

Abstract
Circular resequencing (CirSeq) is a novel technique for efficient and highly accurate next-generation sequencing (NGS) of RNA virus populations. The foundation of this approach is the circularization of fragmented viral RNAs, which are then redundantly encoded into tandem repeats by ‘rolling-circle’ reverse transcription. When sequenced, the redundant copies within each read are aligned to derive a consensus sequence of their initial RNA template. This process yields sequencing data with error rates far below the variant frequencies observed for RNA viruses, facilitating ultra-rare variant detection and accurate measurement of low-frequency variants. Although library preparation takes ~5 d, the high-quality data generated by CirSeq simplifies downstream data analysis, making this approach substantially more tractable for experimentalists.
INTRODUCTION
A fundamental challenge in interpreting NGS data is distinguishing true genetic variation from sequencing error. The problem is twofold: (i) average sequencing error rates for NGS are relatively high1,2, and (ii) the quantity of data generated by these technologies is so large that even very small error probabilities result in substantial numbers of sequencing errors. In addition, intrinsic error of reverse transcription, second-strand synthesis and PCR amplification during library preparation contribute another substantial pool of errors, which, when sequenced at high quality, are indistinguishable from true genetic variation. For single-genome sequencing, these errors can be corrected by using many reads to define a consensus. For populations, however, reads over the same region of the genome most often originate from different individuals, and, without knowing the individual from which each read is derived, it is not possible to remove errors using a consensus approach.
To identify individuals from within a population, several groups have developed molecular barcoding approaches3–6 in which each molecule is tagged with a unique sequence identifier before amplification. When the amplified, barcoded molecules are sequenced, reads that contain the same barcode are grouped together. Consensus sequences are then derived for groups with three or more reads. A major drawback of this approach is its low efficiency owing to uneven sampling of barcodes; the majority of barcodes are sampled either less than or many more than three times5. In addition, barcoded reads are not true independent copies of the original template molecule, as most copies are templated by earlier copies. Consequently, errors in early rounds of amplification can propagate, making them more likely to appear multiple times in a barcode group and, as a result, causing the consensus sequence to deviate from the sequence of the original template molecule. This effect is especially problematic for populations of RNA molecules that must go through a cDNA intermediate before amplification, and thus any errors introduced by reverse transcription will be present in all of the amplified copies.
To address these limitations, we have developed a method called CirSeq, which facilitates the efficient collection of highly accurate sequence data from populations7. In this method, outlined in Figure 1, RNA is fragmented and circularized to generate templates for rolling-circle reverse transcription, which yields cDNA arrays of tandemly repeated copies. Because these copies are physically linked, sequences derived from the same template are inherently grouped together, eliminating the need for barcodes. Given that the length of each circular template is at most one-third of the sequencing read length, this method also ensures that each sequencing read contains precisely enough copies to build a consensus sequence. In addition, because each copy is directly templated by the circularized RNA, consensus sequences are guaranteed to derive from true independent copies. The independence of these copies is crucial in reducing sequencing error rates, as this allows the estimated error probabilities in each copy to be directly multiplied, driving estimated error rates of consensus sequences down orders of magnitude. This marked improvement in accuracy reduces the level of sequencing error (as low as one error in 1012 bases with Illumina sequencing) far below the estimated mutation rates of most organisms, enabling not only the detection of ultra-rare genetic variants within populations but also the accurate measurement of their frequencies.
Figure 1.

Schematic of CirSeq. True genetic variants are represented as orange and green circles. Other colors represent enzymatic and sequencing errors. (Steps 1–18) Full-length viral genomic RNA is processed into short (85–100 nt) circular RNAs. No mutations are introduced during this process. (Steps 19–24) Rolling-circle reverse transcription yields tandem copies of the circular RNA template. Reverse transcriptase introduces nontemplated mutations into the tandem copies. (Steps 25–53) Tandem-copy cDNAs are cloned to generate a library of dsDNA molecules containing sequencing platform–specific adapter sequences. Additional nontemplated mutations are accumulated by enzymatic error during cloning. (Analysis) Sequenced reads are computationally processed using an algorithm that identifies and aligns tandem repeats within each sequencing read. A consensus of the aligned reads, which excludes sequencing and enzymatic errors accumulated in this process, can be used for experiment-specific analysis.
We previously demonstrated7, using populations of poliovirus, a positive-sense RNA virus, how this advancement in the ability to measure variant frequencies enables large-scale measurement of the impact of genetic variants on viral fitness. These measurements were consistent with the known genetic and biochemical properties of this virus, and they also revealed structurally contiguous regions of viral proteins that were clearly tuned by evolution but have no known functional roles, highlighting the potential of this powerful new genetic approach to guide studies of the molecular biology and evolution of viruses and their hosts. This protocol describes in detail the preparation of tandem-repeat libraries from purified viral RNA. For preparation from DNA, Lou et al.8 describe a complementary method.
Overview of CirSeq
The CirSeq protocol is shown in Figure 1. In Steps 1–18, purified viral RNA is chemically fragmented by Zn2+ to produce RNA in a low-molecular-weight range. To ensure that sequencing reads contain approximately three copies of each template, fragmented RNAs are size-selected such that they are no more than one-third of the sequencing read length. These size-selected RNAs are then 5′ phosphorylated and circularized. In Steps 19–24, the circularized RNA is reverse-transcribed using random primers. The tandem-repeat cDNAs generated by this rolling-circle reverse transcription are then cloned in Steps 25–53 to generate a library compatible with Illumina sequencing. First, the cDNAs are converted to dsDNA. These dsDNAs are blunted to remove 3′ overhangs created during second-strand synthesis, and then dA overhangs are added to improve the efficiency of adapter ligation. After adapter ligation, libraries are size-selected to remove adapter dimers and to select molecules in the appropriate size range to ensure that each sequencing read will contain at least three copies of its template. Finally, this size-selected library is amplified and size-selected once again to ensure that it is completely free of adapter dimers. Once the library is sequenced, the data must be bioinformatically processed to generate consensus sequences that fully map to the reference genome. The procedural details for bioinformatically processing the sequencing data are not provided in this protocol; however, data processing is discussed in Experimental design below.
Applications of the method
Our work has focused on detecting and measuring the frequencies of ultra-rare genetic variants in RNA virus populations. Although initially we validated CirSeq using purified poliovirus RNA7, we have also successfully applied this method to other positive- and negative-sense RNA viruses. In addition, we believe that this protocol will be well suited to the phylotyping of microbial communities using 16S rRNA sequences and to the analysis of transcriptional error and RNA editing in organisms with an available reference genome sequence.
Limitations of the method
Although CirSeq facilitates highly accurate population sequencing, this protocol may not be suitable for some viral sequencing applications, particularly sequencing of clinical isolates, owing to its demand for large quantities of purified viral RNA. For applications in which the quantity or purity of viral RNA is limiting, conventional NGS approaches may be more appropriate; however, the quality of those sequencing data will diminish the capacity to confidently identify ultra-rare variants and to quantify their changes in frequency. In addition, because processing of CirSeq data requires mapping reads to a reference genome to resolve the 3′→5′ ligation junction generated in Step 18 (see Experimental design for further discussion), CirSeq is not compatible with de novo sequencing or analysis of populations with unknown constituents.
Experimental design
Sample choice
Three major considerations in choosing an appropriate sample are: the genome length of the organism to be sequenced; the quantity of genetic material that can be obtained; and the purity of that material. First, organisms with large genomes (>0.5–1 million nt) may require an impractically large quantity of data to obtain accurate measurements of low-frequency variants (see ‘Analysis of variant frequencies’ below). For these organisms, CirSeq may be ideal for SNP discovery, but the range of variant frequencies attainable will be markedly limited. Second, because CirSeq requires fragmentation and size selection of samples, most of the input RNA is lost to improperly sized fragments. Because of this, we recommend starting with at least 1 μg of purified target RNA to ensure that enough size-selected molecules are obtained to produce a highly complex and representative library. Although lower amounts of starting material can be used, we find that low quantities of size-selected RNA are challenging to handle. Finally, the purity of the RNA sample (i.e., the proportion of the target genome to total material) can substantially affect the quantity of data that must be acquired—low purity requires more sequencing reads to adequately cover the target genome—as well as the amount of input material needed to generate a representative library.
RNA fragment length
The length of size-selected RNA after fragmentation should be no less than 85 nt and no more than one-third of the length of the sequencing read. We have found that libraries produced using RNA fragments shorter than 85 nt cause severe biases in sequencing coverage depth across viral genomes (Fig. 2), which can impede the identification of rare variants in poorly covered regions. We recommend performing sequencing with read lengths of 300 nt, and thus the maximum RNA fragment length we recommend is 100 nt when using Illumina’s MiSeq platform, which currently supports this read length. Although it is not currently supported to 300 nt, we have also had success using the HiSeq 1500/2500 in Rapid mode. Future increases in maximum read length, and thus maximum RNA fragment length, may require further optimization of RNA ligation conditions to ensure that circular ligation remains favorable.
Figure 2.
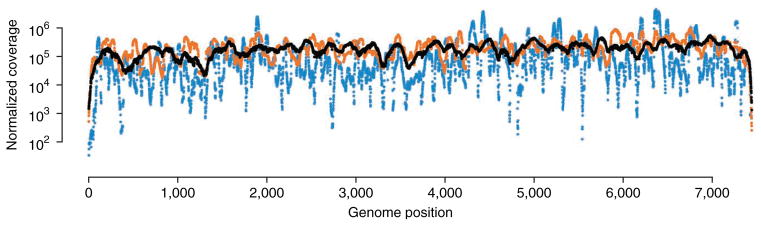
Analysis of coverage from libraries produced with different-sized RNA fragments. Blue, black and orange points denote the coverage depth at each genome position for 30 nt, 90 nt and partially degraded fragments, as shown in Figure 4b, respectively. Short (30 nt) and partially degraded RNA fragments reduce the uniformity of coverage as compared with longer (90 nt) RNA fragments. 30- and 90-nt coverage data were obtained from Acevedo et al.7.
Data processing
Before experiment-specific analysis, reads acquired using CirSeq need to be processed to generate consensus sequences that reflect the length and order of nucleotides in the initial RNA fragments. By using the CirSeq algorithm (available at http://andino.ucsf.edu/CirSeq with specific up-to-date instructions, which are not provided in this protocol) we developed previously7, we first identify the periodicity of each set of tandem repeats by determining the most common distance between identical subsequences within each read. Next, reads are broken into repeats with a length equal to the periodicity defined for that read and aligned. In a typical sequencing experiment, we are able to assemble >85% of reads into consensus sequences with repeats having at least 85% identity. Because rolling-circle reverse transcription is initiated with random primers, the start site of transcription is, in most cases, offset from the 3′→5′ RNA ligation junction, resulting in consensus sequences with blocks of sequence out of order with respect to the reference genome. To resolve these 3′→5′ junctions, we map consensus sequences directly to the reference genome using Bowtie2 (ref. 9). With this approach, the longest block of contiguous sequence in the consensus maps to the reference genome, whereas the shorter block remains unmapped. Finally, we transfer the unmapped block to the opposite end of the consensus sequence to produce a sequence that should now map in its entirety to the reference genome.
A small number of consensus sequences, particularly those with variants near the 5′ or 3′ end of the initial RNA fragment, are difficult to correctly map using the data processing procedure detailed above. These sequences typically contain multiple unmapped regions following the initial alignment to the reference genome or remaining unmapped nucleotides following the rearrangement of sequence blocks. To resolve these consensus sequences, our algorithm aligns every possible rotation of these consensus sequences to the reference genome. The best of these alignments with no unmapped nucleotides is selected as the final consensus sequence used for experiment-specific computational analysis.
Analysis of variant frequencies
Although the accuracy of variant detection is independent of genome coverage, the accuracy of variant frequency measurement is not; the lower the desired variant frequency, the greater the coverage required. We use a binomial distribution to model sampling error of variant frequencies measured by CirSeq (Fig. 3a). The level of tolerable experimental error for downstream analyses sets the range of frequencies that can be used or the depth of coverage that must be attained to accurately measure frequencies in the desired range (Fig. 3b). In addition, variant frequencies must be higher than the estimated error probability used as a threshold for data analysis. For RNA viruses, we typically use an estimated error probability threshold of 10−6, as most variants are present at frequencies between 10−4 and 10−6 (ref. 9). For populations with variants at lower frequencies, that threshold should be adjusted accordingly; however, a more stringent threshold will reduce the total amount of usable data.
Figure 3.

Analysis of variant frequency error. (a,b) Correlation of two sets of technical replicates with 10 million reads each is plotted, with color representing levels of measurement error (a) estimated using a binomial model or total coverage (b) observed at the genome position corresponding to each variant. Estimation of error using a binomial model accurately corresponds to the extent of correlation observed for variant frequencies in technical replicates. This error model is a function of the variant frequency and the coverage depth obtained for each position. Data were obtained from Acevedo et al.7.
MATERIALS
REAGENTS
Viral RNA, 1–5 μg (see Reagent Setup)
RNA fragmentation reagents (Ambion, cat. no. AM8740)
RNaseZap RNase decontamination solution (Ambion, cat. no. AM9780)
Acrylamide/Bis, 40% (wt/vol) solution, 19:1 (Bio-Rad, cat. no. 161-0144) ! CAUTION Acrylamide/Bis is toxic. Wear personal protective equipment and avoid inhalation.
Urea (Bio-Rad, cat. no. 161-0731)
TEMED (Invitrogen, cat. no. 15524-010) ! CAUTION TEMED is flammable and toxic. Wear personal protective equipment and handle it in a fume hood.
Ammonium persulfate (Sigma-Aldrich, cat. no. A3678)
Tris base (Fisher Scientific, cat. no. BP152)
Boric acid (Fisher Scientific, cat. no. BP168) ! CAUTION Boric acid is toxic. Wear personal protective equipment and avoid inhalation.
EDTA, disodium salt (Sigma-Aldrich, cat. no. E5134)
Formamide (Fisher Scientific, cat. no. BP227) ! CAUTION Formamide is toxic. Wear personal protective equipment and avoid inhalation.
Sodium hydroxide (Fisher Scientific, cat no. BP359) ! CAUTION Sodium hydroxide is caustic. Wear personal protective equipment and avoid inhalation.
SYBR Gold nucleic acid gel stain, 10,000× (Invitrogen, cat. no. S-11494)
Sodium acetate, 3 M, pH 5.5 (Ambion, cat. no. AM9740)
TE buffer, 1× (Promega, cat. no. V6231)
SDS (Invitrogen, cat. no. 15525-017) ! CAUTION SDS is toxic. Wear personal protective equipment and avoid inhalation.
-
Glycogen, RNA grade (Thermo Scientific, cat. no. R0551)
▴ CRITICAL In our experience, glycogen from other vendors can result in lower yield of nucleic acids after ethanol precipitation.
Ethanol, 200 proof (Gold Shield) ! CAUTION Ethanol is flammable and toxic. Wear personal protective equipment.
T4 polynucleotide kinase, 10,000 U/ml (New England Biolabs, cat. no. M0201S/L)
T4 RNA ligase 1, 10,000 U/ml, supplied with 10× T4 RNA ligase buffer and 10 mM ATP (New England Biolabs, cat. no. M0204S/L)
Phenol:chloroform:isoamyl alcohol (25:24:1; Invitrogen, cat. no. 15593-031) ! CAUTION Phenol:chloroform:isoamyl alcohol is toxic and corrosive. Wear personal protective equipment and handle it in a fume hood.
Isoamyl alcohol (Fisher Scientific, cat. no. A393) ! CAUTION Isoamyl alcohol is flammable and toxic. Wear personal protective equipment and handle it in a fume hood.
Random hexamers, 50 μM (Invitrogen, cat. no. N8080127)
dNTP mix, 10 mM total (Bioline, cat. no. BIO-39053)
SuperScript III reverse transcriptase, 200 U/μl (Invitrogen, cat. no. 18080-044)
RNase H, 2 U/μl (Invitrogen, cat. no. 18021-071)
NEBNext mRNA second strand synthesis module (New England Biolabs, cat. no. E6111S/L)
NEBNext end repair module (New England Biolabs, cat. no. E6050S/L)
NEBNext dA-tailing module (New England Biolabs, cat. no. E6053S/L)
NEBNext quick ligation module (New England Biolabs, cat. no. E6056S/L)
Phusion high-fidelity DNA polymerase (New England Biolabs, cat. no. M053S/L)
Bromophenol Blue (Sigma-Aldrich, cat. no. B6131)
Xylene cyanol (Affymetrix, cat. no. 23513)
Perfect RNA Markers, 0.1–1 kb (EMD Millipore, cat. no. 69924)
Low-molecular-weight DNA ladder, supplied with 6× gel loading dye (New England Biolabs, cat. no. N3233S/L)
TruSeq indexed adapters and PCR primer cocktail (Illumina, cat. no. FC-121-4001) or equivalent oligonucleotides ▴ CRITICAL Indexed adapter oligonucleotides that are ordered separately should be annealed before use.
Library quantification kit, Illumina/Universal (Kapa Biosystems, cat. no. KK4824)
Sequencing kits, MiSeq reagent kit v2 (300 cycles; Illumina, cat. no. MS-102-2002) or 5× TruSeq rapid SBS kit—HS (50 cycle) and TruSeq rapid SR cluster kit—HS (Illumina, cat. nos. FC-402-4002 and GD-402-4001)
EQUIPMENT
Plastic wrap (Fisher Scientific, cat. no. 01810)
Aluminum foil (Alcan, cat. no. 1851-SE)
Parafilm (VWR International, cat. no. 52858-000)
Single-edge razor blades (Fisher Scientific, cat. no. 17-989-001)
Tubes, 15 ml (VWR International, cat. no. 89039-666)
Tubes, 50 ml (VWR International, cat. no. 89039-658)
Corning Costar Spin-X centrifuge tube filters, cellulose acetate membrane, pore size 0.22 μm, sterile (Sigma-Aldrich, cat. no. CLS8160)
-
Microcentrifuge tubes (Denville Scientific, cat. no. C2170)
▴ CRITICAL Use ultra-clear, low-retention tubes to minimize sample loss during nucleic acid precipitation.
Thin-wall PCR strip tubes and caps (VWR, cat. nos. 89091-884 and 89091-886)
Microcentrifuge (Eppendorf Centrifuge 5424, maximum speed 21,130g)
Nanofuge with strip tube attachment (Denville Scientific Mini Mouse)
Vortex mixer (Fisher Scientific Vortex Genie 2)
Orbital shaker (Bellco standard orbital shaker)
Rotator (Bellco Rotamix)
Vertical electrophoresis system, approximate gel dimensions of 7.3 × 8.3 cm (length × width; Bio-Rad Mini-PROTEAN Tetra Cell including gel-casting stand and frames, short and 1.0-mm spacer glass plates and 1.0-mm five-well combs)
Power source (Owl Scientific Plastics OSP-105)
Thermal cycler (Bio-Rad C1000 Touch thermal cycler)
qPCR system (Bio-Rad CFX Connect real-time system)
UV lamp (FisherBiotech FB-UVM-80, peak emission at 312 nm)
Sequencer (Illumina MiSeq or HiSeq 1500/2500)
REAGENT SETUP
Viral RNA, 1–5 μg
A specific method of viral RNA purification should be determined for each viral system, taking into account the total RNA yield and purity; see Experimental design for further discussion. We have successfully prepared sequencing libraries using viral RNA purified by poly(A) purification (MicroPoly(A)Purist, Ambion, cat. no. AM1919), oligo-capture and virion purification with no difference in performance using this protocol. We typically prepare libraries starting with at least 1 μg of viral RNA of at least 50% purity in no more than a 9-μl volume.
EDTA, 0.5 M (pH 8)
Dissolve 93.05 g of EDTA and 10.14 g of sodium hydroxide in 400 ml of nuclease-free water. After the chemicals have dissolved, bring the final volume of the solution to 500 ml with nuclease-free water. The solution can be stored at room temperature (22–25 °C) indefinitely.
TBE, 10×
Dissolve 108 g of Tris base, 55 g of boric acid and 7.5 g of EDTA in 800 ml of nuclease-free water. After the chemicals have dissolved, bring the final volume of the solution to 1 liter with nuclease-free water. The solution can be stored at room temperature. If a precipitate forms, the solution should be discarded and a fresh batch should be prepared.
Gel solutions
For 12.5% (wt/vol) urea-PAGE mix, combine 25 g of urea, 5 ml of 10× TBE, 15.6 ml of 40% (wt/vol) acrylamide/Bis solution and nuclease-free water up to 50 ml. For 10% (wt/vol) urea-PAGE mix, combine 25 g of urea, 5 ml of 10× TBE, 12.5 ml of 40% (wt/vol) acrylamide/Bis solution and nuclease-free water up to 50 ml. For 7.5% (wt/vol) PAGE mix, combine 5 ml of 10× TBE, 9.4 ml of 40% (wt/vol) acrylamide/Bis solution and nuclease-free water up to 50 ml. The urea-PAGE mixes can be warmed to 37 °C to allow the urea to dissolve more quickly. The gel solutions can be stored at room temperature, protected from light, for at least 1 month.
Denaturing dye, 2×
Add 25 μl of 10% (wt/vol) SDS, 2.5 mg of bromophenol blue and 2.5 mg of xylene cyanol to 9 ml of formamide. After the chemicals have dissolved, bring the final volume of the solution to 10 ml with formamide.
RNA elution buffer
Add 10 ml of 3 M sodium acetate, pH 5.5, 50 μl of 10% (wt/vol) SDS and 100 μl of 0.5 M EDTA (pH 8), to 30 ml of nuclease-free water. Adjust the final volume to 50 ml with nuclease-free water.
PROCEDURE
Preparation of size-selected RNA fragments ● TIMING 18–19 h
▴ CRITICAL Perform Steps 1–23 under RNase-free conditions. To ensure RNase-free conditions, use RNase-free reagents and pretreat equipment and work surfaces with RNaseZap.
▴ CRITICAL Volumes of 3 M sodium acetate and 100% ethanol used after phenol:chloroform:isoamyl alcohol extractions assume that at least 95% of the aqueous phase is used for precipitations. 3 M sodium acetate and 100% ethanol should be added at a one-tenth volume and 2.5 volumes, respectively, of the extracted nucleic acid solution. We recommend performing extraction and precipitation of nucleic acid solutions in the smallest possible volume, as nucleic acids are recovered more efficiently at higher concentrations. We recommend performing extractions in PCR tubes, which, in our experience, makes extracting small volumes much easier than in 1.5-ml tubes.
▴ CRITICAL Timing is based on the preparation of four samples, and it includes the time required for overnight incubations.
-
1|
Pretreat a five-well comb, gel plates and an electrophoresis tank with RNaseZap. Rinse them with nuclease-free water and allow them to dry.
-
2|
Combine 6.25 ml of 12.5% (wt/vol) urea-PAGE mix, 37.5 μl of ammonium persulfate (APS) and 3.75 μl of TEMED in a 15-ml tube. Mix it thoroughly by inversion, and pour it between gel plates. After all of the bubbles have risen to the surface, insert a five-well comb. Allow the gel to polymerize for 15–20 min.
-
3|
Place the polymerized gel into the electrophoresis tank, and fill the upper and lower reservoirs with 1× TBE submerging the top and bottom of the gel. Prerun the gel for 15–30 min at 300 V.
-
4|
While the gel is prerunning, bring 1–5 μg of purified viral RNA to a volume of 9 μl with nuclease-free water in a PCR tube. Add 1 μl of 10× fragmentation buffer. Mix the sample, briefly spin it and incubate it at 70 °C for 7.5 min in a thermal cycler.
? TROUBLESHOOTOOTING
-
5|
Add 1 μl of Stop Solution and 11 μl of 2× denaturing dye. Mix and place the sample on ice.
-
6|
Prepare RNA marker by combining 900 ng of Perfect RNA marker (0.1–1 kb) with an equal volume of 2× denaturing dye.
-
7|
Denature the sample and marker at 95 °C for 5 min in a thermal cycler.
-
8|
Place the sample and marker on ice for at least 2 min or until ready for loading.
-
9|
Turn off the current and flush the wells of the gel by forcefully pipetting 1× TBE into each well.
▴ CRITICAL STEP Flushing the wells should be done immediately before loading the marker and sample to remove excess urea that diffuses from the gel into the wells. Failure to flush the wells may result in poor resolution of samples.
-
10|
Load the marker and the sample and run the gel at 300 V until the upper dye (xylene cyanol) front is ~1 cm from the bottom of the gel.
-
11|
Dilute 1.5 μl of SYBR Gold stain with 15 ml of 1× TBE in the lid of a sterile tip box. Carefully separate the gel plates and transfer the gel into the diluted stain. Cover the tip box lid with aluminum foil and gently agitate it for 10 min.
-
12|
Remove the gel from the stain and place it on a sheet of plastic wrap. Illuminate the gel with a hand-held UV lamp. By using a razor blade, excise an ~2-mm slice of gel containing fragments between 85 and 100 bases.
▴ CRITICAL STEP The fragment length can affect sequencing outcomes. Fragments <85 nt can increase disparities in coverage depth across the genome (Fig. 2), whereas fragments >100 nt (for 300-nt sequencing reads), when read three times in tandem, may exceed the sequencing read length. For read lengths >300 nt, the maximum fragment length may be adjusted to one-third of the read length.
-
13|
Crush the gel slice on a piece of Parafilm using a razor blade until the gel forms a fine paste. Transfer the gel paste into a 1.5-ml tube and add 360 μl of RNA elution buffer. Incubate the gel slurry overnight at 4 °C with constant agitation.
RNA circularization and tandem repeat generation ● TIMING 5–6 h
-
14|
Transfer the gel slurry into a Spin-X tube and centrifuge it at 4,000g for 2 min at room temperature. To precipitate the RNA fragments, combine the eluate with 2 μl of glycogen, 40 μl of 3 M sodium acetate and 1 ml of 100% ethanol in a 1.5-ml tube. Mix the sample thoroughly and incubate it at room temperature for 20 min.
▴ CRITICAL STEP Spin-X columns are provided with 2-ml Dolphin tubes. In our experience, pellets tend to stick poorly to the wall of these tubes, which leads to frequent loss of the nucleic acid pellet during aspiration of the supernatant (Step 15). We recommend transferring the supernatant to a 1.5-ml ultra-clear, low-retention tube.
-
15|
Centrifuge the sample at 21,130g at 4 °C for 30 min. Remove the supernatant.
-
16|
To wash the pellet, add 250 μl of 70% (vol/vol) ethanol and centrifuge it at 21,130g at 4 °C for 2 min. Remove the supernatant and briefly air-dry the pellet.
? TROUBLESHOOTING
-
17|
Dissolve the pellet in 14 μl of nuclease-free water and transfer it to a PCR tube. Heat-denature the sample at 95 °C for 5 min in a thermal cycler, and then place it on ice for 2 min.
-
18|
When the sample has cooled to 4 °C, add the components listed below. Mix the sample thoroughly, spin it briefly and incubate it at 37 °C for 30 min in a thermal cycler.
Component Amount (μl) Final T4 RNA ligase buffer (10×) 2 1× ATP, 10 mM 2 1 mM T4 RNA ligase 1, 10 U/μl 1 10 U T4 polynucleotide kinase, 10 U/μl 1 10 U ▴ CRITICAL STEP To prevent chemical fragmentation of the RNA by Mg2+, do not add the buffer to the sample until the tube has cooled.
-
19|
Add 20 μl of phenol:chloroform:isoamyl alcohol. Vortex and spin until the organic and aqueous phases have separated (~30–60 s). Transfer the aqueous phase to a 1.5-ml tube.
-
20|
Add 2.2 μl of 3 M sodium acetate and 55.5 μl of 100% ethanol. Mix the sample thoroughly and incubate it at room temperature for 20 min.
-
21|
Repeat Steps 15 and 16.
▴ CRITICAL STEP It is not necessary to add glycogen, as the glycogen added to the size-selected RNA fragments in Step 14 is still present in the sample. We recommend only adding glycogen after the gel purification steps.
-
22|
Dissolve the pellet in 9 μl of nuclease-free water and transfer it to a PCR tube. Add the components listed below. Denature the sample at 65 °C for 5 min, and then place it on ice for 2 min.
Component Amount (μl) Finala dNTPs, 10 mM 2 1 mM Random hexamers, 50 ng/μl 2 5 ng/μl aRefers to the final concentration of the completed reaction mix after remaining components added at Step 23. -
23|
Add the components listed below. Mix the sample thoroughly, spin it briefly and incubate it at 25 °C for 10 min in a thermal cycler.
Component Amount (μl) Final First-strand synthesis buffer, 5× 4 1× DTT, 0.1 mM 1 5 μM SuperScript III, 200 U/μl 2 400 U -
24|
Increase the incubation temperature to 42 °C. After 2 min, add 1 μl of RNase H diluted to 0.008 U/μl.
Continue incubating at 42 °C for an additional 30 min.
▪ PAUSE POINT Samples can be stored at −20 °C in nuclease-free conditions for at least 7 d.
Library cloning ● TIMING 24–25 h
-
25|
Cool the components listed below to <16 °C, and add them to the sample. Mix the sample thoroughly, spin it briefly and incubate it at 16 °C for 2.5 h in a thermal cycler.
Component Amount (μl) Final Nuclease-free water 64 — NEBNext second-strand synthesis reaction buffer, 10× 10 1× NEBNext second-strand synthesis enzyme mix 5 — -
26|
Add 100 μl of phenol:chloroform:isoamyl alcohol. Vortex and spin until the organic and aqueous phases have separated. Transfer the aqueous phase to a 1.5-ml tube.
? TROUBLESHOOTOOTING
-
27|
Add 11 μl of 3 M sodium acetate and 278 μl of 100% ethanol. Mix thoroughly and incubate at room temperature for 20 min.
-
28|
Repeat Steps 15 and 16.
-
29|
Dissolve the pellet in 85 μl of nuclease-free water and transfer it to a PCR tube. Add the components listed below. Mix the sample thoroughly, spin it briefly and incubate it at 20 °C for 30 min in a thermal cycler.
Component Amount (μl) Final NEBNext end-repair buffer, 10× 10 1× NEBNext end-repair enzyme mix 5 — -
30|
Repeat Steps 26–28.
-
31|
Dissolve the pellet in 42 μl of nuclease-free water and transfer it to a PCR tube. Add the components listed below. Mix the sample thoroughly, spin it briefly and incubate it at 37 °C for 30 min in a thermal cycler.
Component Amount (μl) Final NEBNext dA-tailing reaction buffer, 10× 5 1× Klenow fragment (3′→5′ exo−) 3 — -
32|
Add 50 μl of phenol:chloroform:isoamyl alcohol. Vortex and spin until the organic and aqueous phases have separated. Transfer the aqueous phase to a 1.5-ml tube.
-
33|
Add 5.5 μl of 3 M sodium acetate and 139 μl of 100% ethanol. Mix the sample thoroughly and incubate it at room temperature for 20 min.
-
34|
Repeat Steps 15 and 16.
-
35|
Dissolve the pellet in 22.5 μl of nuclease-free water and transfer it to a PCR tube. Add the components listed below. Mix the sample thoroughly, spin it briefly and incubate it either at 20 °C for 15 min or at 16 °C overnight in a thermal cycler.
Component Amount (μl) Final NEBNext Quick Ligation reaction buffer, 5× 10 1× NEBNext T4 DNA ligase 5 — TruSeq indexed adapter 12.5 — ▴ CRITICAL STEP For indexed adapters synthesized or purchased separately, anneal the adapters at a concentration of 15 μM (each). Add 1–2.5 μl of annealed adapters to the ligation reaction for a final adapter concentration of 0.3–0.75 μM.
▪ PAUSE POINT Samples can be stored at −20 °C in nuclease-free conditions for at least 7 d.
Size selection of libraries ● TIMING 22–23 h
-
36|
Repeat Steps 2 and 3 using 10% (wt/vol) urea-PAGE mix.
-
37|
While the gel is polymerizing and prerunning, repeat Steps 32–34 with the adapter-ligated sample from Step 35.
-
38|
Dissolve the pellet in 5 μl of nuclease-free water and add 5 μl of 2× denaturing dye.
-
39|
Prepare the DNA marker by combining 100 ng of low-molecular-weight DNA ladder with an equal volume of 2× denaturing dye.
-
40|
Repeat Steps 7–9.
-
41|
Load the marker and sample and run the gel at 300 V until the lower dye (bromophenol blue) front runs off the bottom of the gel.
-
42|
Dilute 1.5 μl of SYBR Gold stain with 15 ml of 1× TBE into the lid of a sterile tip box. Carefully separate the gel plates and transfer the gel into the diluted stain. Cover the tip box lid with aluminum foil and gently agitate it for 10 min.
-
43|
Remove the gel from the stain and place it on a sheet of plastic wrap. Illuminate the gel with a hand-held UV lamp. Use a razor blade to excise a slice of gel containing adapter-ligated fragments between 450 and 600 bases.
▴ CRITICAL STEP Excised fragments should be no shorter than 435 bases to account for the combined length of the indexed adapters (135 bases) and at least 300 bases of the tandem-repeat insert.
? TROUBLESHOOTING
-
44|
Repeat Steps 13–16 using 1× TE as the gel elution buffer.
-
45|
Dissolve the pellet in 20 μl of nuclease-free water. Retain an aliquot for the determination of sample concentration (Step 54).
PAUSE POINT Samples can be stored at −20 °C in nuclease-free conditions for at least 2 years.
Amplification and purification of libraries ● TIMING 22–23 h
-
46|
Combine 5 μl of the size-selected library and the following components. Mix it thoroughly and spin it briefly.
Component Amount (μl) Final Nuclease-free water 62 — Phusion high-fidelity buffer, 5× 20 1× dNTPs, 10 mM 2 0.2 mM PCR primer cocktail 10 — Phusion high-fidelity DNA polymerase, 2 U/μl 1 2 U ▴ CRITICAL STEP For PCR primers synthesized or purchased separately, use them at a final concentration of 0.5 μM each.
-
47|
Thermal-cycle the PCR mix from Step 46 using the following parameters:
Cycle Denature Anneal Extend 1 98 °C for 30 s 2–16 98 °C for 10 s 65 °C for 30 s 72 °C for 30 s 17 72 °C for 5 min -
48|
Repeat Steps 26–28.
-
49|
While the sample is precipitating, combine 6.25 ml of 7.5% (wt/vol) PAGE mix, 37.5 μl of APS and 3.75 μl of TEMED in a 15-ml tube. Mix thoroughly by inversion and pour between gel plates. After all of the bubbles have risen to the surface, insert a five-well comb. Allow the gel to polymerize for 15–20 min.
-
50|
Dissolve the pellet in 10 μl of nuclease-free water and add 2 μl of 6× gel loading dye.
-
51|
Load 100 ng of low-molecular-weight DNA ladder and the sample and run the gel at 150 V until the lower dye (bromophenol blue) front runs off the bottom of the gel.
-
52|
Repeat Steps 42–44.
-
53|
Dissolve the pellet in 40 μl of nuclease-free water. Retain an aliquot for the determination of sample concentration (Step 54).
▪ PAUSE POINT Samples can be stored at −20 °C in nuclease-free conditions for at least 2 years.
Library quantification, sequencing and analysis ● TIMING variable
-
54|
Quantify the concentrations of gel-purified sample after adapter ligation (from Step 45) and amplification (from Step 53) using the library quantification kit according to the manufacturer’s instructions.
▴ CRITICAL STEP The total number of molecules detected in the adapter-ligated sample should exceed the number of sequencing reads desired by at least several fold. This requirement reduces the probability of sampling amplified molecules derived from the same RNA template multiple times.
-
55|
Sequence the amplified, purified library on an Illumina HiSeq or MiSeq according to the manufacturer’s instructions.
-
56|
Analyze the sequencing data by generating consensus sequences and mapping to a known reference sequence. Mapped reads can be rearranged to account for differences in the starting point of the consensus sequence and the 3′–5′ RNA ligation junction. Details on data processing are discussed in Experimental design, and CirSeq computational analysis tools using Bowtie2 (ref. 9) can be obtained from http://andino.ucsf.edu/CirSeq.
? TROUBLESHOOTOOTING
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table.
| Step | Problem | Possible cause | Solution |
|---|---|---|---|
| 4 | Fragment size range does not overlap 85–100 nt | Non-optimal fragmentation time or temperature | Increase or decrease the fragmentation time or temperature to reduce or increase the fragment size range, respectively |
| 16 | No pellet is visible | Low yield of size selected RNA | Start with more viral RNA or optimize the fragmentation time |
| RNA degradation | Prepare new reagents and treat equipment with RNaseZAP | ||
| Poor-quality glycogen | Use a different glycogen supplier; we have found Thermo Scientific glycogen to perform best | ||
| 26 | Excessive foam after phase separation | Typical for extraction from second-strand synthesis reactions | Add a drop of isoamyl alcohol, mix and spin. Repeat until foam dissipates |
| Use PhaseLock gel (5Prime, cat. no. 2302800) to improve phase separation | |||
| 43 | No DNA is visible | Too little circular RNA is used | Increase the quantity of circular RNA in the reverse-transcription reaction |
| RNA degradation | Prepare new reagents and treat equipment with RNaseZAP |
● TIMING
Steps 1–13, preparation of size-selected RNA fragments: 18–19 h; hands-on time: 2–3 h
Steps 14–24, RNA circularization and tandem repeat generation: 5–6 h; hands-on time: 5–6 h
Steps 25–35, library cloning: 24–25 h; hands-on time: 8–9 h
Steps 36–45, size selection of libraries: 22–23 h; hands-on time: 6–7 h
Steps 46–53, amplification and purification of libraries: 22–23 h; hands-on time: 6–7 h
Steps 54–56, library quantification, sequencing and analysis: several days to weeks, depending on access to a sequencer and the quantity of data to be analyzed
ANTICIPATED RESULT
The sample results were generated using poly(A)-purified RNA from poliovirus-infected HeLaS3 cells, as previously described7. We typically obtain 8–10 μg of poly(A) RNA from one confluent 10-cm dish, of which 60–80% is poliovirus genomic RNA. In our hands, yields of RNA fragments after size selection are 50–100 ng depending on the amount of starting material—we typically start with 2–4 μg. Although it is not part of our standard protocol, preparations can be checked by denaturing PAGE or by a Bioanalyzer for fragment length and concentration. Fragments should migrate in a tight band between 85 and 100 bases (Fig. 4a) without signs of degradation (Fig. 4b).
Figure 4.
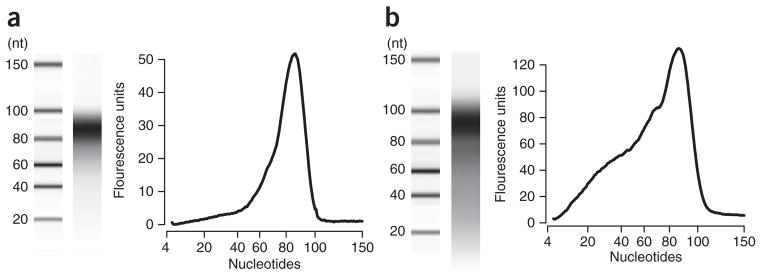
Bioanalysis of size-selected fragmented RNA. (a,b) Digital gels (left) and fluorescence traces (right) of typical (a) and poor (b) purifications of fragmented RNA analyzed using a Bioanalyzer 2100. Size-selected RNA should migrate in a tight band with an average size of no less than 85 nt. Degradation of size-selected RNA fragments below this range (b) can result in poor yield of tandem repeat cDNA, thus reducing the number of unique molecules in the library, and it can distort coverage depth across the viral genome (Fig. 2).
After size selection of the adapter-ligated library and purification of the amplified library, libraries are typically at concentrations of 0.2 and 30 nM, respectively, where the concentration of the adapter-ligated library is approximately linearly dependent on the quantity of RNA used as starting material. Because we generally collect 20–30 million reads per sample, these concentrations are unlikely to result in multiple sampling of molecules derived from the same initial RNA template.
With 20–30 million reads, we generally obtain ~200,000-fold coverage of the poliovirus genome, which is nearly 7,500 nt in length, using a sequence quality threshold of one error in 106 bases. Less coverage should be expected when a higher threshold sequence quality is used. Coverage generally varies across the genome by one order of magnitude (Fig. 2), excluding the genome ends, which is typical of RNA-seq experiments. We have found that the uniformity of coverage can be markedly affected by RNA fragment length, in that A-rich regions are heavily enriched when fragments are <85 bases in length (Fig. 2). Although this coverage bias does not affect the quality of the sequencing data, it does reduce the number of variants detected with accurate frequency measurements.
Acknowledgments
We thank S. Taguwa and P. Ambrose for critical reading of the manuscript, and G. Schroth, M. Harrison, P. Wassam and T. Collins for technical advice. This work was financially supported by a National Science Foundation graduate research fellowship to A.A., and by National Institute of Allergy and Infectious Diseases (NIAID) grants AI091575, AI36178 and AI40085, as well as a Defense Advanced Research Projects Agency (DARPA) Prophecy grant to R.A.
Footnotes
AUTHOR CONTRIBUTIONS
R.A. and A.A. conceived and designed the experiments; A.A. performed the experiments; and R.A. and A.A. wrote the manuscript.
COMPETING FINANCIAL INTEREST
The authors declare no competing financial interests.
References
- 1.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 2.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short-read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119–122. doi: 10.1038/nmeth.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci USA. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci USA. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acevedo A, Brodsky L, Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686–690. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lou DI, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc Natl Acad Sci USA. 2013;110:19872–19877. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]