 7 lysine (K) which can attach to proteins and which control the effect of the ubiquitin on the proteins K6, 11, 27, 29, 33, 48, and 63 [41, 45, 46]. The location and number of ubiquitin is critical, as they will signal for different outcomes. For example, K48 monoubiquitination is a signal for proteasomal degradation, while K63 polylinkages are used in signal transduction pathways [23, 47, 48]. Exhaustive and extensive reviews are available for every aspect of ubiquitination, with recent research attempting to elucidate the roles of lesser studied lysine chain links such as K11, which seems to be involved in cell cycle regulation, K29 which seems to form a foundation for further K48 linkages, and linear linkages via LUBAC, which bear similarity to K63 polyubiquitin chains, but do not bind similarly to NEMO and can promote proteasomal degradation [49, 50]. As its name suggests, ubiquitin is found everywhere in multiple cell types, including immune cells, hepatocytes, skeletal muscle and even vascular smooth muscle cells (VSMCs) [51, 52]. Ubiquitination has even been linked with nitric oxide production through the ubiquitin binding domain of the GTP cyclohydrase GTPCH1, which catalyzes the rate limiting step of tetrahydrobopterin (BH4), the key component in Nitric Oxide Synthase’s manufacture of nitric oxide [53]. This indirect control of vascular tone by ubiquitin regulation is yet another example of the importance of ubiquitin in every somatic system. Thus, it is clear that with such a large number of possible combinations of lysine linkages, dozens of E2 and E3 enzymes to provide specificity and a proteasomal system that can discriminate between linkage types, the process of ubiquitination is a critical part of cellular homeostasis and reactivity to the extracellular environment. Given the importance of the process of appending ubiquitins, it stands to reason that the process of ubiquitin removal would act as a key regulator of cellular homeostasis, a role that CYLD fills.
2.3. Prominent Diseases Featuring CYLD
The most prominent disease caused by a functional deletion or inherited mutation of CYLD is cylindromatosis. These benign tumors grow profusely from the sweat glands around the head, face and neck. Known as Brooke-Spiegler syndrome, the phenotypic manifestation of hereditary cylindroma was mentioned in the clinical literature as far back as 1982, with the first English language journal paper being published 9 years later [54, 55]. There is also some evidence that CYLD is responsible for Paget’s disease of the bones, which is marked by fragility due to affected osteoclast differentiation [16].
Neuroscientific studies have indicated that CYLD accumulates in the postsynaptic density (PSD) and is thought to be involved in removing K-63 from PSD proteins, preventing their autophagic regulation [56]. This accumulation and lack of recycling may cause neurodegeneration [56]. CaM-KII (calmodulin-dependent protein kinase II) mediates this recruitment of CYLD in the brain [31].
Cancer seems to be the overwhelming disease mediated by a lack of CYLD enzymatic activity. This is deemed to be due to CYLD’s role in checking inflammation which has been demonstrated in many types of cancer [17, 18, 34, 35, 57–60]. Since NF-kB-mediated inflammation is widely known to cause the generation of tissue damaging reactive oxygen species, inflammatory cytokine cascades and recruitment of immune cells (which release factors like granzymes and superoxides), it is logical that CYLD’s ability to cut off NF-kB activation would limit subsequent damage. For an in depth look at NF-kB and its myriad targets, regulation and pathways affected, an excellent review by Hayden is recommended [61].
3. CYLD FUNCTION
3.1. CYLD and NF-kB: The Chief Pathway
CYLD’s main role in the cell is to regulate the TNFR-mediated activation of NF-kB (Fig. 1). NF-kB is kept under constant suppression by IkB until it is phosphorylated by IKK (which contains catalytic subunits named IKKα and IKKβ) [46]. The IKK protein has a catalytic subunit named IKKγ (a.k.a, NEMO), which is polyubiquitinated in a K63-linked fashion to signal it to phosphorylate IkB, which then releases p50 and p65 to translocate to the nucleus where they upregulate transcription of inflammatory factors [7, 46]. CYLD can deubiquitinate NEMO, preventing it from causing phosphorylation of IkB, thereby killing the signal [4, 8, 62, 63]. For a more detailed treatment of this process, a review by Harhaj and Dixit is recommended [46]. Since NF-kB is a transcription factor that controls so many other aspects of cellular homeostasis (apoptosis, inflammation, growth, etc.), regulation by CYLD on NF-kB becomes an important mediator for the immune system, development, and cell death.
Fig. 1.

The canonical TNFR to Nf-kB pathway.
B. CYLD in Immune System Regulation and Response to Infectious Diseases (Fig. 2B)
CYLD plays a critical role in the regulation of immune cell maintenance and differentiation. CYLD can regulate recruitment of Lck to Zap70 to control T cell development and NF-kB suppression by CYLD prevents errant B cell activation [62, 63]. T cell regulation by CYLD is extensively reported in the literature, with PKC θ/β activation from the T cell receptor opposing the negative regulation of activation by CYLD [64]. Additionally, splice variants of CYLD (CYLD 7/8, aka “short” CYLD, where a deletion of exons 7 and 8 affect catalytic action) have been shown to result in an increase in FoxP3+ T regulatory cells that lack suppressive capacity [65]. This splice variant lacks binding sites for NEMO and TRAF2 removing the variant’s ability to regulate the NF-kB-mediated activation of B cells [66]. CYLD is assisted in regulatory activities by the action of a de-SUMOylator called SNEP6, which removes the Small Ubiquitin-like Modifier SUMO-2/3 on NEMO so that CYLD can deubiquitinate it [67]. This allows for transduction of the TLR signal in septic infection and subsequent activation of the immune system. It has also been reported that even dendritic cells, professional Antigen Presenting Cells (APCs) which are descended from monocytes or bone marrow, are controlled by CYLD. A report showed that murine primary cells with a non-catalytic CYLD isoform (CYLD 7/8) increased allogenic T cell stimulation and had no toleragenic activity, while treatment of these cells with the NF-kB inhibitor dexamethasone restored toleragenic activity [68]. CYLD can thus be said to reach almost every cell in the immune system. This extends the CYLD paradigm into the tight control of immune cells by regulating their inflammatory cascades and modes of action.
CYLD has also been shown to extensively regulate the response to pathogens by modulating the inflammatory cascade in immune cells. For example, CYLD has been proven to negatively regulate TLR2 by TRAF6 and TRAF7 and an antiviral response in an H. influenza co-infection model [11, 69]. In E. coli-induced pneumonia, CYLD has been shown to negatively regulate the immune response by blocking PAMP (Pathogen Associated Molecular Pattern)-induced NF-kB activation [21]. The PAMP-mediated pathway is readily verified in CYLD’s negative regulation of lung and ear inflammation in H. influenza in mice [22]. The net result of this negative regulation is increased tissue damage in the lungs by hindering the immune system’s control of the pathogens. However, CYLD can also inhibit S. pneumonia-induced lung damage by reducing expression of pneumolysin-induced PAI-1, which is dependent on MKK3-p38 kinase activation [20]. This seemingly contradictory information indicates that CYLD has a dual role in infection response by the immune system, with the degree of CYLD’s action determining whether or not an immune activating inflammation threshold is reached. Loss of CYLD function in mice causes extensive fibrosis of the lungs after S. pneumoniae infection [19]. This was discovered to be due to CYLD’s inhibition of TGFβ signaling via deubiquitination of Smad3. CYLD can inhibit ubiquitination of the RIG1 cytoplasmic viral RNA sensor and also downregulate antiviral Interferon production by controlling IKK activation [69]. This downregulates inflammation and, thusly, activation of the immune system. This is in addition to indications that NF-B, which enhances HIV Long Terminal Repeat transcription by NF-kB/NFAT sites that flank the LTR, is able to drive transcription of viral LTRs when CYLD action was removed in T cell lines, indicating that at least some viral strategies include control of NF-kB and CYLD may be the key to stopping the hijacking of NF-kB in these cases [70]. CYLD impairs production of fibrin in Listeria infection in mice by inhibiting NF-kB-mediated activation of IL-6 production and IL-6’s activation of the STAT3 pathway that leads to fibrosis [71]. Since fibrosis in L. monocytogenes infection is considered to be protective, this is yet another instance of CYLD’s negative regulation of the immune system causing harm by blocking necessary inflammation that provokes a protective response. Taken together, while the immune response itself is intimately controlled by CYLD, there are other factors (such as cytokine milieu and presence or absence of T regulatory cells) that eventually determine whether or not the inflammation is detrimental (tissue damage, edema, etc.) or beneficial (proper immune response to control the infection or injury).
3.3. Cancer and Other Diseases Resulting from CYLD Misregulation
The TNFR-mediated NF-kB pathway is responsible for a myriad of maladies that stem from inflammation-induced tissue damage and errant immune activation which result in the release of reactive oxygen species and tissue damaging components (e.g., granzymes). CYLD is best known as a major negative regulator of this pathway. There are several excellent reviews that detail the involvement of CYLD with NF-kB activation, including an extensive review by Chen on NF-kB and IKK [46, 72, 73]. After initial details of the negative regulation of NF-kB by CYLD emerged, it was found that TRAF3, TRAF5 and TRAF6 do not interact with CYLD but a TRAF-interacting protein (TRIP) does [7, 12]. This shows that, although CYLD is a keystone of NF-kB regulation, alternate mechanisms exist to suppress inflammation and that indirect and alternate regulation could be important factors in TNFR signaling. From this information, multiple mechanisms of inflammation affected by regulation of CYLD are possible. Proteasomal inhibitors can accumulate CYLD and impair RANKL-induced NF-kB expression in an osteoclast-like cell line [74]. Our lab has found that CYLD may play a key role in IgA-induced nephropathy via regulating inflammation that damages nephrons [75]. STAT3, the Signal Transducer and Activator of Transcription, can activate micro RNAs miR-21 and miR-181b-1, suppressing CYLD transcription and causing a transformation to cancer cells via NF-kB-mediated inflammation in MCF10A cells [60]. Upregulation may also have an effect, as CYLD has been shown to prevent apoptotic resistance in two liver cancer cell lines by downregulating NF-kB [76]. In mice, Tak1 (an MLK family kinase important in response to IL-1 that helps activate NF-kB) can be effectively regulated by a complex of CYLD and the Itch E3 Ligase [77, 78]. Overexpression of CYLD in lung cancer cell lines increases the action of TRAIL by NF-kB inhibition, thus promoting apoptosis [79]. Regulation of CYLD can be achieved by suppressive regulatory factors such as the phosphodiesterase PDE4B (which regulates CYLD by activating JNK2 but not JNK1). For example, inhibiting PDE4B in an HMEEC cell line caused an upregulation of CYLD and a concomitant reduction in inflammation [80]. When considered as a whole, the most important aspect of CYLD is the regulation of inflammation, regardless of initial cause. By linking multiple pathways of activation and response to the hub of inflammation, CYLD acts not only as an oncogene, but also a master regulator of critical pathways that result in diverse cellular effects.
3.4. CYLD in Cellular Proliferation, Signaling, and Development (Fig. 2C)
CYLD, while being a master regulator of inflammation, has also been found to play a major role in cellular development and proliferation. Initially, investigations were focused on the role of CYLD in immune cell development as previously discussed [62, 63]. However, it was soon discovered that CYLD had far reaching regulatory effects in multiple cellular pathways. CYLD can act upon Plk1 (Polo-Like Kinase 1) to regulate HeLa cell entry into mitosis [81]. This is notable because Plk1 is a key stabilizer of kinetochore and microtubule formation [82]. CYLD can also regulate micro-tubule development in Hela and CV1 cells by direct interaction with tubulin via its Cap-Gly domains [83]. Angiogenesis is regulated by CYLD modulation of the migration of vascular endothelial cells, which is microtubule dependent [84]. Mitotic spindle formation is a critical step in the cell cycle and CEP192 (Centrosomal Protein of 192kD) interacts directly with CYLD to form the mitotic spindle, further bolstering the proof of CYLD’s regulatory role in cell division [85]. Collectively, this evidence strongly suggests that CYLD has a direct influence on the formation of microtubules, which can then affect migration and cellular division. Additional evidence has also shown CYLD involvement in the regulation of RhoA, JNK and Akt, which are key modulators of proliferation [86–88]. RhoA, in particular, was found to be controlled by CYLD through action on LARG (Leukemia Associate RhoGEF) [86]. Basal cell carcinoma, one of the most common cancers in humans, was shown to downregulate CYLD transcript by the action of the Snail transcription factor, which is in turn activated by the Kruppel zinc finger gene GLI1 [32, 35]. This downregulation affected the keratinocyte cancer cells by increasing their proliferation, which could no longer be controlled by CYLD. It was demonstrated that Notch downstream element Hes-1(Hairy and Enhancer of Split-1) could repress CYLD expression to increase NF-kB activity in mouse primary cell T Acute Lymphoblastic Leukemia [36, 89]. Repression of CYLD expression in this case helps maintain the disease. Knockout of CYLD in tumor cells can prevent K63-linked deubiquitination of Dvl [Dishevelled], which enhances Wnt/β-catenin signaling [90]. Since the Wnt/β-catenin pathway strongly regulates proliferation, it is logical that CYLD exerts strong pressure on Dvl, thereby regulating cell proliferation in a direct manner [91]. Dvl, as a master proliferation gene, also acts as a keystone for spindle formation. CYLD-mediated deubiquitination of Dvl can regulate spindle formation by both stabilization of microtubules [using the aforementioned Cap-Gly domains] and by promoting a dynactin complex formation with Dishevelled and NuMA [Nuclear Mitotic Apparatus protein] [88]. Apparently, Dishevelled binding to its dynaction-NuMA complex becomes more favorable after deubiquitination, indicating yet another critical cellular pathway regulated by CYLD. Eversince the discovery of ubiquitin’s ability to act as both a proteasomal marker and a signaling transduction facilitator, its role in multiple pathways has been extensively catalogued [25, 47, 49, 73, 92, 93]. Since K63-linked ubiquitination is an important transducer of cellular signaling, it then logically follows that the removal of ubiquitin also affects signal pathways. In view of the evidence presented, it is not surprising to find that CYLD also functions as a fundamental regulator of eukaryotic cell development [93].
3.5. CYLD in Necrosis (Fig. 2C)
It is important to include the regulation of cellular death in CYLD’s list of actions. Although necrosis was the classic term in medical literature for cellular death, as far back as 1972 the concept of a differential cell death was known to exist [94]. It was later found that certain environmental changes or exposures could induce this type of death, with starved cells or irradiated cells undergoing a type of death dubbed “apoptosis” [95, 96]. This type of cell death is thought to serve as both protective and necessary in physiological development by safely disposing of damaged or superfluous cells although it can be detrimental as a component of aging or injury in the brain and other tissues [97–99]. Necrosis, on the other hand, is considered to be highly inflammatory and undesirable in all cases. CYLD plays a strong role in activation of necrosis. This leads to the question as to how the cell chooses to undergo an apoptotic or necrotic death, which has grave implications in the prognosis of cancer and developmental disorders. Scrutiny of CYLD’s mode of necrotic regulation gives valuable insight on possible targeting therapies to induce preferential apoptotic death by controlling CYLD transcription or translation.
Necrosis is a tightly regulated process. Upon activation of the TNFα receptor, RIPK1, a signaling kinase, is ubiquitinated and activated to interact with IKK proteins in the NF-kB pathway (as discussed previously). CYLD can deubiquitinate RIPK1, freeing it to be phosphorylated and complex with RIPK3 (Complex IIb) to form the necrosome [100–104]. Conversely, TNFR signaling associated with TRADD and TRAF, along with properly ubiquitinated RIPK1 and IAP (Inhibitor of Apoptosis Proteins) form a complex called “complex I” that moves forward to NF-kB activation and protection from cell death [105, 106]. The critical step in the formation of the functional necrosome is thought to be CYLD-mediated deubiquitination of RIPK1. The necrosome itself is an NP-40 insoluble protein aggregate that shreds the cell membrane, but other intracellular effects such as depletion of ATP (due to repair enzymes using up the available pool) or loss of mitochondrial membrane potential also occur [101, 102, 107]. Unlike apoptosis, where the death can be considered a controlled “implosion”, the process of necrosis could be likened to an “explosion” of the cell. An extensive review by Kung and colleagues details the differences in the process and possible biomarkers (including cell rupture, lack of caspase activity and depleted ATP) to distinguish between necrosis and late stage apoptosis [101]. Necrosis is associated with heavy membrane swelling, as opposed to blebbing in apoptosis [101]. In addition to non-fragmentation of chromatin material, necrosis is also marked by release of cellular products such as heat shock proteins, cyclophilin A and LDH (Lactate Dehydrogenase) [101, 108]. Model systems where TNFα and a pancaspase inhibitor are used to treat cells have proven to be a reliable inducer of necrosis. Also, Necrostatin-1 (an allosteric inhibitor for RIPK1) treatment is able to halt the entire process, thereby demonstrating the reliance of the necrotic process on the formation of the necrosome [106, 109]. Caspases can play a critical role in this process, as the cell will preferentially choose apoptosis over necrosis by the favorable action of the pro-apoptotic caspases. It has been reported in several papers that caspase blockage with inhibitors such as zVAD-FMK (an allosteric pancaspase inhibitor) can protect against TLR/RIPK3-mediated necrosis in microglial cells, but treatment of TNFα and pancaspase inhibitors almost always results in cell death [110]. This action is due to CYLD and its close ties with TNFR. TNFa-induced necrotic signaling can be blocked in some chronic leukemias by the action of LEF1 (Lymphoid Enhancer binding Factor 1), which acts on the Wnt/βCatenin pathway to downregulate CYLD at the transcriptional level [34]. CYLD has been shown to play an integral role in the formation of the necrosome by the deubiquitination of RIPK1, but is itself acted upon by caspase 8, which cleaves it and renders it unable to process RIPK1 [40]. The required action of caspase 8 in cleaving CYLD to halt its deubiquitination of RIPK1 indicates that treatment with a pancaspase inhibitor would not only remove the action of caspase 8, but would remove the action of other pro-apopotic caspases, forcing the cell into necrosis. The intimate involvement of CYLD in necrotic death was elucidated in a landmark paper by Moquin and colleagues that detailed CYLD-mediated regulation of TNFα-induced necrosis by deubiquitination of RIPK1 in MEF and L929 cells, while, conversely, CYLD downregulation by Toll-Like Receptors protects mouse macrophages from necrosis [107, 111]. These reports proved that CYLD induction of necrosis is not limited to a single cell type but that the CYLD-RIPK1 pathway is probably conserved throughout the body. Additionally, both RIPK1 and RIPK3 are deemed to be separate entities that can form a necrosome, versus RIPK3 being a backup for RIPK1. Linker-mann and colleagues reported that RIPK3-deficient mice are protected against TNFα-zVAD-mediated necrosis but that Necrostatin-1 increased death in TNFα-treated RIPK3 knockout mice [that did not also receive caspase inhibitors] but these RIPK3 knockout mice could survive a TNFα challenge without caspase inhibitors and without Necrostatin-1 [106]. This indicates that there is a separate mode of action that prevents simple blockage of RIPK1 from being equivalent to silencing of RIPK3. Further studies are needed to evaluate the role of CYLD in the regulation of RIPK3, but current evidence does not indicate that CYLD can regulate RIPK3 in the same manner as RIPK1. Thus, the role of CYLD as a regulator of necrosis is well supported by its role as the master switch of NF-kB (which induce apoptosis) or in necrosis by the RIPK1 switch. The negative regulation of CYLD by caspase 8 and the subsequent prevention of necrosis leads to further questions as to whether other forms of CYLD regulation (such as phosphorylation or silencing) can force cells to choose between necrosis or apoptosis. Additionally, since necrosis is often found in reperfusion/ischemic injuries, there is the possibility that negative regulation of CYLD in ischemic tissue could provide protection against necrosis [112].
4. CYLD AND THE CARDIOVASCULAR SYSTEM: A NEW FRONTIER
4.1. The Cardiovascular System
The vascular system is a complex network of specialized organs that distribute blood, nutrients, and growth factors to every part of the body, in addition to serving as a conduit for immune cells. The blood vessels consist of three main layers: the innermost intima (endothelial cells lining the blood vessels), the media (a sheath composed of smooth muscle cells that maintain tone), and the outermost adventitia (a tough, fibrous collagen-rich layer that protects and supports the vessel). The pathology and progression of vascular disease and several intriguing areas of ongoing research will be discussed in the context of current knowledge and potential discoveries of the role of CYLD in this system.
Even as far back as 1907, when Duval reported that the toxins of B. mallei caused vascular lesions in rabbits, it was known that injury of some kind would result in the formation of lesions within the vascular structures that suffered the insult [113]. Fast forward to the present where it is now known that multiple types of injuries and insults, such as necrosis, bacterial toxins, mechanical injury (e.g., balloon denudation), hypoxia and lipid-mediated atherosclerosis, can form lesions in the vascular system [114–120]. Multi-stage atherosclerosis, in particular, has been identified as a major cause of lesions in blood vessels, especially those closely associated with the heart, such as the coronary artery and aortic arch [115]. Its pathology is extremely complex, with multiple cell types, inflammation and agents interacting to form a lesion which may be stable at first and then rupture later. Such lesions also may be found in the major cardiovascular areas, including aortic, brain and kidney vessels.
Within the medial layer reside smooth muscle cells and a small population of resident stem cells. The dominant theory in vascular biology is that, upon injury, a large percentage of affected, mature smooth muscle cells (Myosin Heavy Chain and actin positive) will revert to an immature, secretory and synthetic phenotype in a process known and described as “dedifferentiation”. However, a recent discovery by Shi and colleagues has challenged the orthodoxy in the field of vascular lesion formation with a report that proves the presence and action of a small population of resident stem cells that are activated and then differentiated into synthetic or contractile smooth muscle cells [121]. This may be linked to various developmental processes and microenvironmental factors that CYLD is known to affect.
4.2. Effect of CYLD in the Cardiovascular System (Fig. 3)
Fig. 3.
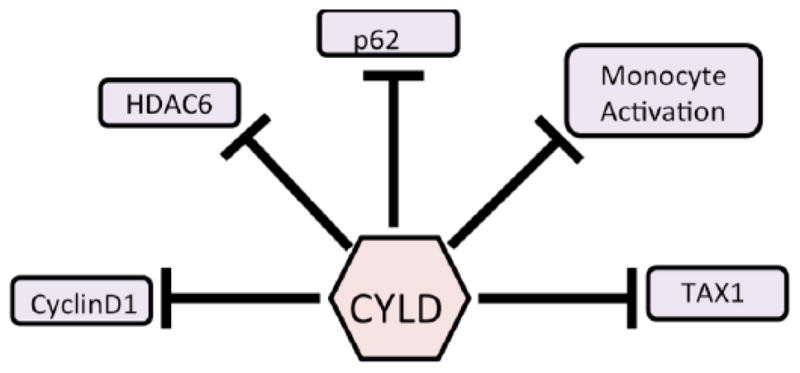
CYLD’s regulation of factors known to play an important role in cardiovascular disease.
As stated above, the arteries consist of three main layers: the innermost intima, the medial layer and the outermost adventitia [122]. These layers form from unique populations of progenitor cells during the developmental stage. The heart, on the other hand, consists mainly of cardiomyocytes and fibroblasts, with smaller populations of vascular smooth muscle cells and endothelial cells [123]. It is in the intimal layer of the vessels and the cardiomyocytes in which CYLD may prove to mediate lesion formation and cardiac dysfunction.
Empirical evidence of CYLD’s involvement in vascular disease is scarce, with very few reports that provide extensive mechanistic studies into the role of CYLD in the cardiovascular system. Our lab has previously enumerated CYLD’s role in modulating tubulointerstitial inflammation in IgA nephropathy, which is a hallmark of end-stage kidney disease [75]. Apparently, knockout of CYLD did not affect albumin or peroxide-related cell death in human epithelial HK-2 cells, but it did increase ICAM-1 and JNK levels upon TNFα stimulation. Another report dealt with the vascular smooth muscle cells (VSMCs) in the aortic arch, thought to be involved in lesion formation [13]. An adenoviral knockdown of CYLD showed a notable decrease in TNFα-induced inflammatory cytokines Mcp-1, IL-6 and ICAM-1. Furthermore, CYLD knockdown also suppressed downstream proinflammatory kinases such as MAPK, ERK, JNK and p38. In both cases, the inflammatory response and CYLD’s direct control of that response were critical, but an interesting observation that CYLD expression was increased in injured coronary artery afflicted with neointimal hyperplasia reinforces the idea of CYLD’s involvement in formation of lesions. For a good starting point in this new CYLD/vascular system connection, a review by Takami and colleagues is recommended [124].
Although there is some initial evidence to link CYLD to the formation of vascular lesions through both the inflammatory and cell cycle pathways, much work remains to be done in this area.
4.3. Conclusions and Future Directions in Cardiovascular Research (Fig. 4)
Fig. 4.
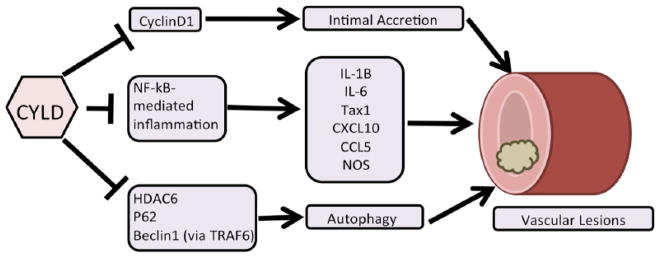
CYLD in cardiovascular disease model. CYLD may affect any or all of these pathways to regulate the formation of vascular lesions.
CYLD has been extensively studied in the immune system as well as areas involving cell signaling, development and death. Numerous pathways such as Wnt/β-catenin, NF-kB and MAPK are affected by CYLD. Although the details of CYLD’s regulation of these important pathways has been well elucidated in the literature, little is known regarding CYLD’s role in the cardiovascular system.
The precise mechanism of vascular lesion formation is still unknown. However, emerging evidence has implicated a potential role of CYLD. A report of polyubiquitin as a discriminatory marker for synthetic versus contractile VSMCs, along with a subsequent finding that angiocidin is regulated by polyubiquitination, has raised the possibility that CYLD is intimately involved in the vascular lesion process, thus revealing a potential therapeutic target [42,52]. Indeed, CYLD is upregulated in injured carotid arteries of rats, reduces cyclin D1 levels, and is capable of suppressing vascular lesion formation. However, it must be kept in mind that since CYLD is associated with normal cellular cycling and development, that its involvement in cardiovascular disease is not as simple as detecting the presence or absence of CYLD as a biomarker.
A potential area may be the downregulation of Cyclins by CYLD. This could be of critical importance, since Cyclins are known to be highly expressed in newly developing hearts, but are normally downregulated in adults. However, abnormally high levels of Cyclin D1 are known to contribute to intimal accretion [125]. Accordingly, this provides clear evidence that CYLD is involved in the atherosclerotic process via the regulation of a developmentally essential protein, and presents a possible direction for further study. Another potential area is inflammation (from infection or injury) which is a major player in vascular lesions as evidenced in the literature concerning activation of NF-kB-mediated inflammatory signaling. The inflammatory reports discussed above also apply here. Indirect lack of control by CYLD can result in a rampant immune reaction which damages endothelial and intimal cells and releases additional inflammatory cytokines (e.g., IL-1B). This cycle of recruitment, inflammation, damage, recruitment results in a large lesion on a blood vessel. A few reports corroborate this, as TRAF6 (controlled by IL-1B, TLR2 and CYLD) was found to immortalize lymphocytes thereby sustaining the damage they can do without the check of CYLD-mediated apoptosis [126]. Artery damaging inflammation in Human T-Cell Associated Leukemia Virus 1 cases is due to the action of viral protein Tax1, which was shown to directly interact with CYLD to inhibit Tax1’s ubiquitination and activation [127]. IL-1B has itself been found to induce IRF-1, which not only plays a critical role in Nitric Oxide Synthase (NOS) activity, but also upregulates CXCL10 and CCL5 to recruit monocytes (IRF-1 is K63 polyubiquitinated]) [128]. While the recruited monocytes result in release of inflammatory cytokines, they will be negatively regulated by CYLD to reduce inflammatory damage to the vascular system. Ventilator-induced pulmonary injury in mice has been reported to increase levels of Akt, which protects against the damage [129]. Interestingly, Akt is polyubiquitinated by TRAF6 in a K63-linked manner, providing a possible target for CYLD, while UCHL1 (controlled by DNA damage-inducible GADD45a demethylation of the UCHL1 promoter) removes K48-linked ubiquitin, preventing Akt degradation [129]. An earlier report from our lab demonstrated that UCHL1 deubiquitination can itself negatively regulate TNFα-mediated VSMC proliferation by suppressing Erk [130]. Such complex roles for ubiquitin [degrading markers versus active signal transducers] were detailed in a review by Willis and colleagues in 2011, where deubiquitination of key regulatory inflammation genes like TRAF2 and Bcl3 as well as the control of autophagy as a driving force behind cardiovascular disease are discussed [45]. Thusly, autophagy may be a third area of potential study. The Willis review states that aggregation of damaged proteins can damage the vascular system and that proteasomal destruction is not effective against large amounts of protein aggregates but that autophagy exists to recycle these errant proteins and this is mediated by protein aggregate sensor receptors p62, HDAC and NBR1 [45]. CYLD interacts with p62 directly and CYLD can directly inactivate HDAC6, thereby controlling autophagy [131]. While proteasomal clearance of damaged cardiovascular proteins was known earlier from canine experiments, a subsequent report linked desmin-related cardiomyopathy to accretion of misfolded proteins and activation of the autophagy pathway [132, 133]. In addition to the aforementioned ubiquitin-regulated p62 and NBR1, autophagy has extensive regulatory elements, such as HO1, HMGB1 and Beclin-1, which interact via CYLD- associated TRAF6 [134–139]. In this manner, CYLD can affect the autophagic pathway that clears damaged protein aggregates and reduces the chances of lesion formation. Thus, by its ability to control the cell cycle, inflammation and autophagic factors that contribute to the formation of lesions, the role of CYLD as a master regulator in vascular disease is evident. Future research that focuses on these aspects of CYLD will allow for new understanding of the mechanisms of vascular disease.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11(4):441–3. doi: 10.1038/ng1295-441. [DOI] [PubMed] [Google Scholar]
- 2.Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25(2):160–5. doi: 10.1038/76006. [DOI] [PubMed] [Google Scholar]
- 3.Poblete Gutiérrez P, Eggermann T, Höller D, et al. Phenotype diversity in familial cylindromatosis: a frameshift mutation in the tumor suppressor gene CYLD underlies different tumors of skin appendages. J Invest Dermatol. 2002;119(2):527–31. doi: 10.1046/j.1523-1747.2002.01839.x. [DOI] [PubMed] [Google Scholar]
- 4.Brummelkamp TR, Nijman SMB, Dirac AMG, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424(6950):797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 5.Regamey A, Hohl D, Liu JW, et al. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J Exp Med. 2003;198(12):1959–64. doi: 10.1084/jem.20031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424(6950):793–6. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 7.Kovalenko A, Chable-bessia C. The tumour suppressor CYLD negatively regulates NF-kB signalling by deubiquitination. Nature. 2003:801–5. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 8.Saito K, Kigawa T, Koshiba S, et al. The CAP-Gly domain of CYLD associates with the proline-rich sequence in NEMO/IKKgamma. Structure. 2004;12(9):1719–28. doi: 10.1016/j.str.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Lian F, Cockerell CJ. Cutaneous appendage tumors: familial cylindromatosis and associated tumors update. Adv Dermatol. 2005;21:217–34. doi: 10.1016/j.yadr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Massoumi R, Paus R. Cylindromatosis and the CYLD gene: new lessons on the molecular principles of epithelial growth control. Bioessays. 2007;29(12):1203–14. doi: 10.1002/bies.20677. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida H, Jono H, Kai H, Li J-D. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem. 2005;280(49):41111–21. doi: 10.1074/jbc.M509526200. [DOI] [PubMed] [Google Scholar]
- 12.Besse A, Campos AD, Webster WK, Darnay BG. TRAF-interacting protein (TRIP) is a RING-dependent ubiquitin ligase. Biochem Biophys Res Commun. 2007;359(3):660–4. doi: 10.1016/j.bbrc.2007.05.149. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, Lv J, Han L, et al. A pro-inflammatory role of deubiquitinating enzyme cylindromatosis (CYLD) in vascular smooth muscle cells. Biochem Biophys Res Commun. 2012;420(1):78–83. doi: 10.1016/j.bbrc.2012.02.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trompouki E, Tsagaratou A, Kosmidis SK, et al. Truncation of the catalytic domain of the cylindromatosis tumor suppressor impairs lung maturation. Neoplasia. 2009;11(5):469–76. doi: 10.1593/neo.81424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blake PW, Toro JR. Update of cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling. Hum Mutat. 2009;30(7):1025–36. doi: 10.1002/humu.21024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sundaram K, Shanmugarajan S, Rao DS, Reddy SV. Mutant p62P392L stimulation of osteoclast differentiation in Paget’s disease of bone. Endocrinology. 2011;152(11):4180–9. doi: 10.1210/en.2011-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi M, Jono H, Shinriki S, et al. Clinical significance of CYLD downregulation in breast cancer. Breast Cancer Res Treat. 2014;143(3):447–57. doi: 10.1007/s10549-013-2824-3. [DOI] [PubMed] [Google Scholar]
- 18.Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125(4):665–77. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 19.Lim JH, Jono H, Komatsu K, et al. CYLD negatively regulates transforming growth factor-β-signalling via deubiquitinating Akt. Nat Commun. 2012;3:771. doi: 10.1038/ncomms1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim JH, Stirling B, Derry J, et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity. 2007;27(2):349–60. doi: 10.1016/j.immuni.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Lim JH, Ha U-H, Woo C-H, Xu H, Li J-D. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol. 2008;10(11):2247–56. doi: 10.1111/j.1462-5822.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- 22.Lim JH, Jono H, Koga T, et al. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS One. 2007;2(10):e1032. doi: 10.1371/journal.pone.0001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komander D, Lord CJ, Scheel H, et al. The structure of the CYLD USP domain explains its specificity for Lys63-linked polyubiquitin and reveals a B box module. Mol Cell. 2008;29(4):451–64. doi: 10.1016/j.molcel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 24.Weissman Institute. Cited 2014 May 5. Available from: www.genecards.org.
- 25.Komander D. CYLD tidies up dishevelled signaling. Mol Cell. 2010;37(5):589–90. doi: 10.1016/j.molcel.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Chu Y, Soberon V, Glockner L, et al. A20 and CYLD do not share significant overlapping functions during B cell development and activation. J Immunol. 2012;189(9):4437–43. doi: 10.4049/jimmunol.1200396. [DOI] [PubMed] [Google Scholar]
- 27.Reiley W, Zhang M, Wu X, Granger E, Sun S. Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol Cell Biol. 2005;25(10):3886–95. doi: 10.1128/MCB.25.10.3886-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu P, Shi KJ, An JJ, et al. The LEF1/CYLD axis and cIAPs regulate RIP1 deubiquitination and trigger apoptosis in selenite-treated colorectal cancer cells. Cell Death Dis. 2014;5:e1085. doi: 10.1038/cddis.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirchhofer D, Vucic D. Protease activity of MALT1: a mystery unravelled. Biochem J. 2012;444(2):e3–5. doi: 10.1042/BJ20120414. [DOI] [PubMed] [Google Scholar]
- 30.Liang G, Ahlqvist K, Pannem R, Posern G, Massoumi R. Serum response factor controls CYLD expression via MAPK signaling pathway. PLoS One. 2011;6(5):e19613. doi: 10.1371/journal.pone.0019613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thein S, Tao-Cheng J-H, Li Y, Bayer KU, Reese TS, Dosemeci A. CaMKII mediates recruitment and activation of the deubiquitinase CYLD at the postsynaptic density. PLoS One. 2014;9(3):e91312. doi: 10.1371/journal.pone.0091312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massoumi R, Kuphal S, Hellerbrand C, et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med. 2009;206(1):221–32. doi: 10.1084/jem.20082044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takiuchi T, Nakagawa T, Tamiya H, et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells. 2014;19(3):254–72. doi: 10.1111/gtc.12128. [DOI] [PubMed] [Google Scholar]
- 34.Liu P, Xu B, Shen W, et al. Dysregulation of TNFα-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26(6):1293–300. doi: 10.1038/leu.2011.357. [DOI] [PubMed] [Google Scholar]
- 35.Kuphal S, Shaw-Hallgren G, Eberl M, et al. GLI1-dependent transcriptional repression of CYLD in basal cell carcinoma. Oncogene. 2011;30(44):4523–30. doi: 10.1038/onc.2011.163. [DOI] [PubMed] [Google Scholar]
- 36.D’Altri T, Gonzalez J, Aifantis I, Espinosa L, Bigas A. Hes1 expression and CYLD repression are essential events downstream of Notch1 in T-cell leukemia. Cell Cycle. 2011;10(7):1031–6. doi: 10.4161/cc.10.7.15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia J, Chen L, Jian W, et al. MicroRNA-362 induces cell proliferation and apoptosis resistance in gastric cancer by activation of NF-κB signaling. J Transl Med. 2014;12:33. doi: 10.1186/1479-5876-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S-J, Jung H-J, Lim C-J. Reactive Oxygen Species-Dependent Down-Regulation of Tumor Suppressor Genes PTEN, USP28, DRAM, TIGAR, and CYLD Under Oxidative Stress. Biochem Genet. 2013;51(11–12):901–15. doi: 10.1007/s10528-013-9616-7. [DOI] [PubMed] [Google Scholar]
- 39.Sun S-C. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17(1):25–34. doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Donnell MA, Perez-Jimenez E, Oberst A, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13(12):1437–42. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goncharov T, Niessen K, de Almagro MC, et al. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013;32(8):1103–14. doi: 10.1038/emboj.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dimitrov S, Sabherwal Y, Raymond DD, L’Heureux DZ, Lu Q, Tuszynski GP. Endothelial apoptotic activity of angiocidin is dependent on its polyubiquitin binding activity. Br J Cancer. 2005;93(6):662–9. doi: 10.1038/sj.bjc.6602773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun. 1978;81(4):1100–5. doi: 10.1016/0006-291x(78)91249-4. [DOI] [PubMed] [Google Scholar]
- 44.Driscoll J, Goldberg AL. The proteasome (multicatalytic protease) is a component of the 1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J Biol Chem. 1990;265(9):4789–92. [PubMed] [Google Scholar]
- 45.Willis MS, Townley-tilson WHD, Kang EY, Jonathon W, Patterson C. Sent to Destroy: The Ubiquitin Proteasome System Regulates Cell Signaling and Protein Quality Control in Cardiovascular Disease. Circ Res. 2011;106(3):463–78. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harhaj EW, Dixit VM. Regulation of NF-κB by deubiquitinases. Immunol Rev. 2012;246(1):107–24. doi: 10.1111/j.1600-065X.2012.01100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Tran. 2009;37(Pt 5):937–53. doi: 10.1042/BST0370937. [DOI] [PubMed] [Google Scholar]
- 48.Varadan R, Assfalg M, Haririnia A, Raasi S, Pickart C, Fushman D. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J Biol Chem. 2004;279(8):7055–63. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao S, Ulrich HD. Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc Natl Acad Sci USA. 2010;107(17):7704–9. doi: 10.1073/pnas.0908764107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bremm A, Freund SMV, Komander D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol. 2010;17(8):939–47. doi: 10.1038/nsmb.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medina R, Wing SS, Goldberg aL. Increase in levels of polyubiquitin and proteasome mRNA in skeletal muscle during starvation and denervation atrophy. Biochem J. 1995;307 (Pt 3):631–7. doi: 10.1042/bj3070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adam PJ, Weissberg PL, Cary NR, Shanahan CM. Polyubiquitin is a new phenotypic marker of contractile vascular smooth muscle cells. Cardiovasc Res. 1997;33(2):416–21. doi: 10.1016/s0008-6363(96)00220-9. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Zhu H, Zou M-H. Non-covalent interaction between polyubiquitin and GTP cyclohydrolase 1 dictates its degradation. PLoS One. 2012;7(9):e43306. doi: 10.1371/journal.pone.0043306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delfino M, D’Anna F, Ianniello S, Donofrio V. Multiple hereditary trichoepithelioma and cylindroma (Brooke-Spiegler syndrome) Dermatologica. 1991;183(2):150–3. doi: 10.1159/000247658. [DOI] [PubMed] [Google Scholar]
- 55.Korting HC, Konz B. Coincidence of multiple cylindroma and trichoepitheliomas (Brooke-Spiegler-Syndrome) Hautarzt. 1982;33(1):34–46. [PubMed] [Google Scholar]
- 56.Dosemeci A, Thein S, Yang Y, Reese TS, Tao-Cheng J-H. CYLD, a deubiquitinase specific for lysine63-linked polyubiquitins, accumulates at the postsynaptic density in an activity-dependent manner. Biochem Biophys Res Commun. 2013;430(1):245–9. doi: 10.1016/j.bbrc.2012.10.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alameda JP, Fernández-Aceñero MJ, Moreno-Maldonado R, et al. CYLD regulates keratinocyte differentiation and skin cancer progression in humans. Cell Death Dis. 2011;2:e208. doi: 10.1038/cddis.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nikolaou K, Tsagaratou A, Eftychi C, Kollias G, Mosialos G, Talianidis I. Inactivation of the deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell. 2012;21(6):738–50. doi: 10.1016/j.ccr.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 59.Pannem RR, Dorn C, Ahlqvist K, Bosserhoff AK, Hellerbrand C, Massoumi R. CYLD controls c-MYC expression through the JNK-dependent signaling pathway in hepatocellular carcinoma. Carcinogenesis. 2014;35(2):461–8. doi: 10.1093/carcin/bgt335. [DOI] [PubMed] [Google Scholar]
- 60.Iliopoulos D, Jaeger Sa, Hirsch Ha, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39(4):493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laplantine E, Fontan E, Chiaravalli J, et al. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009;28(19):2885–95. doi: 10.1038/emboj.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hadian K, Griesbach RA, Dornauer S, et al. NF-κB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-κB activation. J Biol Chem. 2011;286(29):26107–17. doi: 10.1074/jbc.M111.233163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reiley WW, Zhang M, Jin W, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol. 2006;7(4):411–7. doi: 10.1038/ni1315. [DOI] [PubMed] [Google Scholar]
- 65.Jin W, Reiley WR, Lee AJ, et al. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem. 2007;282(21):15884–93. doi: 10.1074/jbc.M609952200. [DOI] [PubMed] [Google Scholar]
- 66.Thuille N, Wachowicz K, Hermann-Kleiter N, et al. PKCθ/β and CYLD are antagonistic partners in the NFκB and NFAT transactivation pathways in primary mouse CD3+ T lymphocytes. PLoS One. 2013;8(1):e53709. doi: 10.1371/journal.pone.0053709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reissig S, Hövelmeyer N, Weigmann B, et al. The tumor suppressor CYLD controls the function of murine regulatory T cells. J Immunol. 2012;189(10):4770–6. doi: 10.4049/jimmunol.1201993. [DOI] [PubMed] [Google Scholar]
- 68.Hövelmeyer N, Wunderlich FT, Massoumi R, et al. Regulation of B cell homeostasis and activation by the tumor suppressor gene CYLD. J Exp Med. 2007;204(11):2615–27. doi: 10.1084/jem.20070318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, Chen W, Wang Q, Li L, Wang C. Negative regulation of TLR inflammatory signaling by the SUMO-deconjugating enzyme SENP6. PLoS Pathog. 2013;9(6):e1003480. doi: 10.1371/journal.ppat.1003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bros M, Dexheimer N, Besche V, et al. Mutated cylindromatosis gene affects the functional state of dendritic cells. Eur J Immuno. 2010;40(10):2848–57. doi: 10.1002/eji.200939285. [DOI] [PubMed] [Google Scholar]
- 71.Zhang M, Wu X, Lee AJ, et al. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008;283(27):18621–6. doi: 10.1074/jbc.M801451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manganaro L, Pache L, Herrmann T, et al. Tumor suppressor cylindromatosis (CYLD) controls HIV transcription in a NF-κB dependent manner. J Virol. 2014;88(13):7528–40. doi: 10.1128/JVI.00239-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nishanth G, Deckert M, Wex K, Massoumi R, Schweitzer K, Naumann M, et al. CYLD enhances severe listeriosis by impairing IL-6/STAT3-dependent fibrin production. PLoS Pathog. 2013;9(6):e1003455. doi: 10.1371/journal.ppat.1003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011;21(1):22–39. doi: 10.1038/cr.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246(1):95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ang E, Pavlos NJ, Rea SL, et al. Proteasome inhibitors impair RANKL-induced NF-kappaB activity in osteoclast-like cells via disruption of p62, TRAF6, CYLD, and IkappaBalpha signaling cascades. J Cell Physiol. 2009;220(2):450–9. doi: 10.1002/jcp.21787. [DOI] [PubMed] [Google Scholar]
- 77.Cui T-G, Ichikawa T, Yang M, Dong X, Li J, Cui T. An emerging role of deubiquitinating enzyme cylindromatosis (CYLD) in the tubulointerstitial inflammation of IgA nephropathy. Biochem Biophys Res Commun. 2009;390(2):307–12. doi: 10.1016/j.bbrc.2009.09.119. [DOI] [PubMed] [Google Scholar]
- 78.Urbanik T, Köhler BC, Boger RJ, et al. Down-regulation of CYLD as a trigger for NF-κ B activation and a mechanism of apoptotic resistance in hepatocellular carcinoma cells. Int J Oncol. 2011:121–31. [PubMed] [Google Scholar]
- 79.Hornbeck PV, Kornhauser JM, Tkachev S, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40(Database issue):D261–70. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahmed N, Zeng M, Sinha I, et al. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat Immunol. 2011;12(12):1176–83. doi: 10.1038/ni.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deng LL, Shao YX, Lv HF, Deng HB, Lv FZ. Over-expressing CYLD augments antitumor activity of TRAIL by inhibiting the NF-κB survival signaling in lung cancer cells. Neoplasma. 2012:18–29. doi: 10.4149/neo_2012_003. [DOI] [PubMed] [Google Scholar]
- 82.Komatsu K, Lee J-Y, Miyata M, et al. Inhibition of PDE4B suppresses inflammation by increasing expression of the deubiquitinase CYLD. Nat Commun. 2013;4:1684. doi: 10.1038/ncomms2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stegmeier F, Sowa ME, Nalepa G, Gygi SP, Harper JW, Elledge SJ. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci USA. 2007;104(21):8869–74. doi: 10.1073/pnas.0703268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu D, Davydenko O, Lampson MA. Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J Cell Biol. 2012;198(4):491–9. doi: 10.1083/jcb.201205090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gao J, Huo L, Sun X, et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283(14):8802–9. doi: 10.1074/jbc.M708470200. [DOI] [PubMed] [Google Scholar]
- 86.Gao J, Sun L, Huo L, Liu M, Li D, Zhou J. CYLD regulates angiogenesis by mediating vascular endothelial cell migration. Blood. 2010;115(20):4130–7. doi: 10.1182/blood-2009-10-248526. [DOI] [PubMed] [Google Scholar]
- 87.Gomez-Ferreria MA, Bashkurov M, Mullin M, Gingras A-C, Pelletier L. CEP192 interacts physically and functionally with the K63-deubiquitinase CYLD to promote mitotic spindle assembly. Cell Cycle. 2012;11(19):3555–8. doi: 10.4161/cc.21574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Y, Sun L, Tala, et al. CYLD regulates RhoA activity by modulating LARG ubiquitination. PLoS One. 2013;8(2):e55833. doi: 10.1371/journal.pone.0055833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reiley W, Zhang M, Sun S-C. Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem. 2004;279(53):55161–7. doi: 10.1074/jbc.M411049200. [DOI] [PubMed] [Google Scholar]
- 90.Yang Y, Liu M, Li D, et al. CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled-NuMA-dynein/dynactin complex formation. Proc Natl Acad Sci USA. 2014;111(6):2158–63. doi: 10.1073/pnas.1319341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Espinosa L, Cathelin S, D’Altri T, et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18(3):268–81. doi: 10.1016/j.ccr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tauriello DVF, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell. 2010;37(5):607–19. doi: 10.1016/j.molcel.2010.01.035. [DOI] [PubMed] [Google Scholar]
- 93.Masckauchán TNH, Shawber CJ, Funahashi Y, Li C-M, Kitajewski J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. 2005;8(1):43–51. doi: 10.1007/s10456-005-5612-9. [DOI] [PubMed] [Google Scholar]
- 94.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 95.Yang W-L, Jin G, Li C-F, et al. Cycles of ubiquitination and deubiquitination critically regulate growth factor-mediated activation of Akt signaling. Sci Signal. 2013;6(257):ra3. doi: 10.1126/scisignal.2003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sheridan JW, Bishop CJ, Simmons RJ. Biophysical and morphological correlates of kinetic change and death in a starved human melanoma cell line. J Cell Sci. 1981;49:119–37. doi: 10.1242/jcs.49.1.119. [DOI] [PubMed] [Google Scholar]
- 98.Neal JV, Potten CS. Effect of low dose ionizing radiation on the murine pericryptal fibroblast sheath: radiation damage in a mesenchymal system in vivo. Int J Radiat Biol Relat Stud Phys Chem Med. 1981;39(2):175–83. doi: 10.1080/09553008114550191. [DOI] [PubMed] [Google Scholar]
- 99.Tatton WG, Chalmers-Redman R, Brown D, Tatton N. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Ann Neurol. 2003;53 (Suppl 3):S61–70. doi: 10.1002/ana.10489. discussion S70–2. [DOI] [PubMed] [Google Scholar]
- 100.Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol. 2003;62(4):329–39. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- 101.Gao Y, Signore AP, Yin W, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25(6):694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 102.Sawai H, Domae N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodide-negative cells. Biochem Biophys Res Commun. 2011;411(3):569–73. doi: 10.1016/j.bbrc.2011.06.186. [DOI] [PubMed] [Google Scholar]
- 103.Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart. Circ Res. 2011;108(8):1017–36. doi: 10.1161/CIRCRESAHA.110.225730. [DOI] [PubMed] [Google Scholar]
- 104.Li J, McQuade T, Siemer AB, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339–50. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3(115):re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 107.Sosna J, Voigt S, Mathieu S, et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci. 2014;71(2):331–48. doi: 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Linkermann A, Bräsen JH, De Zen F, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-α-induced shock. Mol Med. 2012;18:577–86. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moquin DM, McQuade T, Chan FK-M. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One. 2013;8(10):e76841. doi: 10.1371/journal.pone.0076841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12(11):1539–46. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 111.Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013;20(2):185–7. doi: 10.1038/cdd.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Di. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schworer SA, Smirnova II, Kurbatova I, et al. Toll-like Receptor-Mediated Downregulation of the Deubiquitinase CYLD Protects Macrophages from Necroptosis in Wild-Derived Mice. J Biol Chem. 2014;289(20):14422–33. doi: 10.1074/jbc.M114.547547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duval CW. THE EXPERIMENTAL VASCULAR LESIONS PRODUCED BY BACILLUS MALLEI. J Exp Med. 1907;9(3):241–53. doi: 10.1084/jem.9.3.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Doornekamp FN, Borst C, Post MJ. Endothelial cell recoverage and intimal hyperplasia after endothelium removal with or without smooth muscle cell necrosis in the rabbit carotid artery. J Vasc Res. 1996;33(2):146–55. doi: 10.1159/000159143. [DOI] [PubMed] [Google Scholar]
- 117.Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92(5):1355–74. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 118.Schwenke DC, Carew TE. Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arterioscler Thromb Vasc Biol. 1989;9(6):908–18. doi: 10.1161/01.atv.9.6.908. [DOI] [PubMed] [Google Scholar]
- 119.Libby P, Egan D, Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation. 1997;96(11):4095–103. doi: 10.1161/01.cir.96.11.4095. [DOI] [PubMed] [Google Scholar]
- 120.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–25. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 121.Park HS, Hahn S, Choi GH, Yoo YS, Lee JY, Lee T. Muscle-derived stem cells promote angiogenesis and attenuate intimal hyperplasia in different murine vascular disease models. Stem Cells Dev. 2013;22(6):866–77. doi: 10.1089/scd.2012.0391. [DOI] [PubMed] [Google Scholar]
- 122.Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ Res. 1995;77(3):638–43. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- 123.Shi N, Xie W, Chen S-Y. Cell division cycle 7 is a novel regulator of transforming growth factor-β-induced smooth muscle cell differentiation. J Biol Chem. 2012;287(9):6860–7. doi: 10.1074/jbc.M111.306209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Witmer LM. Clinical Anatomy of the Aorta. Athens, OH: The Ohio State University; 2014. pp. 1–15. [Google Scholar]
- 125.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293(3):H1883–91. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 126.Takami Y, Nakagami H, Morishita RK, et al. Potential role of CYLD (Cylindromatosis) as a deubiquitinating enzyme in vascular cells. Am J Pathol. 2008;172(3):818–29. doi: 10.2353/ajpath.2008.070312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hotchkiss A, Robinson J, MacLean J, Feridooni T, Wafa K, Pasumarthi KBS. Role of D-type cyclins in heart development and disease. Can J Physiol Pharmacol. 201;90(9):1197–207. doi: 10.1139/y2012-037. [DOI] [PubMed] [Google Scholar]
- 128.Iha H, Peloponese J-M, Verstrepen L, et al. Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J. 2008;27(4):629–41. doi: 10.1038/emboj.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu X, Zhang M, Sun S-C. Mutual regulation between deubiquitinase CYLD and retroviral oncoprotein Tax. Cell Biosci. 2011;1(1):27. doi: 10.1186/2045-3701-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Harikumar KB, Yester JW, Surace MJ, et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat Immunol. 2014;15(3):231–8. doi: 10.1038/ni.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mitra S, Sammani S, Wang T, et al. Role of growth arrest and DNA damage-inducible α in Akt phosphorylation and ubiquitination after mechanical stress-induced vascular injury. Am J Respir Crit Care Med. 2011;184(9):1030–40. doi: 10.1164/rccm.201103-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ichikawa T, Li J, Dong X, et al. Ubiquitin carboxyl terminal hydrolase L1 negatively regulates TNFalpha-mediated vascular smooth muscle cell proliferation via suppressing ERK activation. Biochem Biophys Res Commun. 2010;391(1):852–6. doi: 10.1016/j.bbrc.2009.11.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wickström SA, Masoumi KC, Khochbin S, Fässler R, Massoumi R. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 2010;29(1):131–44. doi: 10.1038/emboj.2009.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Depre C, Wang Q, Yan L, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114(17):1821–8. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 135.Tannous P, Zhu H, Johnstone JL, et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci USA. 2008;105(28):9745–50. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Banerjee P, Basu A, Wegiel B, et al. Heme oxygenase-1 promotes survival of renal cancer cells through modulation of apoptosis- and autophagy-regulating molecules. J Biol Chem. 2012;287(38):32113–23. doi: 10.1074/jbc.M112.393140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tang D, Kang R, Livesey KM, Cheh C-W, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–92. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wirawan E, Vande Walle L, Kersse K, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010;1(1):e18. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: more than a nonselective pathway. Int J Cell Biol. 2012;2012:219625. doi: 10.1155/2012/219625. (Fig. 1) [DOI] [PMC free article] [PubMed] [Google Scholar]