

Abstract
Objectives
Partial external bile diversion (PEBD) is an established therapy for low-GGT Progressive Familial Intrahepatic Cholestasis (PFIC). This study sought to determine if the dynamics of the cholic acid (CA) and chenodeoxycholic acid (CDCA) pools in low-GGT-PFIC subjects with successful PEBD were equivalent to those achieved with successful liver transplantation (LTX).
Methods
The kinetics of CA and CDCA metabolism were measured by stable isotope dilution in plasma samples in 5 PEBD subjects all with intact canalicular BSEP expression and compared to low-GGT-PFIC subjects with successful LTX. Stomal loss of bile acids was measured in PEBD subjects.
Results
The fractional turnover rate for CA in the PEBD group ranged from 0.5 to 4.2 d−1 (LTX group, range 0.2 – 0.9 d−1, p = 0.076) and for CDCA from 0.7 to 4.5 d−1 (LTX group 0.3 – 0.4 d−1, p = 0.009). The CA and CDCA pool sizes were equivalent between groups; however pool composition in PEBD was somewhat more hydrophilic. The CA/CDCA ratio in PEBD ranged from 0.9 to 19.5, whereas in LTX it ranged from 0.5 to 2.6. Synthesis rates computed from isotope dilution correlated well with timed output for both CA: r2 = 0.760, p = 0.024 and CDCA: r2 = 0.690, p = 0.021.
Conclusions
PEBD results in bile acid fractional turnover rates greater than LTX, pool sizes equivalent to LTX and pool composition that is at least as hydrophilic as produced by LTX.
Keywords: familial cholestasis, surgical treatment of familial cholestasis, bile acid kinetics, liver transplantation
Progressive Familial Intrahepatic Cholestasis (PFIC) comprises autosomal recessive disorders resulting from mutations that impair bile formation. PFIC type 1 (PFIC1) results from mutations in ATP8B1 encoding familial intrahepatic cholestasis 1 (FIC1) (1, 2). PFIC type 2 (PFIC2) results from mutations in ABCB11 encoding bile salt export pump (BSEP) (3, 4). PFIC1 and PFIC2 are classified as low-gamma-glutamyl transferase (GGT) PFIC because serum GGT levels remain normal or only slightly elevated despite persistent cholestasis. In PFIC2 cholestasis results from reduced functional expression of BSEP along the canaliculus (5). How defects in FIC1 expression cause cholestasis is not known. Proposed mechanisms include altered canalicular membrane composition and reduced farnesoid-X receptor signaling leading to reduced BSEP expression (6–10). Low serum GGT activity in ATP8B1 and ABCB11 disease is ascribed respectively to lack of canalicular GGT expression and to lack of elution of GGT from canalicular membranes in the absence of detergent intracanalicular bile acids. It is a defining clinical feature of low-GGT PFIC, a term understood to comprise chronic cholestatic liver disease due to known or presumed gene mutations leading to impairment of bile formation.
Partial external biliary diversion (PEBD) surgery was first reported to improve cholestatic liver disease in low-GGT PFIC well before the genes involved were discovered (11). It is an empiric therapy that works without our knowing how. Despite this gap in knowledge, PEBD has gained popularity and is considered to be first-line therapy for low-GGT PFIC worldwide. PEBD lessens clinical symptoms including pruritus, reduces serum bile acid levels, improves the plasma lipoprotein profile, slows disease progression, and improves liver histopathology in low-GGT PFIC (12–19). If PEBD fails to resolve cholestasis and to halt progression of liver disease, liver transplantation (LTX) is the only recourse.
PEBD performed in a healthy person would substantially reduce the fraction of excreted bile acids subject to enterohepatic recirculation and increase bile acid synthesis to keep up with loss. At steady state such a person with PEBD is expected to show increased bile acid turnover rate and potentially reduced pool size. One hypothesis as to how PEBD improves liver function in low-GGT PFIC is that reduced delivery of bile acids to the intestine (and therefore the recirculation of bile acids to the liver) reduces the demand placed on genetically impaired mechanisms for bile formation. The conundrum is: if incapacity for transport of bile acids is the basis of cholestasis, how could diversion of bile containing little or no bile acids affect such demand? One possible answer is that PEBD improves bile acid transport into the canaliculus. The composition of bile has been assayed in a few low-GGT PFIC patients before and after PEBD (16). The limited data suggest that after PEBD bile contains more bile acids and relatively more hydrophilic bile acids than before diversion. The latter effect may be important because these patients seem to eliminate trihydroxy bile acids better than dihydroxy bile acids (20). These data imply that PEBD improves impaired canalicular bile acid transport mechanisms in low-GGT PFIC patients, but this has never been directly assessed.
Assessment of canalicular bile acid transport is difficult in human subjects because of lack of access to bile. However, bile is readily accessed in patients with PEBD. Analysis of bile composition linked to analysis of kinetics of primary bile acid metabolism, as determined by stable isotope dilution of the primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA), offers a view of canalicular bile acid transport. We hypothesized that successful PEBD results in bile acid pool kinetics that are equivalent to those achieved by successful LTX.
Patients and Methods
Subjects
Low-GGT PFIC subjects were identified and recruited for the study from within our own clinic population and from centers participating in the Childhood Liver Disease Research Network (ChiLDReN). We included subjects 18 months through 25 years of age with the diagnosis of low-GGT PFIC based on gene mutation analysis and/or clinical findings. All subjects had severe clinical cholestasis before PEBD was performed. Mutation analysis for ATP8B1 and ABCB11 permitted classification of most subjects as PFIC1 or PFIC2 (see Table 1). Two PEBD subjects without identifiable mutations were classified clinically as low-GGT PFIC with unknown gene mutation (PFIC-U). Liver biopsy specimens obtained from these two subjects before PEBD were assessed by immunohistochemistry for BSEP (5). Subjects with longstanding (greater than 1 year) successful PEBD were enrolled into the study group (PEBD group). All PEBD subjects had received ursodeoxycholic acid therapy at some time after PEBD, and two were receiving it at the time of recruitment. Therapy was discontinued 1 month prior to study. Subjects with a successful LTX were enrolled into the comparison group (LTX group). All were at least 2 years post-LTX at the time of study. All LTX subjects had failed PEBD therapy, leading to the necessity for LTX. The PEBD was taken down at the time of transplant or in preparation for transplant in 2 subjects who experienced excessive fluid and electrolyte loss with PEBD. The short segment of jejunum used for bile diversion was discarded in all cases. These subjects were considered to be the best bile acid kinetics comparators because they have genetic low-GGT PFIC without hepatic manifestations. Subjects in both groups were required to have no clinical evidence of cholestasis in the year preceding the study.
Table 1.
Characteristics of study subjects
| Subject | Gender | Age (yrs) | Diagnosis | Gene: Nucleotide change Predicted effect on the protein | Immunohisto-chemistry | Serum chemistry | |
|---|---|---|---|---|---|---|---|
| Bilirubin (mg/dL) | Bile acid (uM) | ||||||
| PEBD subjects
| |||||||
| 1 | F | 8 | PFIC1 | ATP8B1: c.923G>T; c.1220G>A; 1982T>C p.G308V; p.S407N; p.I661T^ | BSEP + | 0.4 | 0.8 |
| 2 | F | 19 | PFIC1 | ATP8B1: c.1660G>A (homozygous) p.D554N | BSEP + | 0.6 | 8.8 |
| 3 | F | 1.8 | PFIC1 | ATP8B1: c.923G>T (homozygous) p.G308V | BSEP + | 0.6 | 8.0 |
| 4 | F | 11 | PFIC-U | No mutation identified | BSEP + | 0.8 | 36 |
| 5 | M | 3 | PFIC-U | No mutation identified | BSEP + | 0.7 | 4.5 |
|
| |||||||
| LTX subjects
| |||||||
| 6 | F | 21 | PFIC1 | ATP8B1: c.1982T>C; c.2854C>T, p.I661T; p.R952* | BSEP + | 0.7 | 14 |
| 7 | M | 10 | PFIC2 | ABCB11: c.611+1G>A; c.890A>G; splicing defect; p.E297G | BSEP − | 0.7 | 9.5 |
| 8 | M | 13 | PFIC2 | ABCB11: c.611+1G>A; c.890A>G; splicing defect; p.E297G | BSEP − | 0.5 | 11.5 |
| 9 | F | 7 | PFIC2 | ABCB11: c.2296G>A (homozygous) p.G766R | BSEP − | 0.4 | 11.2 |
| 10 | M | 12 | PFIC2 | ABCB11: c.1460G>C (homozygous) p.R487P | BSEP − | 1.4 | 6.4 |
Except where homozygosity is indicated, mutations are in heterozygous form.
Three heterozygous missense changes detected
The Lurie Children’s Hospital of Chicago Institutional Review Board approved the study protocol. Written informed consent was obtained from parents/legal guardians of minors and from subjects over 18 years of age.
Study Protocol
Baseline serum bilirubin and bile acid concentrations were determined within 2 weeks of study initiation date (Table 1). Studies were performed over a 72-hour period in the Children’s Memorial Hospital outpatient clinical research unit. After an overnight fast a baseline blood sample (time 0) was collected. Then the subjects drank or received by nasogastric tube 50 mg [2,2,4,4-2H]-CA and 50 mg [2,2,4,4-2H]-CDCA dissolved in 80 mL 0.25% NaHCO3. Stable isotopes with 98% isotopic purity were obtained from Isotech (Miamisburg, OH). Blood samples were taken 10, 24, 32, 48, and 72 hours after administration of the bile acid solution. Plasma, separated immediately, was stored at −20 °C until analysis. All PEBD subject bile was decanted from the stomal appliance at the time of blood draw. The volume was recorded and 0.5 ml aliquots were frozen at −20 C for future analysis.
Gas-Liquid Chromatography Electron Capture Negative Chemical Ionization Mass Spectrometry
Duplicate samples of plasma and bile from each collection were subjected to capillary gas-liquid chromatography (GLC) and mass spectrometry (MS) using an Agilent 7890A GC connected to a Agilent 5975C MS detector (Agilent, Amstelveen, Holland). GLC separation was performed on a 10 m × 0.100 mm column, with a film thickness of 0.1 μm (DB-5MS; Agilent). After preparation of pentafluorobezyl/trimethylsilyl conjugates, duplicate 1- to 3-μl samples were injected in the pulsed split less mode at an injector temperature of 320°C and a column temperature of 150°C. The column temperature was programmed to remain at 150°C for 0.5 min, to rise to 320°C at a rate of 60°C/min, and then to remain at 320°C for 4 min. The interface temperature was 320°C. The ion source was operated in the negative ion chemical ionization mode at 200°C. The quadrupole was set at 150°C, applying methane as the moderating gas at a source pressure of 1,600 mTorr. Isotope ratios were determined in the selected ion-monitoring mode on m/z 623.4 (M0) and 627.3 (M4) for CA and on m/z 535.4 (M0) and 539.4 (M4) for CDCA.
Optimal GLC-MS conditions were assessed by examining the effects of MS and selected ion monitoring parameters. Settings were adjusted for each set of samples to obtain at least 20 data points on the GC peak and a peak intensity for the M0 cholate peak at m/z 623.3 between 1 × 104 and 1 × 106 instrument units. Within this range stable isotope ratio values of M1/M0, M2/M0, M3/M0, and M4/M0 were obtained. Injection volume and electron multiplier voltage were adjusted to obtain base peak intensities at the defined intensity plateau, assuring accurate and reproducible M4/M0 isotope ratios. Relative standard deviations for baseline abundance are for CA 1.9% and for CDCA 3.8%.
Calculation of bile acid kinetics
The area ratio M4/M0 was calculated after computerized integration of peak areas of M4 and M0 for CA and CDCA in the mass chromatograms using Masshunter software (Agilent). Enrichment was defined as the increase of M4/M0 after administration of [2H4]CA and [2H4]CDCA and expressed as the natural logarithm of the atom% excess (In APE) value. The decay of ln APE over time was described by linear regression analysis. From this linear decay curve the fractional turnover rate (FTR) and pool size of CA and CDCA were calculated (21–23). The FTR (per day) equals the slope of the regression line. The pool size (milligrams) is determined according to the formula: Pool size = ([D × b × 100]/ea) − D, where D is the administered amount of label (mg), b is the isotopic purity, and a is the intercept on the y-axis of the ln APE-versus-time curve. The CA and CDCA synthesis rates (mg/day) are determined by multiplying pool size and FTR.
Bile composition
The concentrations of bile acid species were measured in duplicate in 10uL bile samples using GLC-MS as described above. Concentrations of CA, CDCA, and others were recorded. The amount of CA and CDCA diverted over the study period was computed as the weighted average (amount per time period) and expressed as mg/day.
Statistics
Because they were not normally distributed, summary data are presented as range and median values and in the figure as box and whisker plots. Differences in kinetic parameters between PEBD and LTX groups were determined by Wilcoxon rank-sum test. Linear correlation analysis was used to determine the closeness of the relationship between bile acid synthesis rates as determined by isotope dilution and the loss of bile acids diverted in PEBD subjects.
Results
Table 1 provides the characteristics of the study subjects. Within the PEBD group, 3 subjects had the genetic diagnosis of PFIC1, and 2 had PFIC-U. The 2 subjects with PFIC-U expressed immunohistochemically demonstrable BSEP, evidence against ABCB11 mutations that abrogated BSEP expression (5). Within the LTX group, 1 subject had the genetic diagnosis of PFIC1 and 4 had that of PFIC2. No subject in either group had serum bilirubin or total serum bile acid concentrations indicative of cholestasis at the time of study (Table 1).
Figure 1 shows representative studies of PEBD and LTX subjects. The FTRs, pool sizes, and synthesis rates for CA and CDCA are calculated from similar plots obtained from the individual subjects. In some PEBD subjects the rapid turnover resulted in very low APE values below the limit of precise measurement with subsequent loss of linearity of the ln(APE) plot beyond 48 hours of study, as shown in Figure 1. In these cases, kinetics parameters were computed from the linear portion of the decay only. Linearity of the decay of ln(APE) was maintained in LTX subjects for the full 72 hours. The calculated kinetics parameters for PEBD and LTX subjects are provided in Table 2. Summary data are provided in Figure 2. The CA FTR in the PEBD group ranged from 0.5 – 4.2 d−1 (median 1.5) as compared to the LTX group, range 0.2 – 0.9 d−1 (median 0.7), Wilcoxon p = 0.076 (Figure 2a). The CDCA FTR in the PEBD group ranged from 0.7 – 4.5 d−1 (median 2.1) as compared to the LTX group, range 0.3 – 0.4 d−1 (median 0.3), Wilcoxon p = 0.009 (Figure 2b). The mean FTRs for CA and CDCA in 5 healthy adult subjects from Everson’s 1987 study were 0.28 ± 0.05 d−1 and 0.17 ± 0.04 d−1, respectively (21). Accordingly, the FTRs of the LTX group, whose members had LTX-related Roux-en-Y choledochoenterostomy and thus lacked a gallbladder and sphincter of Oddi, are somewhat greater than those of normal individuals. In summary, a functioning PEBD appears to result in brisk turnover of both CA and CDCA pools, whereas LTX leaves FTRs in a relatively normal range.
Figure 1.

Representative bile acid kinetic studies of low-GGT PFIC subjects with PEBD and LTX. CA (panel A) and CDCA (panel B) kinetics for Subject 3 with PEBD and Subject 7 with LTX are shown. Measurements (In APE values) at each time point are represented as closed squares for Subject 3 and open circles for Subject 7. Linear regression analysis determined the line of decay of plasma concentration of stable isotope labeled CA and CDCA plotted as lnAPE versus time (hours). Because of the rapid decay of labeled bile acids in PEBD subjects, the regression lines were extended only to the point where reliable measurement could be made.
Table 2.
Kinetics parameters for CA and CDCA acid as determined by stable isotope dilution
| CA | CDCA | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Subject | Diagnosis | Fractional Turnover (d−1) | Pool Size (mg/m2) | Synthesis Rate (mg/d) | Fractional Turnover (d−1) | Pool Size (mg/m2) | Synthesis Rate (mg/d) |
| PEBD subjects | |||||||
|
| |||||||
| 1 | PFIC1 | 0.93 | 1312 | 923 | 2.09 | 565 | 892 |
| 2 | PFIC1 | 4.25 | 365 | 2401 | 2.65 | 418 | 1717 |
| 3 | PFIC1 | 1.86 | 821 | 1081 | 1.9 | 119 | 160 |
| 4 | PFIC-U | 0.49 | 447 | 237 | 0.71 | 48 | 37 |
| 5 | PFIC-U | 1.50 | 1498 | 1463 | 4.48 | 77 | 224 |
|
| |||||||
| LTX subjects | |||||||
|
| |||||||
| 6 | PFIC1 | 0.81 | 428 | 604 | 0.44 | 162 | 124 |
| 7 | PFIC2 | 0.20 | 800 | 183 | 0.26 | 504 | 150 |
| 8 | PFIC2 | 0.38 | 522 | 337 | 0.34 | 402 | 233 |
| 9 | PFIC2 | 0.74 | 282 | 171 | 0.41 | 550 | 185 |
| 10 | PFIC2 | 0.95 | 739 | 1071 | 0.29 | 780 | 345 |
Figure 2.
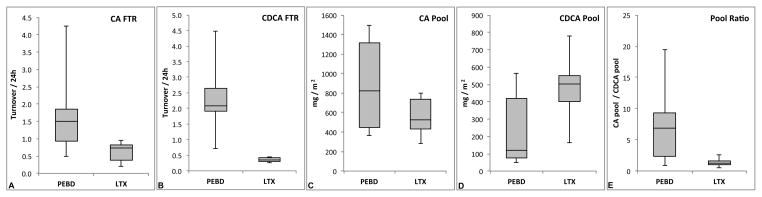
Summary data from kinetics studies are presented as box and whisker plots. A. CA FTR for PEBD and LTX groups. B. CDCA FTR. C. CA pool size. D. CDCA pool size. E. Ratio of CA/CDCA pool sizes.
In this type of analysis the pool size is computed using the y-intercept values extrapolated from the linear regression of the ln(APE). As shown in Table 2, the CA pool size corrected for body surface area for the 5 PEBD subjects ranged from 365 – 1312 mg/m2 (median 821) and the CDCA pool size from 48 – 565 mg/m2 (median 119). In the LTX subjects, the CA pool size ranged from 282 – 800 mg/m2 (median 522) and the CDCA pool size ranged from 162 – 780 mg/m2 (median 504). As shown in Figures 2c and 2d, CA and CDCA pool sizes were similar in PEBD and LTX subjects (CA p = 0.251, CDCA p = 0.175) despite the large ongoing loss of bile acids in the PEBD subjects. Figure 2 shows the ratio of CA to CDCA pool sizes for PEBD and LTX subjects. When pool sizes are measured in normal persons by the same isotope dilution method employed in this study, the CA/CDCA ratio is approximately 1.3 (21). In PFIC patients with cholestasis, the ratio is estimated from serum bile acid concentrations to be approximately 0.1 (20), meaning there is 10 times more CDCA than CA in the bile acid pool. The CA/CDCA ratio in the 5 PEBD subjects ranged from 0.9 – 19.5 (median 6.9), whereas in the 5 LTX subjects it ranged from 0.5 – 2.6 (median 1.3), p = 0.117. These data suggest that PEBD results in a change in the bile acid pool composition to one that is at least as hydrophilic as produced by LTX and far more hydrophilic than in PFIC patients without treatment.
The stomal bile outputs of CA and CDCA in PEBD subjects over the 72-hour study period are provided in Table 3. Daily stomal bile output in these subjects varied greatly, from 114 to 743 ml. The baseline concentration of CA in bile varied from 4.0 to 15.4mM and CDCA from 0.6 to 8.9mM. In every case bile contained more CA than CDCA. Bile from only one subject (#4, PFIC-U) contained measurable amounts of deoxycholic acid (approximately 1% of bile acid output). It is reasonable to conclude that the output of CA and CDCA contributes a large proportion of daily bile acid loss and thus should account for the bulk of bile acid synthesis at steady state. We compared the loss in bile to the synthesis rates computed from the isotope dilution studies to assess these conclusions and the accuracy of the computed values (Table 4). While both synthesis rates and timed outputs of CA and CDCA varied greatly among PEBD subjects, synthesis rates correlated well with timed output for both CA: r2 = 0.760, p = 0.024 and CDCA: r2 = 0.690, p = 0.021. These results validate measurement of bile acid synthesis rates computed from isotope dilution in subjects with large ongoing bile acid losses.
Table 3.
Total bile output and baseline concentrations and timed output of CA and CDCA in bile of PEBD subjects measured over the 72-hour study period
| CA | CDCA | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Subject | Diagnosis | Bile Output (ml/d) | Baseline Concentration (mM) | Timed Output (mg/d) | Baseline Concentration (mM) | Timed Output (mg/d) | |
| 1 | PFIC1 | 242.6 | 12.99 | 1073.9 | 8.09 | 561.1 | |
| 2 | PFIC1 | 742.8 | 12.28 | 3013.2 | 4.45 | 904.2 | |
| 3 | PFIC1 | 273.3 | 4.02 | 384.1 | 0.63 | 52.3 | |
| 4 | PFIC-U | 136.7 | 5.30 | 152.4 | 1.05 | 42.3 | |
| 5 | PFIC-U | 114.0 | 15.45 | 550.1 | 8.91 | 238.2 | |
Table 4.
Synthesis rates and timed output of cholic acid and chenodeoxycholic acid in bile of PEBD subjects measured over the 72-hour study period
| Cholic acid | Chenodeoxycholic acid | ||||
|---|---|---|---|---|---|
|
| |||||
| Subject | Diagnosis | Synthesis Rate (mg/d) | Timed Output (mg/d) | Synthesis Rate (mg/d) | Timed Output (mg/d) |
| 1 | PFIC1 | 923 | 1073 | 892 | 561 |
| 2 | PFIC1 | 2401 | 3013 | 1717 | 904 |
| 3 | PFIC1 | 1081 | 384 | 160 | 52 |
| 4 | PFIC-U | 237 | 152 | 37 | 42 |
| 5 | PFIC-U | 1463 | 550 | 224 | 238 |
Discussion
PEBD improves cholestasis and maintains non-cholestatic liver function including normal serum bile acid concentrations in patients with low-GGT PFIC (11–19). Cholestasis in low-GGT PFIC is thought to result directly or indirectly from genetically determined defects in canalicular bile acid transport (24). It can be assumed therefore that successful PEBD overcomes these defects or their effects on liver function to a degree sufficient to relieve cholestasis. The mechanism by which it does so is unknown. In this study the whole body kinetics of CA and CDCA metabolism in low-GGT PFIC subjects with successful PEBD were compared with those achieved by LTX. The results show that LTX subjects had kinetics parameters that were fairly homogenous and near normal as expected. The median ratio of CA to CDCA in the bile acid pool was 1.3, which is the same as that determined in normal individuals using this technique (21). The kinetics parameters of PEBD subjects showed considerably more variability than those of LTX subjects. In general the CA and CDCA pools were similar in size to those in LTX subjects and their turnover rates were similarly brisk. These findings suggest that the PEBD subjects had capacity for transport of CA and CDCA similar to that achieved by LTX.
The few available data suggest selective retention of dihydroxy bile acids in low-GGT PFIC during cholestasis, producing a highly hydrophobic bile acid pool (20). Serum bile acid levels are markedly elevated in cholestatic low-GGT PFIC patients, averaging around 300μM (normal < 20), with very low CA/CDCA ratios (usually 1:8 – 1:10) (20). Conversely, the concentrations of bile acids in bile of low-GGT PFIC patients are typically in the μM range and are often less than serum concentrations, and CA is the overwhelmingly dominant species in contrast to serum bile acids (16, 20, 25, 26). It should be noted that quantitative analysis of bile composition has not been reported in numbers of genetically proven PFIC1 and PFIC2 patients sufficient to establish whether proportions of components differ substantially between these disorders or from those in low-GGT PFIC patients without identifiable gene mutations (PFIC-U subjects in this study). Despite this limitation, existing data suggest a defect in bile acid export in low-GGT PFIC, in which CDCA is selectively retained (20). Assuming that in cholestatic low-GGT PFIC patients most or all bile acids lie evenly distributed in extracellular fluid (220 ml/kg) outside normal enterohepatic recirculation, the typical 20kg PFIC patient would have a bile acid pool of about 1.3 mmole. If of those bile acids 10% are CA and 90% CDCA (20), the respective bile acid pool sizes are 53mg and 458mg. Comparison with the pool sizes measured in the PEBD subjects demonstrates that PEBD does not deplete body bile acid pools as might be expected. Instead it appears to increase their sizes, with relatively greater increases in the CA pool yielding a generally hydrophilic bile pool.
We postulate that the main effect of PEBD is to cause a shift in bile acid pool composition to a relatively more hydrophilic balance that causes less hepatocyte injury and thereby relieves cholestasis. Logically, the success of PEBD is determined by the capacity of the liver to transport bile acids, particularly hydrophilic ones, and conversely failure of PEBD is likely determined by incapacity for bile acid transport. PEBD therapy is far more likely to fail in patients who have cirrhosis than in those with lesser disease, likely because of failure of excretory function. Another major determinant of success or failure of PEBD is the mutated gene involved and the severity of the mutation. It is generally accepted that PEBD is less likely to be therapeutically effective in PFIC2 than PFIC1. Genotype-phenotype studies will be useful to further clarify this relationship, but one can logically expect very poor response in individuals with severe ABCB11 mutations because of lack of BSEP expression. Since the PEBD group comprised only PFIC1 and PFIC-U subjects whose liver expressed canalicular BSEP, we can only speculate about the action of PEBD in genetic cholestasis with functional BSEP. In PFIC1 a relatively hydrophilic bile acid pool would reduce exposure to hydrophobic bile acids and therefore limit the detergent effect on the canalicular membrane. Atp8b1G308V/G308V mutant mice, which carry the Byler FIC1 mutation, do not develop cholestasis until exposed to exogenous bile acids (27). In these mice introducing the Byler FIC1 mutation reduces asymmetry of the canalicular lipid membrane, decreases expression of canalicular proteins including BSEP, and renders the canalicular membrane more susceptible to hydrophobic bile acid induced injury (10). In addition, the Byler FIC1 mutation may reduce expression and function of the farnesoid-X receptor (8). Dihdroxy bile acids are the most potent of FXR agonists (28). Their binding results in changes in expression of FXR gene targets, including increased BSEP expression. Reduced FXR function owing to FIC1 deficiency might specifically limit canalicular BSEP expression and compound the error in canalicular bile acid transport introduced by FIC1 mutations (7).
A requirement for using isotope the isotope dilution technique to study bile acid kinetics is that the subject’s bile acid pool is in a steady state, or not in flux (29). This steady state does not concern the FTRs relative to normal or comparison subjects, rather whether the rate of disposal is equaled by the rate of synthesis; in other words, the condition in which the pool size itself is stable during the conditions of testing. We have several indications that the PEBD subjects were in steady state during the course of the study despite the relatively high FTRs of their bile acid pools. First, we studied subjects with very stable clinical status, who were required to have had no clinical cholestasis for at least one year prior to study. They all save one had normal levels of serum bile acids and no other evidence of cholestasis at the time of study. Second, neither the plasma bile acid concentrations nor, in the PEBD subjects, the amount of stomal bile acid output varied significantly over the course of the study. Third, the semi-log plots of the decrease of atom-percent excess over time were linear for all individual subjects, as illustrated in Figures 1A and 1B. In a non-steady state condition, this would not be the case: rather, change in FTR would be reflected by deflection of the line and change in the Y-intercept (reflecting the pool size) calculated from the first time points to that computed from later time points. This was not the case in any PEBD or LTX subject, indicating a steady state condition. We should also point out that a condition of being out of steady state, where loss exceeds synthesis or vice versa, cannot persist for long. Indeed, the nature of all biological systems is to quickly find a new steady state after any perturbation. It is quite clear from our data that individuals’ systems find different steady state conditions after PEBD, which resulted in highly variable pool kinetics among the PEBD subjects.
The isotope dilution technique has never before been used to study bile acid kinetics in human subjects with a high rate of loss of bile acids from the pool, or in other words in subjects with high FTRs. The PEBD subjects presented the opportunity to validate the technique because bile acid output could be directly measured. The stomal bile acid losses were precisely measured in each PEBD subject and are taken to be very close estimates of the contribution of PEBD to bile acid turnover. The rate of synthesis would be this amount plus any bile acid loss by another route (i.e. in stool) if the subject was in steady state, as the PEBD subjects were. The calculated synthesis rates correlated with the measured losses, but imperfectly, some higher and some lower than predicted. Steep slopes of FTR regression lines lead to higher y-intercepts and to relative imprecision in the computation of the estimate of pool size. This is what leads to the variances of calculated synthesis rates from measured losses. However, the calculated synthesis rates correlated well with the observed PEBD bile acid outputs in all subjects, which validates the approach. Our findings suggest that the isotope dilution technique could be used to measure bile acid pool kinetics in individuals with large ongoing losses of bile acids, such a persons with ileal bypass or those being administered bile acid bind resins or inhibitors of ileal bile acid transporters.
We studied subjects with bile acid kinetics at steady state, which leaves questions about how that steady state was achieved. Given that the bile in low-GGT PFIC patients undergoing PEBD would contain predominantly CA conjugates, albeit at very low concentrations, we speculate that the initial effect of PEBD is to increase CA FTR. Increasing the FTR of CA would result in increasing its synthesis relative to that of CDCA, and because CA can be excreted, a cycle begins where increasing amounts of CA are lost and synthesized. Meanwhile, CDCA synthesis remains low because it is differentially retained in the hepatocyte. This would ultimately result in a CA rich pool. The pressure to excrete CDCA would be relieved and an increasingly hydrophilic bile acid pool would permit improvement in membrane structure and function, with the effect of relieving cholestasis. This is a speculative but plausible explanation for the effect of PEBD in low-GGT PFIC, at least in individuals with canalicular BSEP expression.
In conclusion, successful PEBD in low-GGT PFIC subjects was associated with bile acid kinetic parameters equivalent to those achieved with successful LTX. Comparing computed values to actual bile acid losses validated the use of stable isotope dilution methodology to assess bile acid pool kinetics in subjects with large ongoing losses.
Acknowledgments
Financial support: NIH grants DK062436, DK062453, UL1RR025741
Contributor Information
Hilary Smith Jericho, Department of Pediatrics, Comer Children’s Hospital, University of Chicago, Chicago, IL.
Elizabeth Kaurs, Department of Pediatrics, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL.
Renze Boverhof, Department of Pediatric Gastroenterology, University of Groningen, Beatrix Children’s Hospital-University Medical Center, Groningen, The Netherlands.
Alex Knisely, Institute of Liver Studies, King’s College Hospital, London, UK.
Benjamin L Shneider, Department of Pediatrics, Children’s Hospital of Pittsburgh, Pittsburgh, PA.
Henkjan J Verkade, Department of Pediatric Gastroenterology, University of Groningen, Beatrix Children’s Hospital-University Medical Center, Groningen, The Netherlands.
Peter F Whitington, Department of Pediatrics, Ann & Robert H. Lurie Children’s Hospital of Chicago, Feinberg School of Medicine of Northwestern University, Chicago, IL.
References
- 1.Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–24. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 2.Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27–38. doi: 10.1002/hep.20285. [DOI] [PubMed] [Google Scholar]
- 3.Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–8. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 4.Strautnieks SS, Byrne JA, Pawlikowska L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–14. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 5.Noe J, Kullak-Ublick GA, Jochum W, et al. Impaired expression and function of the bile salt export pump due to three novel ABCB11 mutations in intrahepatic cholestasis. J Hepatol. 2005;43:536–43. doi: 10.1016/j.jhep.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 6.Chen F, Ananthanarayanan M, Emre S, et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology. 2004;126:756–64. doi: 10.1053/j.gastro.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez L, Jara P, Sanchez-Sabate E, et al. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13:2451–60. doi: 10.1093/hmg/ddh261. [DOI] [PubMed] [Google Scholar]
- 8.Chen F, Ellis E, Strom SC, et al. ATPase Class I Type 8B Member 1 and protein kinase C zeta induce the expression of the canalicular bile salt export pump in human hepatocytes. Pediatr Res. 2010;67:183–7. doi: 10.1203/PDR.0b013e3181c2df16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frankenberg T, Miloh T, Chen FY, et al. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology. 2008;48:1896–905. doi: 10.1002/hep.22431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulusma CC, Groen A, Kunne C, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44:195–204. doi: 10.1002/hep.21212. [DOI] [PubMed] [Google Scholar]
- 11.Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology. 1988;95:130–6. doi: 10.1016/0016-5085(88)90301-0. [DOI] [PubMed] [Google Scholar]
- 12.Arnell H, Papadogiannakis N, Zemack H, et al. Follow-up in children with progressive familial intrahepatic cholestasis after partial external biliary diversion. J Pediatr Gastroenterol Nutr. 2010;51:494–9. doi: 10.1097/MPG.0b013e3181df99d5. [DOI] [PubMed] [Google Scholar]
- 13.Emond JC, Whitington PF. Selective surgical management of progressive familial intrahepatic cholestasis (Byler’s disease) J Pediatr Surg. 1995;30:1635–41. doi: 10.1016/0022-3468(95)90440-9. [DOI] [PubMed] [Google Scholar]
- 14.Halaweish I, Chwals WJ. Long-term outcome after partial external biliary diversion for progressive familial intrahepatic cholestasis. J Pediatr Surg. 2010;45:934–7. doi: 10.1016/j.jpedsurg.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Kalicinski PJ, Ismail H, Jankowska I, et al. Surgical treatment of progressive familial intrahepatic cholestasis: comparison of partial external biliary diversion and ileal bypass. Eur J Pediatr Surg. 2003;13:307–11. doi: 10.1055/s-2003-43570. [DOI] [PubMed] [Google Scholar]
- 16.Kurbegov AC, Setchell KD, Haas JE, et al. Biliary diversion for progressive familial intrahepatic cholestasis: improved liver morphology and bile acid profile. Gastroenterology. 2003;125:1227–34. doi: 10.1016/s0016-5085(03)01199-5. [DOI] [PubMed] [Google Scholar]
- 17.Melter M, Rodeck B, Kardorff R, et al. Progressive familial intrahepatic cholestasis: partial biliary diversion normalizes serum lipids and improves growth in noncirrhotic patients. Am J Gastroenterol. 2000;95:3522–8. doi: 10.1111/j.1572-0241.2000.03370.x. [DOI] [PubMed] [Google Scholar]
- 18.Schukfeh N, Metzelder ML, Petersen C, et al. Normalization of serum bile acids after partial external biliary diversion indicates an excellent long-term outcome in children with progressive familial intrahepatic cholestasis. J Pediatr Surg. 2012;47:501–5. doi: 10.1016/j.jpedsurg.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Porte RJ, Verkade HJ, et al. Partial external biliary diversion in children with progressive familial intrahepatic cholestasis and Alagille disease. J Pediatr Gastroenterol Nutr. 2009;49:216–21. doi: 10.1097/MPG.0b013e31819a4e3d. [DOI] [PubMed] [Google Scholar]
- 20.Jacquemin E, Dumont M, Bernard O, et al. Evidence for defective primary bile acid secretion in children with progressive familial intrahepatic cholestasis (Byler disease) Eur J Pediatr. 1994;153:424–8. doi: 10.1007/BF01983406. [DOI] [PubMed] [Google Scholar]
- 21.Everson GT. Steady-state kinetics of serum bile acids in healthy human subjects: single and dual isotope techniques using stable isotopes and mass spectrometry. J Lipid Res. 1987;28:238–52. [PubMed] [Google Scholar]
- 22.Hulzebos CV, Renfurm L, Bandsma RH, et al. Measurement of parameters of cholic acid kinetics in plasma using a microscale stable isotope dilution technique: application to rodents and humans. J Lipid Res. 2001;42:1923–9. [PubMed] [Google Scholar]
- 23.Stellaard F, Bloks VW, Burgerhof HG, et al. Two time-point assessment of bile acid kinetics in humans using stable isotopes. Isotopes Environ Health Stud. 2011;46:325–36. doi: 10.1080/10256016.2010.503894. [DOI] [PubMed] [Google Scholar]
- 24.Jacquemin E. Progressive familial intrahepatic cholestasis. Genetic basis and treatment. Clin Liver Dis. 2000;4:753–63. doi: 10.1016/s1089-3261(05)70139-2. [DOI] [PubMed] [Google Scholar]
- 25.Tazawa Y, Yamada M, Nakagawa M, et al. Bile acid profiles in siblings with progressive intrahepatic cholestasis: absence of biliary chenodeoxycholate. J Pediatr Gastroenterol Nutr. 1985;4:32–7. doi: 10.1097/00005176-198502000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Jansen PL, Strautnieks SS, Jacquemin E, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–9. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 27.Pawlikowska L, Groen A, Eppens EF, et al. A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum Mol Genet. 2004;13:881–92. doi: 10.1093/hmg/ddh100. [DOI] [PubMed] [Google Scholar]
- 28.Lew JL, Zhao A, Yu J, et al. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J Biol Chem. 2004;279:8856–61. doi: 10.1074/jbc.M306422200. [DOI] [PubMed] [Google Scholar]
- 29.Lindstedt S. The turnover of cholic acid in man: bile acids and steroids. Acta Physiol Scand. 1957;40:1–9. doi: 10.1111/j.1748-1716.1957.tb01473.x. [DOI] [PubMed] [Google Scholar]