

Abstract
During the development of the hematopoietic system, at least 8 distinct lineages are generated in the mouse embryo. Transgenic mice expressing fluorescent proteins at various points in the hematopoietic hierarchy, from hematopoietic stem cell to multipotent progenitors to each of the final differentiated cell types, have provided valuable tools for tagging, tracking, and isolating these cells. In this chapter, we discuss general considerations in designing a transgene, survey available fluorescent probes, and methods for confirming and analyzing transgene expression in the hematopoietic systems of the embryo, fetus, and postnatal/adult animal.
Keywords: hematopoiesis, transgenic mice, knock-in, green fluorescent protein, fluorescent reporter
1. Introduction
Since the discovery and cloning of the first fluorescent protein (FP), wild type green fluorescent protein (wtGFP) from the bioluminescent jellyfish Aequorea victoria (1) and the subsequent creation of spectral variants (2–4), FPs have become indispensable for imaging cellular differentiation and function at high resolution and in real time (reviewed in refs. 3, 4). Gene-specific regulatory elements can be used to drive targeted expression of FP reporters, with spatial- and/or temporal specificity, in virtually any cell type of a transgenic animal. The hematopoietic system has benefitted enormously from this approach, which made it possible to explore the emergence, expansion, migration and differentiation of progenitors for the erythroid, myeloid, and lymphoid lineages. It is now possible to label and track the development of distinct hematopoietic cell types in vivo and to isolate these cells directly, using fluorescence activated cell sorting (FACS). In this chapter, we discuss the steps involved in the generation and analysis of transgenic lines in which fluorescent reporters are expressed in hematopoietic lineages of the mouse, the most genetically tractable model for mammalian development.
1.1 Ontogeny of the mouse hematopoietic system
Hematopoiesis is a precisely orchestrated, stepwise process that leads to the formation of all lineages of the blood (5). Primitive erythroid cells (EryP) are the first hematopoietic lineage to detected in the mouse embryo (reviewed in ref. 6). They are generated late in gastrulation, in the in the blood islands of the yolk sac (YS), along with macrophages and megakaryocytes (7). The first definitive hematopoietic cells, comprising erythroid, megakaryocytic and myeloid lineages, also arise in the YS, shortly after the appearance of EryP (reviewed in refs. 6, 7). Hematopoietic stem cells (HSCs) form in the aorta-gonad-mesonephros (AGM) region of the embryo, in the large arteries and placenta, and, very likely, in the YS(for a review, see ref. 6). They do not differentiate in these sites but instead seed the fetal liver (FL), where they expand and produce progenitors that give rise to definitive erythro-myeloid and lymphoid lineages. Late in gestation, HCSs migrate from the fetal liver to the bone marrow, which becomes the main blood production center in the postnatal animal (8). The general hierarchy of hematopoietic development is shown as a "snapshot" in ref. (9).
1.2 General Considerations in Designing a Transgene
Transgenic mouse lines expressing fluorescent proteins (FPs) are invaluable tools for studying the development of the hematopoietic system. Careful design is necessary to achieve the desired expression of the reporter protein. The main components of a fluorescent reporter transgene are the promoter (and, usually, other upstream regulatory sequences), sequences encoding the fluorescent protein, and splice/polyadenylation signals (Figure 1). A cartoon outlining the most commonly used approach for creation of a transgenic mouse line is presented in Figure 2.
Figure 1. Basic design of a fluorescent transgenic construct.
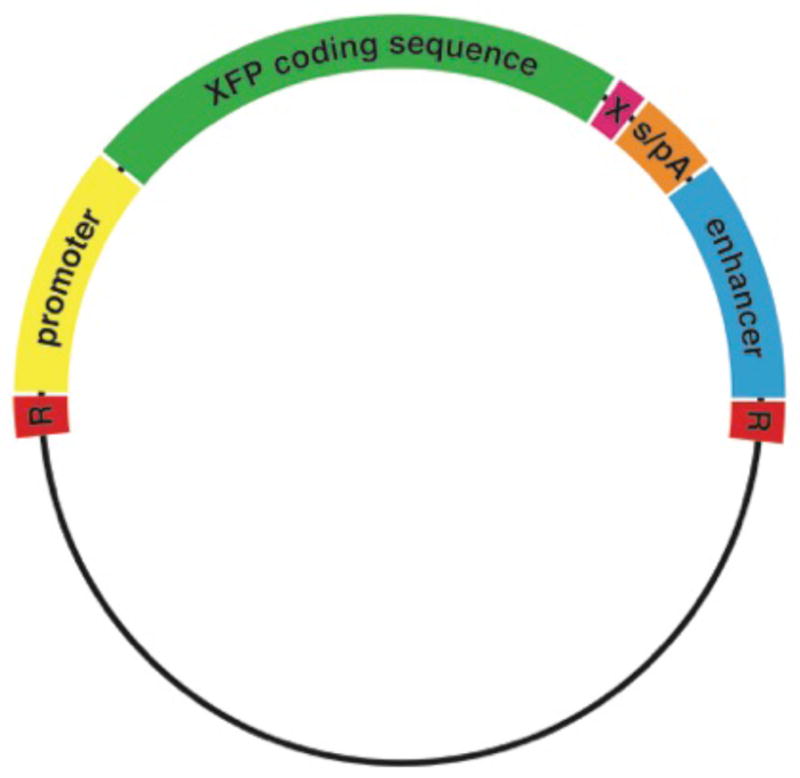
"R" marks the restriction enzyme sites used for removal of the bacterial backbone before transgene injection. s/pA represents the splicing and polyadenylation signals. A stop codon (denoted by "X") should also be included in the construct design. The enhancer may be positioned upstream or downstream from the promoter and sequences encoding the FP and regulatory signals. For additional details, see text.
Figure 2. Basic steps in the generation of a fluorescent transgenic reporter mouse line.

Pronuclear injection of the transgene is shown in this cartoon but transgenic mouse lines can also be generated by blastocyst injection or embryo aggregation with genetically modified ES cells (see text).
1.2.1 Regulatory Elements
The promoter is the region of a gene from which mRNA transcription is initiated and is essential for controlling both the spatial and temporal expression of a transgene. A number of hematopoietic specific promoters have been used successfully to drive expression of fluorescent reporters in different hematopoietic lineages (see Table 1). The transgene construct should include a translational start codon (ATG), a Kozak sequence either upstream from or coupled to the start codon (10), and a translational stop codon (Figure 1). Posttranscriptional regulatory elements may be included to enhance mRNA stability (e.g. see ref. 11).
Table 1.
Transgenic mouse lines expressing fluorescent reporter proteins in the hematopoietic system.
| Gene | Reporter | Lineage labeled | Reference |
|---|---|---|---|
| Gata2 | GFP (Ki) | HSC | (70) |
| Hoxb4 | YFP (Ki) | HSC | (71) |
| Ly-6A | GFP (Tg) | HSC | (72) |
| Bmi1 | GFP (Ki) | HSC§ | (73) |
| Abcg2 | IRES-GFP (Ki) | HSC, erythroid | (74) |
| c-Kit | GFP (Tg) | HSC, progenitors | (75) |
| c-Kit | GFP (CKO) | HSC, progenitors | (76) |
| Runx1 | GFP (Tg) | HSC, progenitors | (77) |
| Pu.1 | IRES-GFP (Ki) | HSC, lymphoid and myeloid progenitors | (78) |
| CD41 | farnesyl-YFP (Tg) | HSC, progenitors, megakaryocytes, platelets | (79) |
| Gfi1B | GFP (Ki) | HSC, erythroid and myeloid progenitors | (80) |
| Etv2 | GFP (Tg) | Hematopoietic and endothelial progenitors | (81) |
| Etv2 | EYFP (Tg) | Hematopoietic and endothelial progenitors | (82) |
| Gata1 | GFP (Tg) | Hemangioblast, EryP, EryD, megakaryocytes | (83) |
| EpoR | GFP-Cre (Ki) | Erythroid progenitors, endothelial | (84) |
| Lysozyme M | EGFP (Ki) | Myelomonocytic cells, including macrophages and granulocytes | (85) |
| MafB | GFP (Tg) | Myelomonocytic lineages of hematopoietic cells, peritoneal macrophages | (86) |
| c-fms | EGFP (Tg) | Macrophages, dendritic cells, myeloid cells | (87) |
| β-globin | ECFP (Tg) | MEP, EryD | (50) |
| miR-144/451 | EGFP (Ki) | EryD | (26) |
| Eklf | GFP (Tg) | EryD | (88) |
| ε-globin | KGFP (Tg); H2B- GFP (Tg) | EryP | (23, 24) |
| γ-globin β-globin |
EGFP DsRed |
EryP EryD |
(89) |
| Langerin | IRES-EGFP (Ki) | Langerhans cells | (90) |
| TCRb | GFP (Tg) | Lymphoid progenitors | (91) |
| Runx1 | IRES-GFP (Ki) | Lymphoid, myeloid, lower levels in erythroid | (29) |
| CD2 | EGFP (Tg) | T lymphoid cells | (92) |
| Rag2 | GFP (Ki) | T and B lymphoid cells | (93) |
| FoxP3 | GFP (Ki) | T regulatory lymphoid cells | (94) |
| Ror(gT) | EGFP (Ki) | T helper 17 lymphoid cells | (95) |
| Pax5 | EGFP (Ki) | Pre-B, B lymphoid cells | (96) |
| Rag1 | GFP (Ki) | B lymphoid cells | (97) |
| Blimp1 | IRES-EGFP (Ki) | B lymphoid cells, plasma cells | (98) |
| CX3CR1 | GFP (Ki) | Macrophages, monocytes, NK cells, dendritic cells, microglia | (99) |
| CD45 | YFP (Ki) | Widespread hematopoietic | (100) |
Abbreviations: Tg, Transgenic; KI, Knock-in; CKO, Conditional Knock-out; GFP, Green fluorescent protein; IRES, Internal ribosomal entry site; EGFP, enhanced GFP; YFP, Yellow Fluorescent Protein; CFP, Cyan Fluorescent Protein; HSC, hematopoietic stem cell; EryP, primitive erythroid; EryD, definitive erythroid; MEP, Megakaryocyte-erythroid progenitor; NK, Natural Killer cells.
GFP expression is highest in hematopoietic stem cells (HSCs) and is downregulated during lineage commitment and differentiation.
Additional regulatory elements should accompany the promoter to drive the desired transgene expression pattern. The most commonly used regulatory elements are enhancers or other upstream regulatory elements (12), an intron, which provides splice donor and acceptor sites, and a polyadenylation signal (Figure 1). The intron may be taken from the same gene as the promoter. Low transgene expression may be significantly increased through the use of a generic intron (13).
It is well documented that prokaryotic sequences in the vector perturb the frequency and extent of transgene expression (14–16). Therefore, restriction enzyme sites flanking the transgene should be included to allow removal of the vector backbone before microinjection.
Following microinjection into the male pronucleus of a fertilized egg (Figure 2), the transgene is inserted randomly, and often in multiple copies, into the genome (16). The neighboring chromatin may influence expression of the transgene, leading to undesired effects such as ectopic expression or even silencing (16, 17). To avoid these effects, chromatin insulators can be used. These DNA elements, together with the proteins that bind to them, impair interactions with neighboring chromatin (18). Enhancers are used to stimulate transcription and may do so in an orientation- and position-independent manner (12). Locus control regions (LCRs) from globin or CD4 genes have been used as enhancers for trangene expression in erythroid- or T cells, respectively (12, 19–21). For example, a minimal human ε-globin promoter combined with a truncated human β-globin LCR, a regulatory element that controls the erythroid-specific expression of all cis-linked globin genes (22), have been used to generate mouse lines expressing GFP in the primitive erythroid lineage (23, 24) (Figure 3A).
Figure 3. Tagging of primitive and definitive erythroid lineages using GFP reporters.

(A) GFP expression in the primitive erythroid cells of an embryonic stage (E)8.5 ε-globin-H2B-GFP embryo (24) (left panel, whole embryo, scale bar 200 μm; right panel, magnified view of yolk sac, scale bar, 50μm). Embryos were photographed on a Zeiss Lumar V12 stereomicroscope equipped with epifluorescence illumination and a NeoLumar S 1.5X FWD 30 mm objective. (B) Wet preparation of green fluorescent erythroid cells from the bone marrow (BM) and peripheral blood (PB) of an adult miR-144/451-GFP knock-in mouse (26). The cells were photographed on a Zeiss Axio Observer Z1 inverted microscope with epifluorescence illumination and a Plan-Apochromat 20X/0.8 objective. Scale bar, 20μm.
We recommend that expression of a newly designed transgene be tested in cultured cells before moving forward with the generation of the transgenic mouse line. This precaution helps to ensure that the transcriptional regulatory elements are functional and that the reporter sequence is translated to a functional protein.
1.2.2 Insertion of Exogenous DNA Into the Genome
The most commonly used approaches for generation of fluorescent reporter mice are microinjection of DNA (plasmid or BAC; see below) into a fertilized egg (Figure 2) and targeted insertion ("knock-in"). Insertion of a transgene into the mouse genome can also be achieved by viral infection of the egg or injection of genetically modified embryonic stem (ES) cells into blastocysts (25).
In the “knock-in” method, DNA sequences are engineered to integrate directly into a defined locus within the genome, using site-specific recombination (16). The knock-in approach avoids problems related to random insertion, as expression of exogenous sequences is controlled by the endogenous regulatory elements of the target gene. A recent example of a knock-in transgenic mouse is the mirR-144/451-GFP line, in which GFP is expressed in adult and fetal liver erythroid cells (26, 27) (Figure 3). A concern for the knock-in approach is that it may result in haploinsufficiency, influencing not only expression of the FP reporter but also the phenotype of the resulting animal. For example, loss of one Runx1 allele affected the distribution of HSCs in the embryo (28). This problem was overcome by linking the sequences encoding the FP to the endogenous gene through an internal ribosomal entry site (IRES) (29) to create a dicistronic fusion mRNA (30).
Conventional transgenes are often too small to accurately reproduce the endogenous expression of the promoter/enhancer elements. Bacterial artificial chromosomes (BAC) accommodate large DNA sequences, allowing cloning of all or most of the endogenous regulatory elements required to recapitulate the normal pattern of gene expression when linked to other sequences (31) such as those encoding a FP.
1.2.3 Alternative Approaches to Drive Hematopoietic Lineage-Specific Expression of Fluorescent Protein Reporters
Inducible expression of fluorescent reporters can be achieved using the CreloxP or FLP-FRT systems. For example, a mouse line in which the fluorescent reporter sequences are preceded by a floxed STOP cassette (e.g. refs. 32, 33, 34) can be mated with a deletor line designed to express Cre recombinase under the control of a promotor active in the cells of interest. Such targeted recombination approaches have been of great utility for lineage tracing studies (for a recent example, see ref. 35).
Depending on the system chosen, recombinase activity may be constitutive or inducible. Inducible recombination activity can be triggered by tamoxifen in the case of a Creestrogen receptor (ER) fusion gene or by doxycycline when Cre expresssion is controlled by a Tetracycline (Tet) responsive element (reviewed in ref. 36). The Mx1-Cre deletor line can be used for targeting of definitive hematopoietic lineages. In this system, Cre expression is activated by injection of interferon or synthetic double-stranded RNA polyinosinic-polycytidylic acid (poly I:C) (37).
1.3 Fluorescent Protein Reporters
A wide range of FPs covering nearly the entire visible spectrum can be used for generating transgenic reporter mice (reviewed in refs. 2, 3, 4). Since the discovery of wtGFP (see Introduction), investigators have sought to generate other FPs FPs with reduced phototoxicity, improved brightness and phosphostability over broad ranges of pH and temperature (2–4). Site-directed mutagenesis of wtGFP has been employed not only for the generation of FPs with improved functionality but also with diverse spectral characteristics, for example cyan FP (CFP), blue FP (BFP), and yellow FP (YFP) (2–4).
FPs with emission peaks in the red and far-red spectra have been especially useful for to live-cell or whole-animal imaging, owing to their long wavelength emission and, consequently, reduced phototoxicity (2, 38). Initially, the tetrameric DsRed was cloned from the nonbioluminescent sea anemone Discosoma striata but was found to be toxic to cells (39). Subsequently, the monomeric variant mRFP was engineered and could be expressed ubiquitously in mice without deleterious effects on development (40).
Directed mutagenesis of mRFP was used to create several variants, including the orange mTomato and the red mStrawberry and mCherry FPs (38). Far-red fluorophores such as mPlum, genetically engineered from a blue chromoprotein of the sea anemone Actinia equina (41), offer deep tissue penetration and reduced autofluorescence. Spectral properties of various FPs are discussed in detail in refs. (2–4).
1.3.1 Fluorescent Fusion Proteins
FPs can be engineered for localization to specific subcellular regions (42). For example, to localize the FP to the nucleus, a nuclear localization signal is incorporated into the construct or the FP is fused to histone H2B sequences. Histone H2B fusion-FPs bind to chromatin and are present through all phases of the cell cycle (43, 44). Unlike cytoplasmic GFP, which is diluted during subsequent cell divisions, H2B-FPs are stably expressed and permit monitoring of both the cell cycle and apoptosis (44). A histone H2B-GFP expressed under the control of a human epsilon-globin promoter and truncated LCR has been used for labeling the nuclei of primitive erythroid cells (24) (Figure 3A). Cell morphology and migration can also be observed by labeling the outer or inner leaflet of the cell membrane with lipid-tagged FP fusions (e.g. containing a glycosylphospatidylinositol (GPI) anchor or a myristoylation sequence, ref. 45).
1.3.2 Photomodulatable FPs
Photoactivatable fluorescent proteins (PAFP) are stimulated by lights of specific wavelengths, intensities and durations, allowing for spatiotemporal labeling of live cells, organelles and molecules (2–4). There are two types of PAFP functions: photoactivation (PA) and photoswitching. Photoactivators convert from a non-fluorescent to a bright fluorescent state and can be either irreversible or reversible. PA-GFP is a GFP variant that is irreversibly converted to an anionic form, resulting in a 100-fold increase in its emission intensity (46). Tetrameric kindling FP (KFP) can be reversibly or irreversibly photoactivated, depending on the intensity of the activating light (4, 47). It converts to a red fluorescent state following exposure to green light and returns to a non-fluorescent state in the absence of stimulation (47). Photoswitchers change their fluorescent state and emit at a different wavelength (such as cyan-green for PS-CFP or green-red for EosFP, Kaede and Kikume Green-Red, KikGR) upon exposure to transient but intense light (3).
1.3.3 General Consideration for Choosing Fluorescent Protein Reporters
When choosing an FP reporter protein, the investigator should consider whether it will be used for multi-color analysis in combination with other FPs or in immunofluorescence studies with a fluorophore of a different color. The availability of fluorescent variants allows the researcher to select combinations that minimize spectral overlap. For example, the combinations of GFP/CFP and GFP/YFP exhibit significant emission overlap, whereas CFP/YFP does not (48). Bright reporters in the red or far-red spectra increase the possibilities of combining different reporters to mark cells of different lineages or to mark different regions of the same cell. Due to the lower phototoxicity of the excitation light required by red or far-red fluorophores, these reporters are more suitable for live imaging studies (49). While imaging or flow cytometric analysis of double transgenic mice expressing FPs with overlapping spectra can be challenging (48), the judicious choice of excitation light and filters will allow optimal separation of reporter signals, as observed for the simultaneous imaging of erythroid cells expressing ECFP and myeloid cells expressing EGFP (50).
1.4 Confirmation and Analysis of Transgene Expression
1.4.1 Mouse background
The choice of genetic background should be carefully considered in planning the generation of a transgenic mouse line. For microinjection, zygotes of mixed or outbred background are often used (16). Microinjection of zygotes from inbred mice is more difficult and embryo viability is lower (16).
Only a relatively small number among the many available inbred strains (e.g. C57BL/6 or 129/Sv) are routinely used to create transgenic or knockin mice. In the context of the present discussion, inbred strains would be desirable if the transgenic animals will be mated with knockout mice known to have a background dependent phenotype or if tissue from the genetically modified animals will be used for HSC or other transplantation studies (51). Genetic background effects (variable penetrance or expressivity) are caused by modifier genes (51). ICR (CD1) mice are the most widely used outbred mouse strain. Unlike inbred mice, ICR mice display inter-individual genetic variation. However, ICR mice are inexpensive, have excellent reproductive and maternal characteristics, and yield relatively large litter sizes (16).
1.4.2 Breeding
Once founders are identified (see section 3.2), the colony should be expanded. Female founders should be bred so that they can give birth to at least one litter before being sacrificed. Male founders should be placed in a cage with two nontransgenic females and plugs checked daily. Ideally, the male founder should plug 6–8 females in the first few weeks (16). The gold standard for assessing transgene integration is germline transmission to the F1 generation. By this metric, founders should transmit the transgene to 50% of their progeny. If transmission is not observed, the founder genotype should be reanalyzed. If the founder is positive for the transgene and germline transmission does not occur, it is likely that the founder is mosaic for the transgene and, therefore, either transmits the transgene through the germline at very low levels or not at all. In certain scenarios, transgenes will integrate at multiple loci resulting in progeny that inherit the transgene at unusually high frequencies (52).
In contrast with mouse lines created by gene targeting, each transgenic founder is distinct because of the random nature of transgene integration. Therefore the decision to eliminate a transgenic mouse line from a colony will be irreversible. To reduce costs, the investigator may choose to maintain an active colony of a few mating pairs or a small number of males that can be mated periodically to produce a younger generation. This is a relatively inexpensive approach but carries the risk that transgene expression may decrease in later generations or as the animals age; this phenomenon has often been seen for globin transgenes (53). Transgene silencing may be avoided through preserving the line as frozen embryos or sperm (so that in vitro fertilization can be performed at a later date) (51). Cryopreservation services are provided by some institutional core transgenic mouse facilities and by mouse suppliers such as Taconic Farms and Charles River Laboratory.
1.5 Analysis of Fluorescent Protein Expression using Microscopy
The fluorescence of embryos, tissues or cells from transgenic reporter mice can be analyzed using epifluorescence or confocal laser scanning microscopy. Confocal microscopes offer several advantages over epifluorescence microscopes. Whereas in epifluorescent microscopy, the entire field is illuminated by light emitted by a mercury or xenon UV lamp, in confocal microscopy, light emitted by the laser is focused through a pinhole, creating point illumination. Out-of-focus signals are thereby eliminated and resolution is increased (54). The confocal microscope images thin sections of the specimen that can be combined using the microscope’s software into accurate 3D reconstructions of the sample. Confocal microscopes also have an increased level of sensitivity due to light detectors that can amplify the signals received from the specimen. Another advantage of confocal microscopy is that it is less invasive, resulting in reduced photobleaching (54). The illumination provided by high-power lasers, combined with their reduced light scattering properties, allows imaging of thick, semitransparent sections, live tissues or embryos (54). Newer model epifluorescent microscopes use LED light sources that are more suitable for live imaging than are classical mercury lamps.
For live imaging of explanted embryos or tissues, temperature and gas composition must be carefully controlled using an environmental chamber. Inverted microscopes are typically used to image live material. For a discussion of imaging mouse embryos using confocal microscopy, see refs. (55–57).
Regardless of the type of microscope used, it is essential to select the appropriate light source and filters for analysis of the fluorescent specimen (54). The identity of the cells expressing the FP may be determined using immunofluorescence, by staining for cell type-specific markers. The staining can be performed on live or fixed cells in solution or on fixed cells deposited on microscope slides. The fixation and permeabilization method should be carefully optimized for each cell type. An overview of different fixation and permeabilization options is reviewed in ref. (54).
1.6 Analysis of Fluorescent Protein Expression using Flow Cytometry
Analytical flow cytometry is a fundamental technique for assessing fluorescence in cells from transgenic FP reporter mice. Once tissues have been dispersed into single cell suspensions, fluorescent reporter expression from a transgene, combined with antibody staining for specific cell surface markers, can be analyzed using a flow cytometer to identify the cell surface characteristics of the component cell populations. Fluorescence Activated Cell Sorting (FACS) is a specialized type of flow cytometry that permits the physical separation of a heterogeneous sample into distinct cell populations based on their fluorescence. The ability to isolate labeled cell populations in a single step provides a valuable tool for elucidation of their developmental potentials, cell cycle and other properties, and for culture ex vivo. For detailed protocols and reviews, see ref. (58).
2. Materials
DNA purification for microinjection
Agarose (Invitrogen; Cat # 16500).
Ethidium bromide (Sigma Aldrich; Cat # E7637).
Injection buffer: 10 mM Tris, pH 7.4, 0.2 mM EDTA.
QIAquick Gel Extraction Kit (Qiagen; Cat # 28704).
Restriction enzymes.
Isolation of genomic DNA
DirectPCR Lysis Reagent (mouse tail) (Viagen; Cat # 102-T).
Proteinase K (Invitrogen; Cat # 25530). Stock solution prepared at 10 mg/ml in TrisHCl 20 mM, pH 8. Aliquots are stored at −20°C.
Polymerase chain reaction (PCR)
Plastic tubes for PCR, 0.2 ml (Denville Scientific, Cat. # C18063).
TaKaRa ExTaq DNA polymerase, supplied with 10X ExTaq buffer and dNTP mix (Clontech, e.g. Cat. # RR001A).
Thermal cycler (available from a variety of companies).
For agarose gel electrophoresis: agarose, gel casting tray, comb with desired number of teeth, power supply, ethidium bromide, DNA size ladder.
Dissecting tools
Dissecting scissors (Roboz Surgical Instrument Inc., Gaithersburg, MD; Cat # RS-6702 and RS-5882) and forceps (Sigma Aldrich, St. Louis, MO; Cat # F4267).
Watchmaker’s forceps, Dumont #5 and #55 (Roboz, FST).
Sterile plastic transfer pipettes, 3 ml (VWR; Cat # 414004-037).
Stereomicroscope with transmitted and reflected light sources (Zeiss, Leica or Nikon).
Glassware and Plasticware
Nunclon tissue culture plates, 24 well (Nunc, Thermo Fisher Scientific; Cat # 142475).
Circular coverslips, 12 mm, no. 1 (Thermo Fisher Scientific; Cat # 12-545-80).
3 and 5 ml syringes (BD Biosciences; Cat # 309657 and # 309646).
Syringe needles, 20G and 25G (BD Biosciences; Cat # 305176 and # 305122).
Polypropylene tubes, 15 and 50 ml (Corning, Lowell, MA; Cat # 430766 and # 430291).
Embryo dissection and cell preparation for flow cytometry
Phosphate buffered saline (PBS) pH 7.4 (GIBCO Invitrogen, Carlsbad, CA; Cat # 10010-023).
Iscove’s Modified Dulbecco’s Medium (GIBCO Invitrogen; Cat # 12440-079).
Fetal Bovine Serum (FBS; Hyclone, Thermo Fisher Scientific).
Dissection medium: IMDM +10% FBS.
Heparin (Sigma Aldrich, St. Louis, MI; Cat # H3149): Dissolve in PBS to 12.5 mg/ml to produce a stock solution (100X).
BD Falcon 40 μm and 70 μm cell strainers (BD Biosciences, San Jose, CA; Cat # 352340 and # 352350).
Cell Dissociation Buffer (GIBCO Invitrogen; Cat # 13150-016).
Collagenase (Sigma Aldrich; Cat # C2674): Stock solution prepared at 100 mg/ml in medium supplemented with 20% serum. Aliquots are stored at −20°C.
Flow cytometry
FACS buffer: heat-inactivated FBS diluted in PBS (see Note 1).
DAPI (4',6-diamidino-2-phenylindole dihydrochloride; Sigma Aldrich; Cat # D9542);
Propidium iodide (Sigma-Aldrich; Cat. # P4170). Dilute powder in distilled water to prepare 1000X stock solution.
Immunostaining and microscopy
4% Paraformaldehyde (PFA) obtained by dilution of 16% PFA (Electron Microscopy Sciences, Cat # 15710) in PBS.
Washing buffers: PBS with 0.05% Tween-20 (v/v) (Sigma Aldrich; Cat # P1379)(PBST); PBST with 0.05% no-fat skim milk powder (Carnation)(PBSMT).
Vectashield with DAPI (Vector Labs, Burlingame, CA; Cat # H-1500) or without (Vector Labs, Burlingame, CA; Cat # H-1400).
Primary and secondary antibodies of choice.
Triton X-100 (Sigma Aldrich; Cat # T8787).
Bovine serum albumin (BSA) (Sigma Aldrich; Cat # B4287).
Blocking buffer: 2% BSA, 0.1% Triton X-100 in PBS.
3. Methods
3.1 DNA Preparation for Microinjection
Transgenes are designed so that the gene to be microinjected can be excised and purified away from plasmid sequences. Prokaryotic sequences from the plasmid do not appear to influence the efficiency of transgene integration but they may impair expression from eukaryotic sequences (14, 15, 16). Therefore, the plasmid backbone sequences should be excised from the transgene construct. It is important to include a final purification step to remove particulate material that may clog the injection needle. The following protocol yields clean DNA for microinjection from plasmids smaller than 20 kb. Plasmids larger than 20 kb require a different purification procedure (59), that will not be discussed here.
Digest plasmid DNA to completion with the appropriate restriction enzyme(s).
Separate the restriction fragments by electrophoresis through an agarose gel (see Note 2).
Using an ultraviolet transilluminator, identify and isolate the band containing the transgene to be microinjected.
Extract and purify the DNA according to the manufacturer’s protocols for the QIAquick Gel Extraction Kit (QIAGEN).
Elute the DNA from the QIAquick column using injection buffer (see Materials section) and measure the DNA concentration using a NanoDrop spectrophotometer (Thermo Scientific).
Pronuclear injection is typically performed in an institutional core transgenic mouse facility. For a detailed protocol, see ref. (60).
At our institution, the purified DNA (1–2 μg, 50–100 ng/μl) is submitted to the Mouse Genetics and Gene Targeting Shared Resource Facility. Filtration and final dilution of the DNA to be used for microinjection is performed by the facility, as is transfer of the injected zygote into pseudopregnant females(16).
3.2 Genotyping
Offspring born from injected zygotes are termed "founders" and are usually screened for the presence of the transgene. Genomic screening is most commonly performed using polymerase chain reaction (PCR) analysis of DNA from biopsied tissue (for comments on primer design, see Note 3). In most cases, the microinjected DNA will be stably integrated at the one-cell stage. However, the foreign DNA may integrate at a later (e.g. 4- or 8-cell) stage, resulting in mosaic expression of the transgene and disruption of germline transmission (16). In addition, silencing of the transgene may occur following integration into or near heterochromatin (see Note 4).
It may be possible to identify founders by microscopic analysis of biopsied tissues (see Note 5).
Preparation of Tissue Samples for Genotyping
Tail tips <0.5 cm may be biopsied from pups ≤ 21 days old without the use of an analgesic. Adult mice must be anesthetized according to federal and Institutional Animal Care and Use Committee (IACUC) regulations. Using scissors cleaned with 70% ethanol, cut a 1cm section of the tail tip of the founder mouse. Be sure to wipe the scissors with 70% ethanol before cutting the next tail, to prevent DNA cross-contamination between samples. Place the tail tip samples into labeled 1.5 ml eppendorf tubes. Samples can be stored frozen at −80°C and DNA isolated at a later date.
For DNA isolation, add 195 μl of DirectPCR Lysis Reagent (mouse tail) to each tube, followed by 5 μl Proteinase K (10 mg/ml).
Incubate the tubes at 55°C using mild agitation for 4 hr or overnight.
Heat inactivate the samples at 85°C for 45 min.
Genomic PCR
For PCR, pipette 1 μl DNA into a clean 0.2 ml tube containing 0.5 μl of 10 μM stocks of forward and reverse primer (see Table 2); 2 μl of 10X ExTaq Buffer containing MgCl2 (supplied with the polymerase), 1 μl of 2.5 mM dNTP mix, 0.2 μl TaKaRa ExTaq DNA polymerase (Clontech), and water to a total volume of 20 μl. For multiple reactions using the same forward and reverse primers, it is advisable to prepare a master mix containing all components except for the genomic DNA. See Table 2 for fluorescent reporter primer sequences used in our laboratory.
Conditions for PCR in a standard thermal cycler: (a) initial denaturation at 95°C for 5 min; (b) 35 cycles of: denaturation at 95°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 1 min; (c) final extension at 72°C for 5 min. Samples are then maintained at 4°C until ready for analysis using standard agarose gel electrophoresis (e.g. ref. 61). Annealing temperature is generally close to the melting temperature for the primers and may need to be optimized. The extension time may also need to be optimized according to the length of the PCR product.
Table 2.
PCR primer pair sequences for commonly used fluorescent reporters
| Reporter | Primer Sequences | Tm | Product size |
|---|---|---|---|
| GFP | Forward - 5’-CAT GAG CAA GGG CGA GGA ACT-3’ Reverse - 5’-CAG CAG CGG TCA CAA ACT CC -3' |
55°C | 750 bp |
| ExFP (for GFP, CFP,YFP) | Forward - 5'-CAC CAT CTT CTT CAA GGA CGA C-3' Reverse - 5'-TTC TCG TTG GGG TCT TTG C-3' |
53°C | 350 bp |
| mCherry | Forward -5’-GAT ACT CGA CC AAG CAA GGG CGA GG-3’ Reverse -5’-CATA ACT CGA GTT ATG TAC AGC TCG TC-3’ |
53°C | 720 bp |
| tdTomato | Forward -5’-ATG GAG GGC TCC ATG AAC G -3’ Reverse - 5’-CCC ATG GTC TTC TTC TGC -3’ |
50°C | 370 bp |
Abbreviations: bp, base pairs; Tm, annealing temperature
3.3 Dissection
Following identification of founders using genomic PCR, expression of the reporter is examined in whole embryos, dissected tissues, peripheral blood or bone marrow using microscopy or flow cytometry.
3.3.1 Embryo dissection
Typically the analysis of hematopoietic lineages in the embryo/fetus is performed from E7.5 through E16.5. Below we present a general protocol for dissection of E9.5 to E14.5 embryos. For more detailed information about dissection of earlier stage embryos, see refs. (16, 62).
Euthanize pregnant mother by CO2 asphyxiation followed by cervical dislocation.
Spray the abdomen of the mouse with 70% ethanol. Pinch the fur on the abdomen and make a midline incision to open the abdominal cavity and expose the uterine horns.
Use a forceps to lift up the ends of the uterine horns and carefully remove the attached connective tissue and fat. Wash off the maternal blood in a 10 cm Petri dish containing PBS supplemented with 5% FBS.
Carefully cut each conceptus and transfer it to a 10 cm Petri dish containing PBS. Rinse thoroughly with PBS to remove maternal blood and then transfer to a Petri dish containing dissection medium.
Using a pair of #5 or #55 watchmaker’s forceps, peel away the uterine tissue, starting from the incision created in step 4.
Gently detach the Reichert’s membrane by holding the embryo with one pair of forceps and removing the membrane with a second pair of forceps.
The embryo is visible inside the yolk sac, with the placenta attached. The placenta can be separated using forceps or fine scissors. Once the placental vessels are cut, the embryonic blood will be released into the medium. Placental dissociation is performed as described in section 3.3.4. The YS can be removed from the embryo by carefully peeling it away using forceps.
3.3.2 Isolation of Peripheral Blood from Embryos
Peripheral blood can be obtained from ~E9.5 onward (63), when the embryonic circulation is well established (64).
Each embryo (YS and placenta intact) is transferred to a well of a 24-well dish containing dissection medium with 0.5% heparin. Fluorescence can be easily evaluated using a fluorescence stereomicroscope.
After selection of the desired embryos, peripheral blood can be collected. Grab the region between the embryo and the placenta using a #5 watchmaker’s forceps and cut umbilical and vitelline vessels with another #5 forceps. Peripheral blood cells will be released into the medium in large numbers.
Allow the embryos to exsanguinate (~10 min). Blood cells will collect at the bottom of the well.
Remove the embryos and debris from each well and resuspend the blood cells in dissection medium, using a P1000 Pipetman.
Filter the blood cell suspension through a 40 μm cell strainer and then collect the cells by centrifugation at 1200 rpm (100 × g) in an Eppendorf microcentrifuge.
Resuspend the cells at the desired concentration in a buffer appropriate for the intended application.
3.3.3 Dissection of Fetal Liver
The FL does not develop a tightly adherent epithelial structure and can be easily dispersed mechanically, allowing simple isolation of hematopoietic cells through ~E16.5.
Carefully dissect the FL from the embryo using a pair of #5 watchmaker’s forceps.
Transfer the FL to a 1.5 ml eppendorf tube containing 0.5 ml dissection medium.
Disperse the FL by pipetting in dissection medium using a P1000 Pipetman until obtaining a homogeneous suspension.
Filter the cell suspension through a 70μm cell strainer, collect the cells by centrifugation at 1200 rpm (100 × g) in an Eppendorf microcentrifuge and resuspend in PBS for cytocentrifugation or in FACS buffer for flow cytometry.
3.3.4 Dissection of Yolk Sac and Placenta
YS and placenta contain endothelial cells with adherent junctions and endodermal cells with tight junctions, therefore requiring more vigorous dissociation steps than for FL prior to isolation of hematopoietic cells.
Dissect the YS (62) and placenta (65) from each embryo at the desired stage.
Place each tissue into an individual 1.5 ml eppendorf tube containing 1 ml collagenase. If two or more YSs or placentae will be collected from embryos at the same stage, they can be pooled in a 15 ml conical tube containing 4 ml collagenase. Dissociation to single cells is more efficient if narrow dissection scissors (Roboz RS-6702) are used to macerate the YS or placental tissue.
Incubate at 37°C for at least 20 min. Shake vigorously every 5 min. until a uniform cell suspension has been obtained (no large clumps remain). Filter the sample through a 70μm cell strainer, collect by centrifugation in an Eppendorf microcentrifuge at 1200 rpm (100 × g) and resuspend in PBS or FACS buffer, as described above.
3.3.5 Isolation of the AGM Region
Dissection of the AGM region has been described in detail, and presented in a video, by others (66).
3.3.6 Isolation of Adult Bone Marrow
Euthanize the adult mouse by CO2 asphyxiation followed by cervical dislocation.
Spray the abdomen of the mouse with 70% ethanol. Make a midline incision to open the abdominal cavity and expose the hind legs.
Remove all the muscle and connective tissue from the bones using scissors and then cut the tibia and femur from the joints.
Cut the ends of the bones and flush the bone marrow with 1 ml buffer of choice (according to the subsequent analysis) into a collection tube stored on ice, using a 25G needle and a 5 ml syringe.
Homogenize the bone marrow suspension by pipetting up and down and pass the cell suspension through a 70 μm cell strainer.
3.4 General Immunofluorescence Protocol
Fix and permeabilize the cells using the appropriate reagents (e.g. 4% PFA in PBS; see Note 6). Wash the cells twice for 5 min in PBS.
Block the cells for 15–30 min in blocking buffer.
Dilute primary antibody in blocking buffer and add 100 μl per slide. Incubate for 1 hr at room temperature.
Wash 3 times for 5 min with PBS + 0.01 % (v/v) Triton X-100.
Dilute secondary antibody in blocking buffer and add 100 μl per slide. Incubate for 45 min.
Wash 3 times for 5 min with PBS + 0.01 % (v/v) Triton X-100. Rinse in PBS and then in water to avoid crystallization of the salts from PBS.
Mount coverslips using Vectashield mounting medium with or without DAPI (see Note 7).
3.5 Flow Cytometry
3.5.1 Labeling of Cells for Flow Cytometry
Count cells using a hemacytometer and dispense 1×106 cells into a 1.5 ml eppendorf tube. Collect by centrifugation at 1200 rpm (100 × g) in an Eppendorf microcentrifuge and aspirate supernatant.
Dilute fluorescently conjugated antibody into 10% FACS buffer (see Note 1). We generally use 2 μg of antibody per 100,000 cells; however, each antibody should be titrated for optimal results. Antibodies conjugated to different fluorochromes should be combined in the same "cocktail" to minimize cell loss due to additional incubation and washing steps.
As EryP do not express Fc receptors, they exhibit low background binding to antibodies. We have not found that treatment of cells with normal mouse serum or FcBlock is necessary. Cells expressing the Fc receptor should be treated with normal mouse serum or purified anti-mouse CD16/CD32 to prevent non-specific binding.
Resuspend cells in a 1.5 ml Eppendorf tube containing 100 μl of the antibody cocktail and incubate on ice in the dark for 20 min, inverting the tubes every 5 min.
Wash with 1 ml 10% FACS buffer and collect cells by centrifugation at 1200 rpm (100 × g) in an Eppendorf microcentrifuge.
If primary antibodies are unconjugated or biotin-conjugated, treat cells with fluorescently conjugated secondary antibodies or streptavidin antibodies in a final volume of 100 μl in 10% FACS buffer. Resuspend the cell pellet in 100 μl of diluted streptavidin. Incubate on ice in the dark for 20 min., inverting the tube every 5 min.
Wash with 1 ml 10% FACS buffer and collect cells by centrifugation at 1200 rpm (100 × g) in an Eppendorf microcentrifuge..
Resuspend the cell pellet in 400 μl of 3% FACS buffer containing DAPI (see Note 8) if a UV laser available or PI if a UV laser is not available. Transfer the cell suspension to a 5 ml round bottom tube (see Note 9).
Analyze using a flow cytometer.
3.5.2 Preparation of Cells for Sorting by FACS
Label cells for FACS as described above in section 3.5.1.
Prepare collection tubes for sorted cells. If the sorted cells are to be cultured, collect into a 5 ml round bottom tube containing 1 ml sterile medium.
FACS instruments are typically operated by trained personnel. The investigator will advise the operator regarding the populations to be sorted.
Tubes containing sorted cells are kept briefly on ice until the cells can be collected by centrifugation. Cell pellets can be stored frozen at -80ºC prior to isolation of RNA.
Acknowledgments
We thank J. Barminko and G. Camprecios for their comments on the manuscript. Work in our laboratory was supported in part by grants to M.H.B. from the National Institutes of Health (RO1 HL62248 and, DK52191, and EB02209), the Roche Foundation for Anemia Research (grant 9699367999, cycle X) and the New York State Department of Health (NYSTEM grant N08G-024).
Footnotes
FACS buffer is PBS containing proteins (e.g. FBS). The proteins in FBS reduce cell loss by decreasing their interaction with plastic during the antibody staining procedure and, more importantly, the plastic tubing of the fluidics system of the flow cytometer. During antibody staining, we routinely use PBS containing 10% heat-inactivated FBS (10% FACS buffer). After the last washing step, the cells are resuspended in PBS containing 3% heat-inactivated FBS (3% FACS buffer).
Some investigators prefer to stain the DNA with crystal violet to avoid toxicity of ethidium bromide.
PCR sequences for common fluorescent reporters such as GFP are available in the literature (refer to Table 2 for primer sequences used in our laboratory). If more than one FP transgenic reporter mouse line is maintained in the colony, it is advisable to use a primer specific for sequences outside the FP coding region. For example, one primer would hybridize with a sequence in the FP coding region while the second would hybridize with an external sequence such as the promoter or 3'-UTR.
Microinjected DNA can be transcriptionally silenced as a result of integration in or near heterochromatin, a process called variegation. Variegation is a heterocellular pattern of gene expression sometimes observed in transgenic mice. It is commonly age-dependent and also results from progressive breeding of the mice (53). This phenomenon is often seen with both the alpha- and beta-globin genes. Variegation may be suppressed if the transgene is linked to certain globin gene enhancer elements (67, 68).
Identification of founders may be possible using direct microscopic analysis of tissue biopsies from pups (up to 21 days old) if transgene expression is sufficiently bright. For example, we genotype Flk1-H2B-YFP transgenic mice (69) by examining ear clips from 10 day-old newborn mice. The Flk1 promoter is active in endothelial cells of the skin at this stage. This simple genotyping approach can be applied to other transgenic fluorescent reporters that are expressed in tissues that are easily collected by biopsy.
Exposure of the FPs to fixation reagents (e.g. formaldehyde, glutaraldehyde, methanol, acetone) denatures protein, leading to loss of fluorescence. Therefore, the fixation method (reagent and time of exposure) must be optimized. When using PFA or glutaraldehyde as fixative, quenching for 15 min with 0.1M glycine (final concentration) following fixation will help to reduce FP denaturation. If fluorescence is lost during the fixation step, the FP can be detected by immunofluorescece after staining with an FP specific antibody.
The use of Vectashield mounting medium with DAPI may result in increased background fluorescence. This may present a particular problem when the intensity of reporter fluorescence is weak. It may, therefore, be desirable to stain with DAPI, followed by washing of the slide and mounting the coverslip using Vectashield without DAPI.
To exclude dead cells, which may bind nonspecifically to antibodies and produce a false positive signal, we routinely resuspend the final cell pellet in FACS buffer containing 0.2 mg/ml DAPI. DAPI is a poorly cell-permeable DNA binding dye that is excited by the violet laser in the flow cytometer. DAPI is soluble in water but not in PBS. Therefore, we prepare a 1000X stock solution in deionized water. The stock is then diluted into FACS buffer.
Sorting time and efficiency varies widely due to concentration of cell suspension and the degree of cell death. A highly concentrated sample of cells and significant amount of cell death will increase the "abortion rate" – the rate at which droplets containing single cells are not selected by the FACS machine to be sorted –resulting in lower recovery of viable sorted cells. The abortion rate can be reduced and sample purity increased by diluting the sample and/or by modifying the cell dissociation technique.
References
- 1.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992 Feb 15;111(2):229–33. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 2.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005 Dec;2(12):905–9. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 3.Nowotschin S, Eakin GS, Hadjantonakis AK. Live-imaging fluorescent proteins in mouse embryos: multi-dimensional, multi-spectral perspectives. Trends Biotechnol. 2009 May;27(5):266–76. doi: 10.1016/j.tibtech.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010 Jul;90(3):1103–63. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- 5.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008 Feb 22;132(4):631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baron MH, Isern J, Fraser ST. The embryonic origins of erythropoiesis in mammals. Blood. 2012 May 24;119(21):4828–37. doi: 10.1182/blood-2012-01-153486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palis J, Yoder MC. Yolk-sac hematopoiesis: the first blood cells of mouse and man. Exp Hematol. 2001 Aug;29(8):927–36. doi: 10.1016/s0301-472x(01)00669-5. [DOI] [PubMed] [Google Scholar]
- 8.Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. 2008 Feb;9(2):129–36. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orkin SH, Zon LI. SnapShot: hematopoiesis. Cell. 2008 Feb 22;132(4):712. doi: 10.1016/j.cell.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol. 1987 Aug 20;196(4):947–50. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 11.Zufferey R, Donello JE, Trono D, Hope TJ. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol. 1999 Apr;73(4):2886–92. doi: 10.1128/jvi.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulger M, Groudine M. Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev Biol. 2010 Mar 15;339(2):250–7. doi: 10.1016/j.ydbio.2009.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Mol Cell Biol. 1991 Jun;11(6):3070–4. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chada K, Magram J, Raphael K, Radice G, Lacy E, Costantini F. Specific expression of a foreign β-globin gene in erythroid cells of transgenic mice. Nature. 1985;314:377–80. doi: 10.1038/314377a0. [DOI] [PubMed] [Google Scholar]
- 15.Townes TM, Lingrel JB, Chen HY, Brinster RL, Palmiter RD. Erythroid-specific expression of human beta-globin genes in transgenic mice. EMBO J. 1985 Jul;4(7):1715–23. doi: 10.1002/j.1460-2075.1985.tb03841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagy A, Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- 17.Henikoff S. Conspiracy of silence among repeated transgenes. Bioessays. 1998 Jul;20(7):532–5. doi: 10.1002/(SICI)1521-1878(199807)20:7<532::AID-BIES3>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 18.Bushey AM, Dorman ER, Corces VG. Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol Cell. 2008 Oct 10;32(1):1–9. doi: 10.1016/j.molcel.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Festenstein R, Tolaini M, Corbella P, Mamalaki C, Parrington J, Fox M, et al. Locus control region function and heterochromatin-induced position effect variegation. Science. 1996 Feb 23;271(5252):1123–5. doi: 10.1126/science.271.5252.1123. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Harju S, Peterson KR. Locus control regions: coming of age at a decade plus. Trends Genet. 1999 Oct;15(10):403–8. doi: 10.1016/s0168-9525(99)01780-1. [DOI] [PubMed] [Google Scholar]
- 21.Festenstein R, Kioussis D. Locus control regions and epigenetic chromatin modifiers. Curr Opin Genet Dev. 2000 Apr;10(2):199–203. doi: 10.1016/s0959-437x(00)00060-5. [DOI] [PubMed] [Google Scholar]
- 22.Forrester WC, Novak U, Gelinas R, Groudine M. Molecular analysis of the human beta-globin locus activation region. Proc Natl Acad Sci U S A. 1989 Jul;86(14):5439–43. doi: 10.1073/pnas.86.14.5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dyer MA, Farrington SM, Mohn D, Munday JR, Baron MH. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development. 2001 May;128(10):1717–30. doi: 10.1242/dev.128.10.1717. [DOI] [PubMed] [Google Scholar]
- 24.Isern J, Fraser ST, He Z, Baron MH. The fetal liver is a niche for maturation of primitive erythroid cells. Proc Natl Acad Sci U S A. 2008 May 6;105(18):6662–7. doi: 10.1073/pnas.0802032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haruyama N, Cho A, Kulkarni AB. Overview: engineering transgenic constructs and mice. Curr Protoc Cell Biol. 2009 Mar;Chapter 19(Unit 19 0) doi: 10.1002/0471143030.cb1910s42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasmussen KD, O'Carroll D. The miR-144/451eGFP allele, a novel tool for resolving the erythroid potential of hematopoietic precursors. Blood. 2011 Sep 15;118(11):2988–92. doi: 10.1182/blood-2011-04-350728. [DOI] [PubMed] [Google Scholar]
- 27.Buza-Vidas N, Cismasiu VB, Moore S, Mead AJ, Woll PS, Lutteropp M, et al. Dicer is selectively important for the earliest stages of erythroid development. Blood. 2012 Sep 20;120(12):2412–6. doi: 10.1182/blood-2011-10-383653. [DOI] [PubMed] [Google Scholar]
- 28.North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, et al. Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity. 2002 May;16(5):661–72. doi: 10.1016/s1074-7613(02)00296-0. [DOI] [PubMed] [Google Scholar]
- 29.Lorsbach RB, Moore J, Ang SO, Sun W, Lenny N, Downing JR. Role of RUNX1 in adult hematopoiesis: analysis of RUNX1-IRES-GFP knock-in mice reveals differential lineage expression. Blood. 2004 Apr 1;103(7):2522–9. doi: 10.1182/blood-2003-07-2439. [DOI] [PubMed] [Google Scholar]
- 30.Mountford PS, Smith AG. Internal ribosome entry sites and dicistronic RNAs in mammalian transgenesis. Trends Genet. 1995 May;11(5):179–84. doi: 10.1016/S0168-9525(00)89040-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vintersten K, Testa G, Naumann R, Anastassiadis K, Stewart AF. Bacterial artificial chromosome transgenesis through pronuclear injection of fertilized mouse oocytes. Methods Mol Biol. 2008;415:83–100. doi: 10.1007/978-1-59745-570-1_5. [DOI] [PubMed] [Google Scholar]
- 32.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC developmental biology. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007 Sep;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 34.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010 Jan;13(1):133–40. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyer SW, Beaudin AE, Forsberg EC. Mapping differentiation pathways from hematopoietic stem cells using Flk2/Flt3 lineage tracing. Cell Cycle. 2012 Sep 1;11(17):3180–8. doi: 10.4161/cc.21279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewandoski M. Analysis of Mouse Development with Conditional Mutagenesis. Handbook of Experimental Therapeutics. 2007;178:231–58. doi: 10.1007/978-3-540-35109-2_10. [DOI] [PubMed] [Google Scholar]
- 37.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995 Sep 8;269(5229):1427–9. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 38.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004 Dec;22(12):1567–72. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 39.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, et al. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002 Jun 11;99(12):7877–82. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu H, Wang G, Li G, Han M, Xu T, Zhuang Y, et al. Ubiquitous expression of mRFP1 in transgenic mice. Genesis. 2005 Jun;42(2):86–90. doi: 10.1002/gene.20129. [DOI] [PubMed] [Google Scholar]
- 41.Shkrob MA, Yanushevich YG, Chudakov DM, Gurskaya NG, Labas YA, Poponov SY, et al. Far-red fluorescent proteins evolved from a blue chromoprotein from Actinia equina. Biochem J. 2005 Dec 15;392(Pt 3):649–54. doi: 10.1042/BJ20051314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snapp E. Design and use of fluorescent fusion proteins in cell biology. Curr Protoc Cell Biol. 2005 Jul;Chapter 21(Unit 21 4) doi: 10.1002/0471143030.cb2104s27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998 Mar 26;8(7):377–85. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 44.Hadjantonakis AK, Papaioannou VE. Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol. 2004 Dec 24;4:33. doi: 10.1186/1472-6750-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhee JM, Pirity MK, Lackan CS, Long JZ, Kondoh G, Takeda J, et al. In vivo imaging and differential localization of lipid-modified GFP-variant fusions in embryonic stem cells and mice. Genesis. 2006 Apr;44(4):202–18. doi: 10.1002/dvg.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Innovation: Photoactivatable fluorescent proteins. Nat Rev Mol Cell Biol. 2005 Nov;6(11):885–91. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]
- 47.Chudakov DM, Belousov VV, Zaraisky AG, Novoselov VV, Staroverov DB, Zorov DB, et al. Kindling fluorescent proteins for precise in vivo photolabeling. Nat Biotechnol. 2003 Feb;21(2):191–4. doi: 10.1038/nbt778. [DOI] [PubMed] [Google Scholar]
- 48.Stadtfeld M, Varas F, Graf T. Fluorescent protein-cell labeling and its application in time-lapse analysis of hematopoietic differentiation. Methods Mol Med. 2005;105:395–412. doi: 10.1385/1-59259-826-9:395. [DOI] [PubMed] [Google Scholar]
- 49.Nowotschin S, Eakin GS, Hadjantonakis AK. Dual transgene strategy for live visualization of chromatin and plasma membrane dynamics in murine embryonic stem cells and embryonic tissues. Genesis. 2009 May;47(5):330–6. doi: 10.1002/dvg.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heck S, Ermakova O, Iwasaki H, Akashi K, Sun CW, Ryan TM, et al. Distinguishable live erythroid and myeloid cells in beta-globin ECFP × lysozyme EGFP mice. Blood. 2003 Feb 1;101(3):903–6. doi: 10.1182/blood-2002-06-1861. [DOI] [PubMed] [Google Scholar]
- 51.Papaioannou VE, Behringer RR. Mouse Phenotypes: A Handbook of Mutation Analysis. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- 52.Conner DA. Transgenic mouse colony management. Curr Protoc Mol Biol. 2005 Aug;Chapter 23(Unit 23):10. doi: 10.1002/0471142727.mb2310s71. [DOI] [PubMed] [Google Scholar]
- 53.Robertson G, Garrick D, Wilson M, Martin DI, Whitelaw E. Age-dependent silencing of globin transgenes in the mouse. Nucleic Acids Res. 1996 Apr 15;24(8):1465–71. doi: 10.1093/nar/24.8.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spector DL, Goldman RD. Basic Methods in Microscopy. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2006. [Google Scholar]
- 55.Jones EA, Baron MH, Fraser SE, Dickinson ME. Dynamic in vivo imaging of mammalian hematovascular development using whole embryo culture. Methods Mol Med. 2005;105:381–94. doi: 10.1385/1-59259-826-9:381. [DOI] [PubMed] [Google Scholar]
- 56.Udan RS, Dickinson ME. Imaging Mouse Embryonic Development. In: Wassarman PM, Soriano PM, editors. Methods in Enzymology. San Diego, CA: Academic Press; 2010. pp. 329–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowotschin S, Ferrer-Vaquer A, Hadjantonakis A-K. Imaging Mouse Development with Confocal Time-Lapse Microscopy. In: Wassarman PM, Soriano PM, editors. Methods in Enzymology. San Diego, CA: Academic Press; 2010. pp. 351–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hawley TS, Hawley R. Flow Cytometry Protocols. 3. Totowa, NJ: Humana Press; 2011. [Google Scholar]
- 59.Krajnc NL, Smrekar F, Cerne J, Raspor P, Modic M, Krgovic D, et al. Purification of large plasmids with methacrylate monolithic columns. J Sep Sci. 2009 Aug;32(15–16):2682–90. doi: 10.1002/jssc.200900260. [DOI] [PubMed] [Google Scholar]
- 60.Ittner LM, Gotz J. Pronuclear injection for the production of transgenic mice. Nat Protoc. 2007;2(5):1206–15. doi: 10.1038/nprot.2007.145. [DOI] [PubMed] [Google Scholar]
- 61.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 62.Baron MH, Mohn D. Mouse embryonic explant culture system for analysis of hematopoietic and vascular development. Methods Mol Med. 2005;105:231–56. doi: 10.1385/1-59259-826-9:231. [DOI] [PubMed] [Google Scholar]
- 63.Fraser ST, Isern J, Baron MH. Maturation and enucleation of primitive erythroblasts during mouse embryogenesis is accompanied by changes in cell-surface antigen expression. Blood. 2007 Jan 1;109(1):343–52. doi: 10.1182/blood-2006-03-006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGrath KE, Koniski AD, Malik J, Palis J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood. 2003 Mar 1;101(5):1669–76. doi: 10.1182/blood-2002-08-2531. [DOI] [PubMed] [Google Scholar]
- 65.Gekas C, Rhodes KE, Mikkola HK. Isolation and visualization of mouse placental hematopoietic stem cells. Current protocols in stem cell biology. 2008 Aug;Chapter 2(Unit 2A 8 1–2A):8–14. doi: 10.1002/9780470151808.sc02a08s6. [DOI] [PubMed] [Google Scholar]
- 66.Morgan K, Kharas M, Dzierzak E, Gilliland DG. Isolation of Early Hematopoietic Stem Cells from Murine Yolk Sac and AGM. J Vis Exp. 2008;16:e789. doi: 10.3791/789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Graubert TA, Hug BA, Wesselschmidt R, Hsieh CL, Ryan TM, Townes TM, et al. Stochastic, stage-specific mechanisms account for the variegation of a human globin transgene. Nucleic Acids Res. 1998 Jun 15;26(12):2849–58. doi: 10.1093/nar/26.12.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robertson G, Garrick D, Wu W, Kearns M, Martin D, Whitelaw E. Position-dependent variegation of globin transgene expression in mice. Proc Natl Acad Sci U S A. 1995 Jun 6;92(12):5371–5. doi: 10.1073/pnas.92.12.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fraser ST, Hadjantonakis AK, Sahr KE, Willey S, Kelly OG, Jones EA, et al. Using a histone yellow fluorescent protein fusion for tagging and tracking endothelial cells in ES cells and mice. Genesis. 2005 Jul;42(3):162–71. doi: 10.1002/gene.20139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suzuki N, Ohneda O, Minegishi N, Nishikawa M, Ohta T, Takahashi S, et al. Combinatorial Gata2 and Sca1 expression defines hematopoietic stem cells in the bone marrow niche. Proc Natl Acad Sci U S A. 2006 Feb 14;103(7):2202–7. doi: 10.1073/pnas.0508928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hills D, Gribi R, Ure J, Buza-Vidas N, Luc S, Jacobsen SE, et al. Hoxb4-YFP reporter mouse model: a novel tool for tracking HSC development and studying the role of Hoxb4 in hematopoiesis. Blood. 2011 Mar 31;117(13):3521–8. doi: 10.1182/blood-2009-12-253989. [DOI] [PubMed] [Google Scholar]
- 72.Ma X, Robin C, Ottersbach K, Dzierzak E. The Ly-6A (Sca-1) GFP transgene is expressed in all adult mouse hematopoietic stem cells. Stem Cells. 2002;20(6):514–21. doi: 10.1634/stemcells.20-6-514. [DOI] [PubMed] [Google Scholar]
- 73.Hosen N, Yamane T, Muijtjens M, Pham K, Clarke MF, Weissman IL. Bmi-1-green fluorescent protein-knock-in mice reveal the dynamic regulation of bmi-1 expression in normal and leukemic hematopoietic cells. Stem Cells. 2007 Jul;25(7):1635–44. doi: 10.1634/stemcells.2006-0229. [DOI] [PubMed] [Google Scholar]
- 74.Tadjali M, Zhou S, Rehg J, Sorrentino BP. Prospective isolation of murine hematopoietic stem cells by expression of an Abcg2/GFP allele. Stem Cells. 2006 Jun;24(6):1556–63. doi: 10.1634/stemcells.2005-0562. [DOI] [PubMed] [Google Scholar]
- 75.Cairns LA, Moroni E, Levantini E, Giorgetti A, Klinger FG, Ronzoni S, et al. Kit regulatory elements required for expression in developing hematopoietic and germ cell lineages. Blood. 2003 Dec 1;102(12):3954–62. doi: 10.1182/blood-2003-04-1296. [DOI] [PubMed] [Google Scholar]
- 76.Kimura Y, Ding B, Imai N, Nolan DJ, Butler JM, Rafii S. c-Kit-mediated functional positioning of stem cells to their niches is essential for maintenance and regeneration of adult hematopoiesis. PLoS One. 2011;6(10):e26918. doi: 10.1371/journal.pone.0026918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bee T, Swiers G, Muroi S, Pozner A, Nottingham W, Santos AC, et al. Nonredundant roles for Runx1 alternative promoters reflect their activity at discrete stages of developmental hematopoiesis. Blood. 2010 Apr 15;115(15):3042–50. doi: 10.1182/blood-2009-08-238626. [DOI] [PubMed] [Google Scholar]
- 78.Nutt SL, Metcalf D, D'Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. 2005 Jan 17;201(2):221–31. doi: 10.1084/jem.20041535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang J, Varas F, Stadtfeld M, Heck S, Faust N, Graf T. CD41-YFP mice allow in vivo labeling of megakaryocytic cells and reveal a subset of platelets hyperreactive to thrombin stimulation. Exp Hematol. 2007 Mar;35(3):490–9. doi: 10.1016/j.exphem.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 80.Vassen L, Okayama T, Moroy T. Gfi1b:green fluorescent protein knock-in mice reveal a dynamic expression pattern of Gfi1b during hematopoiesis that is largely complementary to Gfi1. Blood. 2007 Mar 15;109(6):2356–64. doi: 10.1182/blood-2006-06-030031. [DOI] [PubMed] [Google Scholar]
- 81.Wareing S, Eliades A, Lacaud G, Kouskoff V. ETV2 expression marks blood and endothelium precursors, including hemogenic endothelium, at the onset of blood development. Dev Dyn. 2012 Sep;241(9):1454–64. doi: 10.1002/dvdy.23825. [DOI] [PubMed] [Google Scholar]
- 82.Koyano-Nakagawa N, Kweon J, Iacovino M, Shi X, Rasmussen TL, Borges L, et al. Etv2 is expressed in the yolk sac hematopoietic and endothelial progenitors and regulates Lmo2 gene expression. Stem Cells. 2012 Aug;30(8):1611–23. doi: 10.1002/stem.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishimura S, Takahashi S, Kuroha T, Suwabe N, Nagasawa T, Trainor C, et al. A GATA box in the GATA-1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol Cell Biol. 2000 Jan;20(2):713–23. doi: 10.1128/mcb.20.2.713-723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heinrich AC, Pelanda R, Klingmuller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood. 2004 Aug 1;104(3):659–66. doi: 10.1182/blood-2003-05-1442. [DOI] [PubMed] [Google Scholar]
- 85.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000 Jul 15;96(2):719–26. [PubMed] [Google Scholar]
- 86.Hamada M, Moriguchi T, Yokomizo T, Morito N, Zhang C, Takahashi S. The mouse mafB 5'-upstream fragment directs gene expression in myelomonocytic cells, differentiated macrophages and the ventral spinal cord in transgenic mice. J Biochem. 2003 Aug;134(2):203–10. doi: 10.1093/jb/mvg130. [DOI] [PubMed] [Google Scholar]
- 87.Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003 Feb 1;101(3):1155–63. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 88.Lohmann F, Bieker JJ. Activation of Eklf expression during hematopoiesis by Gata2 and Smad5 prior to erythroid commitment. Development. 2008 Jun;135(12):2071–82. doi: 10.1242/dev.018200. [DOI] [PubMed] [Google Scholar]
- 89.Papadopoulos P, Gutierrez L, van der Linden R, Kong ASJ, Maas A, Drabek D, et al. A dual reporter mouse model of the human beta-globin locus: applications and limitations. PLoS One. 2012;7(12):e51272. doi: 10.1371/journal.pone.0051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005 May;22(5):643–54. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 91.Norris HH, Martin AJ, Lybarger LP, Andersen H, Chervenak DC, Chervenak R. TCRbeta enhancer activation in early and late lymphoid progenitors. Cell Immunol. 2007 Jun;247(2):59–71. doi: 10.1016/j.cellimm.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 92.Singbartl K, Thatte J, Smith ML, Wethmar K, Day K, Ley K. A CD2-green fluorescence protein-transgenic mouse reveals very late antigen-4-dependent CD8+ lymphocyte rolling in inflamed venules. J Immunol. 2001 Jun 15;166(12):7520–6. doi: 10.4049/jimmunol.166.12.7520. [DOI] [PubMed] [Google Scholar]
- 93.Monroe RJ, Seidl KJ, Gaertner F, Han S, Chen F, Sekiguchi J, et al. RAG2:GFP knockin mice reveal novel aspects of RAG2 expression in primary and peripheral lymphoid tissues. Immunity. 1999 Aug;11(2):201–12. doi: 10.1016/s1074-7613(00)80095-3. [DOI] [PubMed] [Google Scholar]
- 94.Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med. 2005 Oct 3;202(7):901–6. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med. 2008 Jun 9;205(6):1381–93. doi: 10.1084/jem.20080034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fuxa M, Busslinger M. Reporter gene insertions reveal a strictly B lymphoid-specific expression pattern of Pax5 in support of its B cell identity function. J Immunol. 2007 Jun 15;178(12):8222–8. doi: 10.4049/jimmunol.178.12.8221-a. [DOI] [PubMed] [Google Scholar]
- 97.Kuwata N, Igarashi H, Ohmura T, Aizawa S, Sakaguchi N. Cutting edge: absence of expression of RAG1 in peritoneal B-1 cells detected by knocking into RAG1 locus with green fluorescent protein gene. J Immunol. 1999 Dec 15;163(12):6355–9. [PubMed] [Google Scholar]
- 98.Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004 Oct 18;200(8):967–77. doi: 10.1084/jem.20040973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000 Jun;20(11):4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang J, Hills D, Taylor E, Pfeffer K, Ure J, Medvinsky A. Transgenic tools for analysis of the haematopoietic system: knock-in CD45 reporter and deletor mice. J Immunol Methods. 2008 Sep 15;337(2):81–7. doi: 10.1016/j.jim.2008.06.001. [DOI] [PubMed] [Google Scholar]