

Abstract
The repertoire of proteins and nucleic acids in the living world is determined by evolution; their properties are determined by the laws of physics and chemistry. Explanations of these two kinds of causality — the purviews of evolutionary biology and biochemistry, respectively — are typically pursued in isolation, but many fundamental questions fall squarely at the interface of fields. Here we articulate the paradigm of evolutionary biochemistry, which aims to dissect the physical mechanisms and evolutionary processes by which biological molecules diversified and to reveal how their physical architecture facilitates and constrains their evolution. We show how an integration of evolution with biochemistry moves us towards a more complete understanding of why biological molecules have the properties that they do.
Both biochemists and evolutionary biologists seek to explain why biological systems work as they do. Evolutionary biology accounts for the characteristics of living systems in terms of their histories; biochemistry explains those characteristics as the product of the fundamental properties of matter and energy. In truth, the why of biological systems lies in the interplay of historical and physical causes, and only a mode of explanation that incorporates both types of analysis can comprehend that interplay.
The common interest of biochemists and evolutionary biologists in ultimate explanations represents fertile ground for work across the disciplines’ boundaries. Because of an accident of history, however, the two fields inhabit largely separate spheres. In the 1950s and 1960s, a group of chemists realized that molecular biology allowed studies of ‘the most basic aspects of the evolutionary process’1. They produced a flurry of papers proposing molecular phylogenetics2,3, the molecular clock4, ancestral protein reconstruction5, the importance of functionally neutral changes in evolution1 and the use of studies of protein function to understand organisms’ adaptation to their environments6,7.
Unfortunately, this early attempt at integrating biochemical and evolutionary reasoning became a casualty in the acrimonious battle between molecular and classical biologists8-10. Chemists such as Zuckerkandl and Pauling dismissed traditional evolutionary biology, writing that what “most counts in the life sciences today is the uncovering of the molecular mechanisms that underlie the observations of classical biology”4. In turn, prominent evolutionary biologists — such as G. G. Simpson, who called molecular biology a “gaudy bandwagon ... manned by reductionists, traveling on biochemical and biophysical roads”11 — were deeply sceptical that studies of molecules could contribute useful insights about evolutionary processes, which (they insisted) took place only at the level of organisms8,10,12.
This tension hardened into a cultural and institutional split as the fields competed for resources and legitimacy. The two groups defined themselves as asking incommensurable questions with different scientific aesthetics: biochemists and molecular biologists dissect the underlying mechanisms by which model systems function, whereas evolutionary biologists analyse how the diversity of living forms in nature came to be8,10,13. At most institutions, biology departments split into separate entities, creating a barrier to interactions between biochemists and evolutionists.
Science has been hobbled as a result. Few biochemists and molecular biologists receive evolutionary training, leading to widespread confusion about evolutionary concepts such as homology14, natural selection15 and the phylogenetic structure of molecular diversity16. Conversely, many evolutionary biologists — even those who specialize in ‘molecular evolution’ — treat molecular sequences as mere strings of letters, the patterns of which carry the traces of historical processes, rather than as functioning objects for which the physical properties determine their behaviour17-19. As a consequence, investigations in either field that are strongly informed by the other have been rare (but see REFS 17,20-24).
Today, the animus of old battles has largely faded. Meanwhile, new experimental strategies have emerged, enabling rigorous work to be carried out at the interface of evolution and the chemistry of biological molecules18,25-27 (BOXES 1,2). Biochemists have begun to leverage evolutionary information to dissect how biological molecules function26,28-30. And evolutionary biologists are studying changes in molecular properties to tackle classic questions in evolutionary biology31-37.
Box 1. Methods for studying the evolutionary trajectories of proteins.

Two interdisciplinary approaches have had key roles in the emergence of present-day evolutionary biochemistry. Both trace in detail the evolutionary processes and biochemical mechanisms by which changes in protein sequence have caused shifts in function or other properties.
The first strategy explicitly reconstructs the historical trajectory that a protein or group of proteins took during evolution (see panel a of the figure). For proteins that evolved new functions or properties very recently, population genetic analyses can identify which genotypes and phenotypes are ancestral and which are derived89,93,153. For more ancient divergences, ancestral protein reconstruction (APR) uses phylogenetic techniques to reconstruct statistical approximations of ancestral proteins computationally, which are then physically synthesized and experimentally studied5,154. Starting from an alignment of modern sequences, the phylogenetic tree is inferred, and statistical methods are used to infer ancestral sequences at the internal nodes of the tree (that is, at the circles in the figure). The maximum likelihood sequences are those with the highest probabilities of yielding all of the sequence data observed in the present world (in the figure, those sequences at the tips of the tree). Genes that encode the inferred ancestral sequences can then be synthesized and expressed in cultured cells; this approach allows the structure, function and biophysical properties of each ‘resurrected’ protein to be experimentally characterized. When statistical reconstructions are ambiguous, multiple plausible ancestral proteins can be studied to determine the robustness of experimental results to uncertainty about the reconstruction. By characterizing ancestral proteins at multiple nodes on a phylogeny, the evolutionary interval (shown by the black box in the figure) during which major shifts in those properties occurred can be identified. Sequence substitutions that occurred during that interval can then be introduced singly and in combination into ancestral backgrounds (see inset box in the figure), allowing the effects of historical mutations on protein structure, function and physical properties to be determined directly.
The second strategy is to use directed evolution to drive a functional transition of interest in the laboratory and then study the mechanisms of evolution (see panel b of the figure)25,26. A library of random variants of a protein of interest is generated and then screened to recover those with a desired property. Selected variants are iteratively re-mutagenized and are subject to selection to optimize the property. Causal mutations and their mechanisms can then be identified by characterizing the sequences and functions of the intermediate states realized during evolution of the protein. The evolutionary process can be manipulated and repeated from various starting points and under different evolutionary conditions, allowing the effects of these factors on evolutionary trajectories and outcomes to be rigorously inferred30,155-157.
Box 2. Charting protein sequence space.
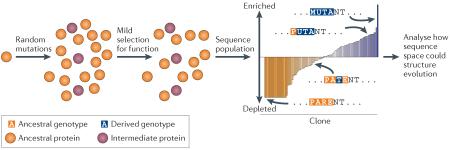
A third evolutionary biochemical approach characterizes a portion of sequence space in detail and explores its evolutionary implications (BOX 3) without explicit reference to a historical trajectory across it. Rather than reconstructing what evolution did in the past (BOX 1), this strategy aims to reveal what it could do, given detailed knowledge of sequence space and fundamental understanding of evolutionary processes. Recent methods for characterizing large libraries of protein variants using deep sequencing make these efforts possible27,50,158-160. An initial protein (see the figure; orange circle) is subjected to random mutagenesis, and weak selection for a property of interest is applied, enriching the library for clones with the property and depleting those without it. The population is then sequenced; the degree of enrichment of each clone allows the direct and epistatic effects of each mutation on the function to be quantitatively characterized.
This approach can reveal the distribution of a property of interest in sequence space and thereby can illuminate the potential of various evolutionary forces to drive trajectories across the space. One study, for example, characterized the fitness effects in a defined environment of all possible point mutants in a nine-amino-acid region of yeast heat shock protein 90 (Hsp90)27. This work revealed the potential for selection and neutral processes to drive the evolution of potential genotypes, both those realized during the evolution of real-world sequences and many more that have never been observed in nature.
A related approach to characterizing sequence space is to shuffle amino acid states between extant proteins and then to characterize the recombinants. This strategy has revealed the fraction of paths between present-day sequences that involve loss or changes in function106,161, has identified trade-offs between protein properties that limit the capacity of selection to optimize both23,35,83 and has determined how the sequence ‘background’ in different evolutionary lineages changes the functional effects of specific mutations34,110.
In this Review, we articulate the paradigm of evolutionary biochemistry, which combines evolutionary analysis with rigorous biophysical and biochemical studies. By simultaneously asking ‘how things work’ and ‘how they got to be that way’, evolutionary biochemistry provides unique insight into how evolution shapes the physical properties of biological molecules and how those properties shape evolutionary trajectories.
We begin by describing this approach and what it can contribute to both biochemistry and evolutionary biology. We then highlight recent work on key questions, such as the evolution of protein stability, the mechanisms of parallel evolution, the biochemical causes and evolutionary consequences of epistasis and the extent to which the paths and outcomes of molecular evolution are predictable or contingent on chance events. We conclude with thoughts about the future of the field. Although we focus on proteins, the concepts and techniques we discuss can also be applied to DNA and RNA evolution38-41. We highlight experimental work, but computational and theoretical explorations have also contributed to the development of the new field42,43. Evolutionary biochemistry is a part of a larger ‘functional synthesis’18 of evolutionary biology with fields that seek mechanistic molecular explanations for biological forms and functions.
Why evolutionary biochemistry?
Evolutionary biochemistry can help both biochemists and evolutionary biologists to understand classic questions in their fields. It also raises new questions about the interplay between evolutionary and physical causes in determining present-day protein properties.
Exploring sequence space
Sequence space provides a rich metaphor to organize thinking about the evolution of biological molecules44-49 and reveals the potential common ground for evolution and biochemistry (BOX 3). Sequence space is a spatial representation of all possible amino acid sequences and the mutational connections between them. Each sequence is a node, and each node is connected by edges to all neighbouring proteins that differ from it by just one amino acid. This space of sequences becomes a genotype–phenotype space when each node is assigned information about its functional or physical properties; this representation serves as a map of the total set of relations between sequence and those properties. As proteins evolve, they follow trajectories along edges through the genotype–phenotype space.
Box 3. Protein sequence space and evolutionary biochemistry.
Protein sequence space is a useful way of understanding the relationship between biochemistry and evolutionary biology. The simplest multidimensional space represents genotypes only (see panel a of the figure). Every possible sequence is a node. Neighbouring nodes that differ by a single point mutation are connected by edges. The example in the figure shows a three-site protein with only two possible states (0 or 1). For a real 200-residue protein, genotype space contains 20200 nodes, which is far more than the number of subatomic particles in the observable universe. In genotype–phenotype space, each sequence is associated with its functional characteristics, which are determined by the biochemical properties of the molecule (see panel b of the figure). Here, three possible states are shown: an ancestral functional state (α; shown in orange), a derived functional state (β; blue) and a non-functional state (grey). An intermediate state between α and β (purple) is also shown. Evolutionary forces that drive proteins across the genotype–phenotype space (see panel c of the figure) show one trajectory beginning at genotype 000 and ending at 111 (see panel c of the figure).
In the simple example shown, none of the nodes accessible from the ancestral genotype 000 improves function β: nodes 001 and 010 are non-functional and thus are unlikely to be populated under selection, whereas 100 has function α. This implies that the first mutational step in the evolutionary trajectory cannot be driven by selection for β. By contrast, the transitions from 100 to 110, and from 110 to 111 both improve function β and thus can be driven by selection for this property. Studies to reveal the physical mechanisms by which any mutational step of interest produces its effects on function can reveal how and why evolution produced proteins with their present-day sequences, physical properties and biological functions.

Biochemistry and evolutionary biology have traditionally addressed different aspects of this map. Biochemists have sought to characterize the structure of the map and its physical determinants: that is, the links among protein sequence, biochemical properties and function. Evolutionary biologists have studied the trajectories that proteins follow across this map and the evolutionary forces that drive them to do so. Evolutionary biochemistry unites these approaches, seeking to reveal how and why proteins evolve across genotype-phenotype space to produce the diversity of proteins found in nature. This agenda can involve diverse strategies, such as explicitly reconstructing historical evolutionary trajectories across sequence space, identifying the biophysical mechanisms for the evolution of new functions and characterizing the biophysical factors that determine the structure of genotype-phenotype space and thereby affect the capacity of evolutionary forces to drive proteins across it (BOXES 1,2).
Evolution for biochemistry
A key goal of biochemistry is to determine how the sequence of a protein determines its physical properties and functions. Specific questions address different facets of this fundamental question: how do protein sequences determine three-dimensional structure? How do proteins fold rapidly and specifically? What is the physical basis of properties such as allostery, specificity, activity or cooperativity? Although these questions can be asked without reference to evolution, they have proved to be hard to answer, because the vast size of protein sequence space makes it impossible to characterize more than a tiny sample of it experimentally, even with modern high-throughput techniques27,50.
Evolutionary analysis is a powerful but underused tool in the biochemist’s kit. Protein evolution has been a massive experiment, conducted in parallel over billions of years, in the diversification and optimization of structure and function. The data from this experiment persist in the patterns of conservation and variation in presentday sequences. Explicit evolutionary analysis therefore provides a powerful and efficient means to interpret these data directly and to identify the key determinants of protein properties. For example, a recent evolutionary study used ancestral protein reconstruction to study the causes of the distinct ligand specificities of two major clades of vertebrate hormone receptors: although the present-day proteins differ at ~70% of their residues, evolutionary analysis identified just two historical substitutions that are sufficient to recapitulate a 70,000-fold shift in hormone preference. Experimental biochemical analysis then revealed the mechanisms by which these two substitutions reshaped a complex hydrogen bond network that determines ligand specificity51.
An evolutionary approach also focuses biochemical investigations on concrete, answerable questions. The classic question ‘how does sequence encode function?’ is intractable, both conceptually and practically, because there are an unimaginably vast number of possible sequences and an infinite number of possible functions. Evolutionary biochemical investigations focus this question by asking how changes in amino acid sequence during evolution changed a specific function or property. Framing studies in this way allows the sequence differences that cause real-world differences between real-world proteins to be identified29.
Finally, an evolutionary approach to biochemistry can illuminate why proteins with certain sequences and physical properties — out of a huge space of possibilities — occur. For example, does evolution optimize protein folding by selecting for fast folding sequences52, or does fast folding naturally arise from sequences that encode a folded structure53? Does selection against misfolding and aggregation due to translational errors explain why certain codons for the same amino acid are observed more frequently than others54-56? Answering these questions requires evaluating the role of an evolutionary force — natural selection in this case — on physical properties, so an approach that synthesizes the two modes of analysis is necessary.
Biochemistry for evolution
Major questions in evolutionary biology are unresolved, including whether phenotypic change is driven by a few large-effect or many small-effect mutations, the role of epistasis in shaping evolutionary pathways and outcomes, and the relative roles of neutral processes and selection in driving evolution. Mainstream molecular evolution seeks to address these issues by looking for statistical sequence signatures of evolutionary processes in molecular sequences without treating the proteins that those sequences encode as functioning physical objects17,18,57,58. This approach is intrinsically limited, because all of the questions described above — and almost any other question about evolutionary processes59 — require us to characterize the genotype–phenotype map. For example, questions about epistasis and effect size distributions are explicit inquiries into the phenotypic consequences of mutations during evolution. Selection also acts on phenotypes, so the topology of the genotype–phenotype space that surrounds two sequences determines what forms of selection could drive a trajectory between them. Further, statistical inferences of evolutionary processes are prone to artefacts caused by other processes, such as changing population size60,61. Without experimental evidence of the functional or phenotypic impacts of mutations to corroborate such signatures, sequence-based statistical inferences remain thin and potentially misleading32,62-64.
Evolutionary biochemistry can help to resolve these problems. What were the functionally important sequence substitutions during evolution? What physical mechanisms mediated their effects? How did the constraints and opportunities imposed by the genotype–phenotype map shape the evolution of the protein? A mechanistic strategy that addresses these questions can lead to a rich and complete account of the evolutionary events, processes and forces by which biological molecules acquired new properties (BOXES 1,2). This kind of approach has now been used to experimentally investigate many classic ideas in evolutionary biology — including adaptation31, parallel evolution32, epistasis33,34, adaptive constraint35, contingency36 and reversibility37 — and then to account for the results in mechanistic terms.
Insights from evolutionary biochemistry
Why are proteins marginally stable?
By directly studying both the genotype–phenotype space and the trajectories across it, evolutionary biochemistry can provide insights into evolutionary history that are individually inaccessible to either field. One example is our understanding of marginal protein stability. Biochemists long ago observed that most proteins are only slightly above the energetic threshold of unfolding65, and they can be further stabilized by simple amino acid replacements66. Many researchers assumed that this ‘marginal stability’ results from natural selection optimizing an intrinsic trade-off between stability and function67-69.
An evolutionary biochemical approach, however, revealed a different explanation for this near-universal property of proteins. Directed evolution studies generated enzymes that were both hyperstable and hyperfunctional70,71, indicating that the trade-off was not obligatory. Computational studies of protein folding and evolution then showed that marginal stability can arise neutrally through mutation–selection balance. If excess stability neither improves nor impairs function, selection will not distinguish between marginal and hyperstable proteins. Because there are many more ways for a protein to be marginally stable than for it to be hyperstable, mutational pressure and genetic drift will then neutrally drive proteins to occupy the most numerous set of states — the sequences with the lowest stabilities — that are compatible with their function72-74.
Thus, marginal stability need not be the optimal result of natural selection; rather, it will naturally arise as proteins evolve across sequence space owing to mutation, drift and the inability of selection to distinguish between hyperstable and ‘stable enough’. This explanation transcended the narrow confines of biochemistry and evolutionary biology and set the stage for future investigations of how changes in protein stability can limit or facilitate evolutionary change54,75-80.
Parallelism and constraints
Studying the physical constraints that shape protein function evolution is another fruitful intersection between evolutionary biology and biochemistry. Proteins must satisfy various constraints, including: rapid and correct folding55,81,82, thermodynamic stability83,84, solubility54 and maintaining specific functions. Only some sequences are compatible with the constraints that are important for each protein. Identifying those constraints and how they map onto sequence space are key questions in biochemistry, but they are hard to answer in the abstract because of the vast size of protein sequence space.
Cases of parallel evolution in nature can provide strong information about molecular constraints. When similar phenotypes independently evolve in different lineages under similar selection pressures, identical or different mutations may be involved each time. Repeated acquisition of the same underlying mutations indicates that constraints strongly limit the set of accessible sequences that can produce the selected phenotype. Dissecting the genetic causes of parallel evolution and identifying the constraints that have shaped this process can therefore reveal the underlying determinants of the physical and functional properties of a protein. It can also shed light on a classic evolutionary question: how repeatable, predictable and deterministic is evolution, and what factors make it that way85-87?
In a remarkable number of cases, parallel evolution has occurred by the repeated acquisition of precisely the same mutations88-93, sometimes in the very same order78,94-99. For example, five lineages of birds that have adapted to high altitudes have independently evolved haemoglobin with high oxygen affinity through the same key substitution at a key protein–protein interface89. Rats and mice exposed to warfarin on different continents have independently evolved the same mutations in the vitamin K epoxide reductase complex90. HIV-1 viral proteins follow predictable evolutionary trajectories in patients treated with anti-retrovirals91. Opsins evolved for deep-water environments with the same set of mutations eight different times32 (FIG. 1a). The malaria-causing parasite has independently evolved resistance to the drug chloroquine five times, always through the same parallel mutation in the binding pocket of a transporter protein92 (FIG. 1b-d).
Figure 1. Parallel evolution due to biophysical constraints.

a | Distribution of mutations observed during the evolution of the visual pigment opsin in fish. Opsin absorbs light at specific wavelengths through its bound retinal (yellow). Mutations can alter its absorption properties by changing the environment of the chromophore. Spheres highlight residues that changed as fish adapted to different light environments. Large-effect mutations acquired in parallel on multiple lineages (green) border the retinal; small-effect, lineage-specific mutations (orange) are more distant32. b-d | Mechanism for the parallel evolution of herbicide resistance via the same mutation in 68 different species of weeds162. b | Crystal structure of one half of the symmetrical multi-subunit complex of photosystem II (PSII). The site of the Ser264Gly mutation, which confers resistance, is shown in green, and the endogenous cofactor plastoquinone is yellow. c | Cofactor plastoquinone forms hydrogen bonds to Ser264 and to His215. d | The herbicide terbutryn (shown in yellow) directly hydrogen bonds only to Ser264. The Ser264Gly mutation abolishes all hydrogen bonds from the side chain of this residue, radically reducing terbutryn binding while only partially compromising plastoquinone binding. No other mutations are known that affect herbicide resistance without having a concomitantly large effect on plastoquinone binding.
In each of these cases, the parallel mutations were of large effect and directly occurred at the functional site of the protein (that is, the catalytic site of enzymes, the binding site for ligand interactions, and so on). Genetic parallelism occurred because these sites — and only these sites — bring about the functional change while satisfying other constraints. For example, 68 different weed species independently accrued the same resistance-inducing point mutation after exposure to triazine herbicides93 (FIG. 1b). The mutation occurs in a subunit of photosystem II (PSII), to which the herbicide binds and then disrupts photosynthesis. A high-fitness phenotype requires resistance to the pesticide to evolve without compromising the essential functions of PSII in photosynthesis. The site at which the parallel mutation occurs is just one of ~2,500 residues in the PSII complex, but it makes the only side-chain hydrogen bond between the protein and the herbicide; additional interactions mediate binding to the endogenous cofactor ligand of the protein. The parallel mutation therefore reduces affinity for the herbicide, but it does not strongly disrupt binding of the cofactor.
Although large-effect parallel mutations often occur, they are rarely sufficient to achieve the full parallel phenotype. Instead, they are almost always accompanied by other mutations of smaller effect, located farther from the functional site100. These ‘secondary’ mutations typically do not occur in parallel but instead affect different sites and/or states among lineages, even when the large-effect mutations are parallel32,89,90,93,101. This distribution implies fewer constraints on their location and mechanisms than on the large-effect mutations97. The underpinnings by which these ‘indirect’ mutations act usually fine-tune the derived function28,102,103 by more subtly optimizing interactions among atoms or by compensating for deleterious effects on stability or other properties caused by the large-effect mutation (or mutations)101,104,105. The lack of parallelism among these smaller-effect mutations indicates weaker constraints and more generic physical mechanisms.
Epistasis: physical and genetic interactions
Another interest shared by evolutionary biologists and biochemists is how interactions among amino acids determine protein functions and their evolution. Protein properties arise from complex physical interactions among residues, leading to strong epistasis in the genetic basis of protein structure106,107, thermodynamic stability99,108,109, substrate specificity36,110, allostery111,112 and function33,34,113,102,114,115. This epistasis makes the genotype–phenotype map rugged, in the sense that different mutational pathways to the same location in sequence space pass through proteins with very different properties. For example, a mutation that in one genetic context enhances some function may radically impair that function if introduced in a different context. Epistasis can therefore profoundly affect the ability of evolutionary forces to drive proteins through genotype–phenotype space, so understanding epistasis from a mechanistic standpoint sheds light on the nature and causes of evolutionary dynamics. Conversely, analysis of the co-evolutionary signal among interacting residues in present-day proteins has revealed ‘rules’ underlying genotype–phenotype space; these rules are sufficient to design foldable proteins de novo and proteins with new functional specificities, which are two particularly challenging goals in biochemistry112,116.
Permissive mutations in evolution
The recent discovery of permissive epistatic mutations, for example, has important evolutionary implications. Permissive mutations have no effect when introduced singly but are required for one or more other mutations to change the function of a protein36. Because permissive mutations are functionally silent, they cannot be driven by selection for the derived function and therefore introduce an element of contingency into the evolutionary process.
In some cases, permissive mutations cause another mutation, which would otherwise have been functionally silent, to have major functional effects28,92,102,117,118. For example, one study characterized the effects of historical mutations in ancestral GFP-like proteins from corals. One historical substitution at a key residue was essential for a shift from green to red fluorescence, but it had no effect unless three other historical substitutions — which by themselves did not change the colour — were introduced first102. Epistasis arose because the side chain of the derived amino acid at the key site is autocatalytically incorporated into the red fluorophore, but this reaction cannot occur unless the other substitutions have tuned the chemistry of the local environment. Thus one consequence of the biophysical architecture of fluorescence in these proteins was to make selection for red fluorescence insufficient to drive the acquisition of that phenotype.
In other cases, permissive mutations allow a protein to tolerate function-switching mutations that would otherwise be strongly deleterious. Mutations that confer new functions often also compromise the stability, solubility and affinity for partner molecules of a protein, among other properties36,79,93,107,114,119. Under most conditions, purifying selection effectively removes nonfunctional or poorly functional variants from a population46, so trajectories to the new function are blocked. Permissive mutations, however, create a genetic background in which the function-switching mutations can be tolerated. Numerous examples of permissive mutations of this type have been documented in natural evolution36,100,114,115. They have also been shown to facilitate the evolution of new enzyme functions in directed evolution experiments76,77,120. In each case, the permissive mutations compensated for deleterious effects that the function-switching mutations in isolation would have caused.
Mechanisms of epistasis
Genetic epistasis arises from physical causes; understanding those causes can illuminate how the architecture of a protein determines the topology of its genotype–phenotype space and thereby affects its evolutionary dynamics. The physical mechanisms underlying permissive mutations fall into two broad classes. The first is nonspecific epistasis between permissive and function-changing mutations; this occurs because of offsetting effects on some global property of a protein. For example, function-switching mutations often compromise stability119,121 (FIG. 2Aa). Stabilizing mutations, however, may buffer a protein against these destabilizing effects, thus opening an evolutionary trajectory to the new function76,77,120,118. For example, bacteria evolved resistance to cephalosporin antibiotics by expansion of the active site of β-lactamase, which breaks down the drug. On their own, these mutations increase the activity of the protein but compromise its stability — leading to low resistance — but high resistance evolved only after a stabilizing mutation distant from the active site also occurred122. Although nonspecific epistasis is usually discussed for thermodynamic stability, it can also arise for other global properties, such as solubility, aggregation and folding rate123.
Figure 2. Molecular mechanisms of evolutionary epistasis.

A | Epistasis mediated by effects on global stability. Aa | The schematic shows the effects on the evolution of a new function (blue) of two interacting mutations (green and purple) with different effects on stability and function. Proteins with stabilities below a given threshold are unstructured and non-functional. Ab | Epistasis in the evolution of bacterial resistance to the antibiotic ceftazidime is mediated by effects on global stability122. Analysis of the major-effect mutations shows that stability modulates resistance. Each platform is an allele; its location along the y axis shows its melting temperature of unfolding (Tm). Bar graphs show the enzymatic activity of each allele (Kcat/KM) relative to the ancestral protein (yellow bar) and antibiotic resistance (blue bar, measured as inverse halo diameter). The E104K and G238S mutations (purple spheres in the structure) confer high enzymatic activity but low resistance because the protein is unstable. The distant mutation M182T (green sphere) confers high stability by addition of a new hydrogen bond but does not change activity. Their combination yields resistance. The antibiotic is shown as yellow spheres. B | Specific epistasis mediated by a direct interaction. Ba | A schematic showing a direct, physical conformational change (blue). Bb | An example of direct epistasis from engineered Streptococcus spp. protein G domains that differ at two residues but have radically different folds106. These residues form a packing interaction only when both are aromatic residues, driving the transition between folds. C | Specific epistasis indirectly mediated by a conformational change36. Ca | The schematic shows how two mutations that do not physically interact can genetically interact in the evolution of a new function. One mutation creates the potential for a new interaction (green), which is realised only if the first residue is repositioned by a conformational change triggered by the other mutation (purple). Cb | An example of conformational epistasis from the evolution of ligand sensitivity in the vertebrate glucocorticoid receptor36. Crystal structure of the ancestral (orange) and derived (blue) forms of the glucocorticoid receptor. Novel specificity for glucocorticoid ligand (yellow) evolved because of the interaction of historical substitutions L111Q (green), which introduces a hydrogen bond acceptor, and S106P (purple), which repositioned the helix on which the L111Q is located (arrows), allowing L111Q to form a novel hydrogen bond with the ligand.
A particularly compelling example of nonspecific permissive epistasis occurred during the evolution of drug resistance in H1N1 influenza114. The antiviral oseltamivir targets the neuraminidase protein of H1N1. A single point mutation in neuraminidase was discovered years ago that could reduce the affinity of the protein for the drug and produce resistance124; however, this mutation never evolved naturally because it also severely compromises the ability of the protein to fold and to reach the surface of the cell, so overall it decreased viral fitness, even in the presence of the drug125. By 2008, however, a resistant strain carrying the mutation became widespread. Careful phylogenetic and experimental analyses showed that two permissive substitutions had taken place in the neuraminidase gene during the previous years. These mutations had no effect on the drug resistance of the virus, but they increased the amount of protein reaching the cell surface. After these permissive mutations were in place, the resistance-inducing mutation could be tolerated and natural selection in the presence of the drug drove it to fixation114. Another recent study reconstructed the precise mutational trajectory taken by a viral coat protein in a different influenza strain and found that numerous permissive stabilizing mutations, which occurred early in the trajectory, allowed the virus to tolerate later destabilizing mutations that — after they could be tolerated — apparently promoted escape from the host’s immune system126.
The second class of permissive mutations acts much more specifically: two or more mutations directly cooperate to change the properties of the protein (FIG. 2Ab). A remarkable recent study identified mutations in an engineered gene that do not affect the conformation of the encoded protein when introduced individually, but when introduced together, they lead to formation of a hydrophobic interaction in the protein that causes a discrete switch to an entirely new fold106 (FIG. 2Ab). Specific epistasis can also occur between mutations that do not contact each other (FIG. 2B), so long as the effect of one depends on the state at a different specific site. For example, two historical substitutions cooperated to alter the hormone specificity of the glucocorticoid receptor. One substitution introduced a hydrogen bond donor on an inward-facing helix — causing no effect on function — but a second mutation shifted the helix, relocating the other site to form a ligand-specific hydrogen bond36 (FIG. 2B). Only when both mutations occur together can specificity be achieved. In both cases, the epistasis is specific in the sense that only one (or a few) possible mutations can interact to open the trajectory to the new form and function of the protein.
The relative frequency of nonspecific versus specific epistasis in evolutionary transitions is an important open question. The two mechanisms have profoundly different implications for the role of chance in protein evolution. A nonspecific permissive mutation might open pathways for many different function-switching mutations and, conversely, a function-switching mutation that interacts nonspecifically could be allowed by many different permissive mutations127,128; the effects of nonspecific epistasis on the ultimate outcomes of evolution may therefore be fairly weak. Specific epistasis, however, suggests that a certain evolutionary transition in function might be allowed by only a small set of permissive mutations, making evolutionary outcomes strongly contingent on the low-probability accumulation of mutations that cannot be fixed by selection for the derived function itself. Determining the relative frequency of the two modes of permissive mutation will require more mechanistic studies of epistatic evolutionary trajectories.
Contingency, predictability and optimality
Chance and determinism in evolution
The previous sections present a puzzle. Many proteins display strong patterns of parallel evolution, amassing the same mutations in response to selection given their physical constraints; protein evolution therefore seems to be predictable and deterministic94. However, mutations that do not alter the function of the protein are often required to open evolutionary paths, suggesting that evolutionary trajectories are often contingent on chance events that are invisible to selection; protein evolution therefore seems to be unpredictable and unlikely to be repeated36. How can these two perspectives be reconciled?
A closer look reveals that these findings are compatible with each other. The set of mutational pathways available to a protein because of epistasis and constraints depends on its position in a neutral network in sequence space129 (FIG. 3). These networks appear to be vast: some protein families contain sequences that have little discernable homology but maintain the same fold130-132 and even the same function133. Further, saturation mutagenesis studies have shown that proteins can tolerate changes at many positions without compromising their conformation or function27,50,134-138. Thus, although proteins within neutral networks by definition have similar folds and functions, they may have different sequences, and the effects of mutations on them may be different.
Figure 3. The position of a protein in its neutral network determines which mutational path it takes to a derived function.

Protein sequences (ovals) are connected by point mutations (arrows). The colours represent functions: ancestral (orange) or derived (blue). Transitional colours represent transitional functions. Nearby sequences in the ancestral neutral network follow the same ‘deterministic’ pathways (dark arrows) when selection for the derived function is applied. Some sequences in the neutral network cannot achieve the derived function without first taking a permissive functionally neutral step through the network (ovals with dashed outlines).
A particularly striking example of the variable effects of mutation on sequences within a neutral network comes from an elegant experiment using the enzyme isopropymalate dehydrogenase (IMDH) of two bacterial species. The two proteins differ at 168 of 365 sites, but their structures are nearly identical, and their enzymatic activities are comparable. The authors substituted each of the 168 residues that differed between the homologues and individually substituted them from one protein into the other. They then characterized the activities of these chimeric enzymes. Thirty-eight per cent of these cross-substitutions radically compromised IMDH activity34. This finding indicates that many amino acid states that are fully compatible with the function of the enzyme in one sequence context are incompatible when introduced into the context of a functionally indistinguishable related protein.
When two similar sequences are subject to selection for some function, they have available to them a largely shared set of mutational trajectories, because they are subject to the same constraints and epistatic interactions. The result is a set of repeatable and apparently deterministic evolutionary outcomes. By contrast, sequences further away from each other in the neutral network may be subject to different constraints and genetic interactions, so a mutational path that produces a selected phenotype in one background may not do so in the other. Under these circumstances, the proteins may follow different evolutionary pathways in response to selection.
Several case studies support the view that different outcomes are realized from different starting points97,139,140. For example, when treated with the antiviral drug nelfinavir, one variant of HIV-1 protease reproducibly acquires the same set of mutations to achieve resistance. A different variant, which differs from the first by only 6 of 99 positions (and which has only a slight difference in affinity for the drug), repeatedly acquires resistance through a different mutation141,142. In another example, six closely related species of Andean ducks that independently colonized highaltitude environments accumulated a key mutation at the same amino acid site, increasing haemoglobin oxygen affinity by destabilizing the deoxy state89. The more distantly related Asian bar-headed goose, by contrast, also evolved increased oxygen affinity, but it did so via a different substitution with a similar physical mechanism21,143. Thus, evolution under natural selection appears to be fairly deterministic when it is launched from similar starting points, but over long periods of time sequences may travel nondeterministically to different nodes in sequence space that have very different evolutionary potentials.
Optimality and ‘design principles’
The interplay between chance moves in the neutral network and the predictable evolution of a given protein sequence in response to selection suggests that few protein properties are likely to represent optimal states. Although natural selection efficiently climbs accessible fitness peaks, the specific uphill pathway that is available — and thus the endpoint that it eventually occupies — is determined by the starting sequence of the protein. Because the starting sequence within a neutral network is determined largely by chance, there is no reason to suppose that the peak that is actually climbed is a globally optimal state.
This perspective means that we should be careful about extracting ‘design principles’ from natural proteins to assist with protein engineering144-146. The sequences and physical properties of natural proteins were not designed, however, so the only ‘design principles’ are those that emerge from the evolutionary process, which may often not produce optimal forms. The forms and functions of proteins are shaped by the peculiarities of their history147: namely, the interplay of common descent, physical and biological constraints, shifting genetic interactions and random mutations, all of which work together to open or close the pathways accessible to selection at any moment in evolution.
Prospects for a new field
The studies we have reviewed reveal a deep interplay between evolutionary processes and the biochemical properties of genotype–phenotype space. Although insights are emerging from evolutionary biochemistry, many questions remain unresolved. A first priority is to conduct many more case studies in order to determine the generality of findings made thus far and to reveal new mechanisms not yet observed.
Future directions for evolutionary biochemistry
Developing new strategies for studying evolutionary biochemistry will also allow entirely new questions at the interface of the fields to be addressed. One exciting avenue is to characterize the effects of the topology of sequence space on historical evolution. This issue can be illuminated by combining historical approaches such as ancestral protein reconstruction with directed evolution and high-throughput methods to assess the functions and evolvability of large mutant libraries of ancestral (or extant) proteins. How big were the neutral networks associated with a given protein property, and how dense are the connections between them? Were the connections uniformly distributed, or did narrow ‘wormholes’ of specific mutational combinations connect otherwise isolated island subnetworks into archipelagos? How many different mutational combinations would have allowed the new function to evolve, and would the physical mechanisms have differed? When permissive mutations were required, how many potentially permissive mutations could there have been, and what physical constraints limited their number? Answering these questions will help to characterize not only the trajectory that evolution did take but also alternative ‘might-have-been’ histories, thus providing direct insight into contingency, determinism and causes of sequence conservation and divergence.
Work thus far in evolutionary biochemistry has primarily addressed evolutionary changes in existing protein properties, such as shifts in ligand–substrate specificity, or bulk properties, such as thermodynamic stability. The mechanisms and dynamics by which new folds, functions, catalytic activities and modes of allosteric regulation originate in the first place have not been experimentally addressed. These phenomena present a rich set of questions with major implications for both biochemistry and molecular evolution.
For evolutionary biology, an exciting goal is to link research across very broad scales, from the specific atom-level mechanisms that mediate the shifts in molecular function to the population genetic processes that drive phenotypic evolution in real-world environments. There have already been some efforts to link population processes in an environmental context to underlying genetic changes and their effects on development and physiology31,148-152. Biochemical studies could extend such studies to the most fundamental level, providing a complete mechanistic linkage of evolutionary change across levels, from mutations in sequence to shifts in protein structure and function, and onwards to changes in phenotype, fitness and the composition of populations31.
Fostering evolutionary biochemistry
Work at the biochemistry–evolution interface often falls through the cracks between institutional programmes that are focused on traditional approaches within each discipline. With funding streams becoming ever tighter, for example, it is hard for even the most compelling interdisciplinary research to compete against mainstream work when grant evaluation processes are organized along disciplinary boundaries. To foster the development of this young field, then, funding agencies should earmark funds to support research in evolutionary biochemistry. One model is the kind of dedicated interdisciplinary funding programs that allowed research efforts in the evolution of development to prosper over the past two decades.
Preparing young scientists to work at the interface is particularly important for the development of the field. We know of no programmes that provide or even encourage graduate training in both protein biochemistry (such as physical chemistry and structural biology) and evolutionary biology (such as population genetics, phylogenetics and molecular evolution). Support from universities and funding agencies to develop interdisciplinary training programmes would help greatly to bridge the intellectual gulf that has separated the fields.
Finally, scientists working at the interface need space to talk to each other. Scientific meetings — both new meetings devoted to evolutionary biochemistry and dedicated sessions within the core meetings of each discipline — could provide such a venue. Evolutionary biochemists also need the opportunity to present their work in the pages of the strongest journals in the fields.
The ultimate goal should be for evolutionary biochemistry to become more than a novelty item within each field but rather a canonical element of each discipline’s body of knowledge, concepts and approaches. Understanding why proteins have the properties they do cannot be achieved by biochemists or evolutionary biologists alone. Achieving this goal requires us to transcend arbitrary historical divisions and to treat proteins as integrated physical and historical wholes.
Acknowledgements
This work was supported by US National Institutes of Health Grants R01-GM081592 , R01-GM104397 and F32-GM090650, as well as by the Howard Hughes Medical Institute. The authors thank A. Drummond, T. Dean and members of the Thornton laboratory for helpful comments.
Glossary
- Biochemistry
The study of the chemical and physical properties of biological molecules and how those properties determine the functions of each molecule. Defined this way, biochemistry also includes structural biology, biophysics and some areas of molecular and computational biology.
- Molecular clock
The hypothesis that, over long timescales, mutations accumulate at a characteristic rate for each gene. For genes with clock-like evolution, the proportion of sequence differences between related genes can be used to estimate the time since they diverged.
- Ancestral protein reconstruction
The use of statistical phylogenetic methods to infer ancestral protein sequences from large alignments of present-day proteins, followed by synthesis, expression and experimental characterization of the ‘resurrected’ ancestral proteins.
- Homology
Similarity due to descent from a shared common ancestral form.
- Protein stability
A thermodynamic description of the difference in free energy between the folded and unfolded states of a protein.
- Parallel evolution
The repeated acquisition of the same phenotype on different lineages under similar forms of selection.
- Epistasis
Dependency of the phenotypic effects of a mutation on the genetic state at other sites in the same or other loci.
- Sequence signatures
Patterns in groups of protein or DNA sequences — such as the relative frequency of synonymous and nonsynonymous mutations or the degree of genetic diversity within and between populations — that are interpreted as reflecting specific evolutionary processes.
- Directed evolution
A laboratory procedure for identifying genotypes with a desired property by iteratively introducing random mutations into a protein and using chemical or biological means to select for variants in which the property is improved.
- Mutation–selection balance
Equilibrium between the accumulation of variation in a population due to ongoing mutation and the removal of variation due to purifying selection.
- Genetic drift
Changes in the frequency across generations of genotypes in populations due to stochastic factors.
- Neutral network
A set of protein sequences that are connected to each other by single amino acid replacements and have similar enough functions and physical properties that selection does not distinguish among them.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Anfinsen C. Molecular Basis of Evolution. John Wiley & Sons; 1959. This is a prescient early attempt by a Nobel-prize-winning biochemist to consider how chemistry might shape protein evolution.
- 2.Florkin M. Biochemical Evolution. Academic Press; 1949. [Google Scholar]
- 3.Zuckerkandl E, Pauling L. Molecules as documents of evolutionary history. J. Theor. Biol. 1965;8:357–366. doi: 10.1016/0022-5193(65)90083-4. [DOI] [PubMed] [Google Scholar]
- 4.Zuckerkandl E, Pauling L. Evolving Genes and Proteins. Bryson; 1965. Two chemists defend the potential contributions of biochemistry to evolutionary knowledge at a 1964 conference that brought molecular biologists and classical evolutionary biologists together.
- 5.Pauling L, Zuckerkandl E. Chemical paleogenetics: molecular ‘restoration studies’ of extinct forms of life. Acta Chem. Scand. 1963;17:S9–S16. [Google Scholar]
- 6.Ingram VM. Gene evolution and the haemoglobins. Nature. 1961;189:704–708. doi: 10.1038/189704a0. [DOI] [PubMed] [Google Scholar]
- 7.Wald G. Phylogeny and ontogeny at the molecular level. Evol. Biochem. 1963;3:12–51. [Google Scholar]
- 8.Dietrich MR. Paradox and persuasion: negotiating the place of molecular evolution within evolutionary biology. J. Hist. Biol. 1998;31:85–111. doi: 10.1023/a:1004257523100. [DOI] [PubMed] [Google Scholar]
- 9.Morgan GJ. Emile Zuckerkandl, Linus Pauling, and the molecular evolutionary clock, 1959-1965. J. Hist. Biol. 1998;31:155–178. doi: 10.1023/a:1004394418084. [DOI] [PubMed] [Google Scholar]
- 10.Aronson JD. ‘Molecules and monkeys’: George Gaylord Simpson and the challenge of molecular evolution. Hist. Philos. Life Sci. 2002;24:441–465. doi: 10.1080/03919710210001714503. [DOI] [PubMed] [Google Scholar]
- 11.Simpson GG. The status of the study of organisms. Am. Scientist. 1962;50:36–45. [PubMed] [Google Scholar]
- 12.Simpson G. Organisms and molecules in evolution. Science. 1964;146:1535–1538. doi: 10.1126/science.146.3651.1535. [DOI] [PubMed] [Google Scholar]
- 13.Dobzhansky T. Biology, molecular and organismic. Am. Zool. 1964;4:443–452. doi: 10.1093/icb/4.4.443. [DOI] [PubMed] [Google Scholar]
- 14.Fitch WM. Homology: a personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/s0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- 15.Gould SJ, Lewontin RC. The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc. R. Soc. Lond. B. 1979;205:581–598. doi: 10.1098/rspb.1979.0086. [DOI] [PubMed] [Google Scholar]
- 16.Baum DA, Smith SD, Donovan SSS. The tree-thinking challenge. Science. 2005;310:979–980. doi: 10.1126/science.1117727. [DOI] [PubMed] [Google Scholar]
- 17.Watt WB. Allozymes in evolutionary genetics: self-imposed burden or extraordinary tool? Genetics. 1994;136:11–16. doi: 10.1093/genetics/136.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dean AM, Thornton JW. Mechanistic approaches to the study of evolution: the functional synthesis. Nature Rev. Genet. 2007;8:675–688. doi: 10.1038/nrg2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilke CO. Bringing molecules back into molecular evolution. PLoS Comput. Biol. 2012;8:e1002572. doi: 10.1371/journal.pcbi.1002572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blundell TL, Wood SP. Is the evolution of insulin Darwinian or due to selectively neutral mutation? Nature. 1975;257:197–203. doi: 10.1038/257197a0. [DOI] [PubMed] [Google Scholar]
- 21.Perutz MF. Species adaptation in a protein molecule. Mol. Biol. Evol. 1983;1:1–28. doi: 10.1093/oxfordjournals.molbev.a040299. This is the first article in the inaugural issue of Molecular Biology and Evolution. It lays out an agenda for experimental studies of protein evolution, using biochemical and structural studies of haemoglobin in a phylogenetic context as a template.
- 22.Malcolm BA, Wilson KP, Matthews BW, Kirsch JF, Wilson AC. Ancestral lysozymes reconstructed, neutrality tested, and thermostability linked to hydrocarbon packing. Nature. 1990;345:86–89. doi: 10.1038/345086a0. [DOI] [PubMed] [Google Scholar]
- 23.Serrano L, Day AG, Fersht AR. Step-wise mutation of barnase to binase. A procedure for engineering increased stability of proteins and an experimental analysis of the evolution of protein stability. J. Mol. Biol. 1993;233:305–312. doi: 10.1006/jmbi.1993.1508. [DOI] [PubMed] [Google Scholar]
- 24.Golding GB, Dean AM. The structural basis of molecular adaptation. Mol. Biol. Evol. 1998;15:355–369. doi: 10.1093/oxfordjournals.molbev.a025932. [DOI] [PubMed] [Google Scholar]
- 25.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nature Rev. Mol. Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peisajovich SG, Tawfik DS. Protein engineers turned evolutionists. Nature Meth. 2007;4:991–994. doi: 10.1038/nmeth1207-991. [DOI] [PubMed] [Google Scholar]
- 27.Hietpas RT, Jensen JD, Bolon DNA. Experimental illumination of a fitness landscape. Proc. Natl Acad. Sci. USA. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. A high-throughput experimental evolution study is presented that directly characterizes the distribution of fitness effects of a very large number of possible mutations in heat shock protein 90 (HSP90).
- 28.Yokoyama S, Yang H, Starmer WT. Molecular basis of spectral tuning in the red- and green-sensitive (M/LWS) pigments in vertebrates. Genetics. 2008;179:2037–2043. doi: 10.1534/genetics.108.090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harms MJ, Thornton JW. Analyzing protein structure and function using ancestral gene reconstruction. Curr. Opin. Struct. Biol. 2010;20:360–366. doi: 10.1016/j.sbi.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brustad EM, Arnold FH. Optimizing non-natural protein function with directed evolution. Curr. Opin. Chem. Biol. 2011;15:201–210. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Storz JF, et al. Evolutionary and functional insights into the mechanism underlying high-altitude adaptation of deer mouse hemoglobin. Proc. Natl Acad. Sci. USA. 2009;106:14450–14455. doi: 10.1073/pnas.0905224106. This multifaceted study links ecological context and population-level variation in haemoglobin allele frequencies to the experimentally measured oxygen affinity of those alleles.
- 32.Yokoyama S, Tada T, Zhang H, Britt L. Elucidation of phenotypic adaptations: molecular analyses of dim-light vision proteins in vertebrates. Proc. Natl Acad. Sci. USA. 2008;105:13480–13485. doi: 10.1073/pnas.0802426105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics. 2010;185:293–303. doi: 10.1534/genetics.109.112458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lunzer M, Golding GB, Dean AM. Pervasive cryptic epistasis in molecular evolution. PLoS Genet. 2010;6:e1001162. doi: 10.1371/journal.pgen.1001162. This elegant experiment demonstrates that functionally equivalent, orthologous proteins can have different tolerances for identical mutations.
- 35.Miller SP, Lunzer M, Dean AM. Direct demonstration of an adaptive constraint. Science. 2006;314:458–461. doi: 10.1126/science.1133479. [DOI] [PubMed] [Google Scholar]
- 36.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317:1544–1548. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bridgham JT, Ortlund EA, Thornton JW. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–519. doi: 10.1038/nature08249. References 36 and 37 describe the first experimental identification of permissive and restrictive mutations, which open and close evolutionary trajectories despite being functionally neutral themselves; this paper also reports the first X-ray crystallographic structures of reconstructed ancestral proteins.
- 38.Berkhout B, Klaver B, Das A. Forced evolution of a regulatory RNA helix in the HIV-1 genome. Nucl. Acids Res. 1997;25:940–947. doi: 10.1093/nar/25.5.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burch CL, Chao L. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature. 2000;406:625–628. doi: 10.1038/35020564. [DOI] [PubMed] [Google Scholar]
- 40.Hayden EJ, Ferrada E, Wagner A. Cryptic genetic variation promotes rapid evolutionary adaptation in an RNA enzyme. Nature. 2011;474:92–95. doi: 10.1038/nature10083. [DOI] [PubMed] [Google Scholar]
- 41.Cheng N, Mao Y, Shi Y, Tao S. Coevolution in RNA molecules driven by selective constraints: evidence from 5S rRNA. PLoS ONE. 2012;7:e44376. doi: 10.1371/journal.pone.0044376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldstein RA. The structure of protein evolution and the evolution of protein structure. Curr. Opin. Struct. Biol. 2008;18:170–177. doi: 10.1016/j.sbi.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Zeldovich KB, Shakhnovich EI. Understanding protein evolution: from protein physics to Darwinian selection. Annu. Rev. Phys. Chem. 2008;59:105–127. doi: 10.1146/annurev.physchem.58.032806.104449. [DOI] [PubMed] [Google Scholar]
- 44.Wright S. Proceedings of the Sixth International Congress of Genetics; 1932.pp. 356–366. [Google Scholar]
- 45.Dobzhansky T. Genetics and the Origin of Species. Columbia Univ. Press; 1937. [Google Scholar]
- 46.Smith JM. Natural selection and the concept of a protein space. Nature. 1970;225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 47.Gavrilets S. Evolution and speciation on holey adaptive landscapes. Trends Ecol. Evol. 1997;12:307–312. doi: 10.1016/S0169-5347(97)01098-7. [DOI] [PubMed] [Google Scholar]
- 48.McGhee GR. The Geometry of Evolution: Adaptive Landscapes and Theoretical Morphospaces. Cambridge Univ. Press; 2006. [Google Scholar]
- 49.Carneiro M, Hartl DL. Colloquium paper: adaptive landscapes and protein evolution. Proc. Natl Acad. Sci. USA. 2009;107:1747–1751. doi: 10.1073/pnas.0906192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fowler DM, et al. High-resolution mapping of protein sequence-function relationships. Nature Meth. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harms MJ, et al. Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. Proc. Natl Acad. Sci. USA. 2013 doi: 10.1073/pnas.1303930110. http://dx.doi.org/10.1073/pnas.1303930110. This paper presents an evolutionary biochemical study that uses ancestral reconstruction to identify two historical substitutions that cause a massive historical shift in binding specificity in the steroid receptors. It then follows up with detailed biophysical investigations of the mechanism of the transition.
- 52.Gruebele M. Downhill protein folding: evolution meets physics. Comp. Rend. Biol. 2005;328:701–712. doi: 10.1016/j.crvi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Rose GD, Fleming PJ, Banavar JR, Maritan A. A backbone-based theory of protein folding. Proc. Natl Acad. Sci. USA. 2006;103:16623. doi: 10.1073/pnas.0606843103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc. Natl Acad. Sci. USA. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geiler-Samerotte KA, et al. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc. Natl Acad. Sci. USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serohijos AWR, Rimas Z, Shakhnovich EI. Protein biophysics explains why highly abundant proteins evolve slowly. Cell Rep. 2012;2:249–256. doi: 10.1016/j.celrep.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hughes AL. Looking for Darwin in all the wrong places: the misguided quest for positive selection at the nucleotide sequence level. Heredity. 2007;99:364–373. doi: 10.1038/sj.hdy.6801031. [DOI] [PubMed] [Google Scholar]
- 58.Barrett RDH, Hoekstra HE. Molecular spandrels: tests of adaptation at the genetic level. Nature Rev. Genet. 2011;12:767–780. doi: 10.1038/nrg3015. [DOI] [PubMed] [Google Scholar]
- 59.Lewontin RC. Genetic Basis of Evolutionary Change. Columbia Univ. Press; 1974. [Google Scholar]
- 60.Eyre-Walker A. Changing effective population size and the McDonald-Kreitman test. Genetics. 2002;162:2017–2024. doi: 10.1093/genetics/162.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nielsen R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 62.Timpson N, Heron J, Smith GD, Enard W. Comment on papers by Evans et al. and Mekel-Bobrov et al. on evidence for positive selection of MCPH1 and ASPM. Science. 2007;317:1036–1036. doi: 10.1126/science.1141705. [DOI] [PubMed] [Google Scholar]
- 63.Zhuang H, Chien M-S, Matsunami H. Dynamic functional evolution of an odorant receptor for sex-steroid-derived odors in primates. Proc. Natl Acad. Sci. USA. 2009;106:21247–21251. doi: 10.1073/pnas.0808378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hopkins R, Levin DA, Rausher MD. Molecular signatures of selection on reproductive character displacement of flower color in Phlox drummondii. Evolution. 2012;66:469–485. doi: 10.1111/j.1558-5646.2011.01452.x. [DOI] [PubMed] [Google Scholar]
- 65.Pace CN. The stability of globular proteins. Crit. Rev. Biochem. 1975;3:1–43. doi: 10.3109/10409237509102551. [DOI] [PubMed] [Google Scholar]
- 66.Fersht AR, Serrano L. Principles of protein stability derived from protein engineering experiments. Curr. Opin. Struct. Biol. 1993;3:75–75. [Google Scholar]
- 67.Tang KES, Dill KA. Native protein fluctuations: the conformational-motion temperature and the inverse correlation of protein flexibility with protein stability. J. Biomol. Struct. Dynam. 1998;16:397–411. doi: 10.1080/07391102.1998.10508256. [DOI] [PubMed] [Google Scholar]
- 68.Dunker AK, Obradovic Z. The protein trinity— linking function and disorder. Nature Biotech. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 69.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nature Rev. Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 70.Giver L, Gershenson A, Freskgard PO, Arnold FH. Directed evolution of a thermostable esterase. Proc. Natl Acad. Sci. USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnold FH, Wintrode PL, Miyazaki K, Gershenson A. How enzymes adapt: lessons from directed evolution. Trends Biochem. Sci. 2001;26:100–106. doi: 10.1016/s0968-0004(00)01755-2. [DOI] [PubMed] [Google Scholar]
- 72.Taverna DM, Goldstein RA. Why are proteins marginally stable? Proteins Struct. Function Genet. 2002;46:105–109. doi: 10.1002/prot.10016. [DOI] [PubMed] [Google Scholar]
- 73.Goldstein RA. Computational Science — ICCS 2004. 2004. pp. 718–727. [Google Scholar]
- 74.Bloom JD, Raval A, Wilke CO. Thermodynamics of neutral protein evolution. Genetics. 2007;175:255–266. doi: 10.1534/genetics.106.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Godoy-Ruiz R, Perez-Jimenez R, Ibarra-Molero B, Sanchez-Ruiz JM. Relation between protein stability, evolution and structure, as probed by carboxylic acid mutations. J. Mol. Biol. 2004;336:313–318. doi: 10.1016/j.jmb.2003.12.048. [DOI] [PubMed] [Google Scholar]
- 76.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc. Natl Acad. Sci. USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. A directed evolution experiment is presented here that shows how increasing the stability of a protein makes it more ‘evolvable’ by offsetting the destabilizing effects of function-switching mutations.
- 77.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. This is a direct demonstration in a laboratory evolution experiment that epistasis can arise directly from stability thresholds.
- 78.Couñago R, Wilson CJ, Peña MI, Wittung-Stafshede P, Shamoo Y. An adaptive mutation in adenylate kinase that increases organismal fitness is linked to stability-activity trade-offs. Protein Eng. Des. Sel. 2008;21:19–27. doi: 10.1093/protein/gzm072. [DOI] [PubMed] [Google Scholar]
- 79.Tokuriki N, Stricher F, Serrano L, Tawfik DS. How protein stability and new functions trade off. PLoS Comput. Biol. 2008;4:e1000002. doi: 10.1371/journal.pcbi.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilke CO, Drummond DA. Signatures of protein biophysics in coding sequence evolution. Curr. Opin. Struct. Biol. 2010;20:385–389. doi: 10.1016/j.sbi.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Godoy-Ruiz R, et al. Natural selection for kinetic stability is a likely origin of correlations between mutational effects on protein energetics and frequencies of amino acid occurrences in sequence alignments. J. Mol. Biol. 2006;362:966–978. doi: 10.1016/j.jmb.2006.07.065. [DOI] [PubMed] [Google Scholar]
- 82.Worth CL, Gong S, Blundell TL. Structural and functional constraints in the evolution of protein families. Nature Rev. Mol. Cell Biol. 2009;10:709–720. doi: 10.1038/nrm2762. [DOI] [PubMed] [Google Scholar]
- 83.Schreiber G, Buckle AM, Fersht AR. Stability and function: two constraints in the evolution of barstar and other proteins. Structure. 1994;2:945–951. doi: 10.1016/s0969-2126(94)00096-4. [DOI] [PubMed] [Google Scholar]
- 84.Zeldovich KB, Chen P, Shakhnovich EI. Protein stability imposes limits on organism complexity and speed of molecular evolution. Proc. Natl Acad. Sci. USA. 2007;104:16152–16157. doi: 10.1073/pnas.0705366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gould SJ. Wonderful Life: The Burgess Shale and the Nature of History. W. W. Norton & Company; 1990. [Google Scholar]
- 86.Losos JB, Jackman TR, Larson A, Queiroz K, de Rodriguez-Schettino L. Contingency and determinism in replicated adaptive radiations of island lizards. Science. 1998;279:2115–2118. doi: 10.1126/science.279.5359.2115. [DOI] [PubMed] [Google Scholar]
- 87.Blount ZD, Borland CZ, Lenski RE. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc. Natl Acad. Sci. USA. 2008;105:7899–7906. doi: 10.1073/pnas.0803151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rokas A, Carroll SB. Frequent and widespread parallel evolution of protein sequences. Mol. Biol. Evol. 2008;25:1943–1953. doi: 10.1093/molbev/msn143. [DOI] [PubMed] [Google Scholar]
- 89.McCracken KG, et al. Parallel evolution in the major haemoglobin genes of eight species of Andean waterfowl. Mol. Ecol. 2009;18:3992–4005. doi: 10.1111/j.1365-294X.2009.04352.x. [DOI] [PubMed] [Google Scholar]
- 90.Pelz H-J, et al. The genetic basis of resistance to anticoagulants in rodents. Genetics. 2005;170:1839–1847. doi: 10.1534/genetics.104.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menéndez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 2010;85:210–231. doi: 10.1016/j.antiviral.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 92.Martin RE, et al. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science. 2009;325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- 93.Powles SB, Yu Q. Evolution in action: plants resistant to herbicides. Annu. Rev. Plant Biol. 2010;61:317–347. doi: 10.1146/annurev-arplant-042809-112119. [DOI] [PubMed] [Google Scholar]
- 94.Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 95.Lozovsky ER, et al. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc. Natl Acad. Sci. USA. 2009;106:12025–12030. doi: 10.1073/pnas.0905922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brown KM, et al. Compensatory mutations restore fitness during the evolution of dihydrofolate reductase. Mol. Biol. Evol. 2010;27:2682–2690. doi: 10.1093/molbev/msq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Costanzo MS, Brown KM, Hartl DL. Fitness trade-offs in the evolution of dihydrofolate reductase and drug resistance in Plasmodium falciparum. PLoS ONE. 2011;6:e19636. doi: 10.1371/journal.pone.0019636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Counago R, Chen S, Shamoo Y. In vivo molecular evolution reveals biophysical origins of organismal fitness. Mol. Cell. 2006;22:441–449. doi: 10.1016/j.molcel.2006.04.012. This is a laboratory demonstration of the capacity of biophysical constraints to cause the parallel accumulation of identical mutations in independent lineages.
- 99.Miller C, et al. Experimental evolution of adenylate kinase reveals contrasting strategies toward protein thermostability. Biophys. J. 2010;99:887–896. doi: 10.1016/j.bpj.2010.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Davis BH, Poon AFY, Whitlock MC. Compensatory mutations are repeatable and clustered within proteins. Proc. R. Soc. B. 2009;276:1823–1827. doi: 10.1098/rspb.2008.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Summers RL, Nash MN, Martin RE. Know your enemy: understanding the role of PfCRT in drug resistance could lead to new antimalarial tactics. Cell. Mol. Life Sci. 2012 doi: 10.1007/s00018-011-0906-0. http://dx.doi.org/10.1007/s00018-011-0906-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Field SF, Matz MV. Retracing evolution of red fluorescence in GFP-like proteins from faviina corals. Mol. Biol. Evol. 2010;27:225–233. doi: 10.1093/molbev/msp230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tokuriki N, et al. Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme. Nature Commun. 2012;3:1257. doi: 10.1038/ncomms2246. [DOI] [PubMed] [Google Scholar]
- 104.Nijhuis M, et al. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS. 1999;13:2349–2359. doi: 10.1097/00002030-199912030-00006. [DOI] [PubMed] [Google Scholar]
- 105.Maisnier-Patin S, Andersson DI. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 2004;155:360–369. doi: 10.1016/j.resmic.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 106.Alexander PA, He Y, Chen Y, Orban J, Bryan PN. A minimal sequence code for switching protein structure and function. Proc. Natl Acad. Sci. USA. 2009;106:21149–21154. doi: 10.1073/pnas.0906408106. This is an amazing demonstration of epistasis in protein folding, in which a mutation that is merely destabilizing in some genetic backgrounds drives a transition to an entirely different fold in another background.
- 107.Lynch VJ, May G, Wagner GP. Regulatory evolution through divergence of a phosphoswitch in the transcription factor CEBPB. Nature. 2011;480:383–386. doi: 10.1038/nature10595. [DOI] [PubMed] [Google Scholar]
- 108.Green SM, Shortle D. Patterns of nonadditivity between pairs of stability mutations in staphylococcal nuclease. Biochemistry. 1993;32:10131–10139. doi: 10.1021/bi00089a032. [DOI] [PubMed] [Google Scholar]
- 109.LiCata VJ, Ackers GK. Long-range, small magnitude nonadditivity of mutational effects in proteins. Biochemistry. 1995;34:3133–3139. doi: 10.1021/bi00010a001. [DOI] [PubMed] [Google Scholar]
- 110.O’Maille PE, et al. Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nature Chem. Biol. 2008;4:617–623. doi: 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature Struct. Mol. Biol. 2002;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 112.Lee J, et al. Surface sites for engineering allosteric control in proteins. Science. 2008;322:438–442. doi: 10.1126/science.1159052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Poelwijk FJ, de Vos MGJ, Tans SJ. Tradeoffs and optimality in the evolution of gene regulation. Cell. 2011;146:462–470. doi: 10.1016/j.cell.2011.06.035. [DOI] [PubMed] [Google Scholar]
- 114.Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–1275. doi: 10.1126/science.1187816. This analysis of historical viral evolution data unequivocally identifies permissive mutations that preceded function-switching mutations.
- 115.Tungtur S, Meinhardt S, Swint-Kruse L. Comparing the functional roles of nonconserved sequence positions in homologous transcription repressors: implications for sequence/function analyses. J. Mol. Biol. 2010;395:785–802. doi: 10.1016/j.jmb.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Skerker JM, et al. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bloom JD, Romero P, Lu Z, Arnold F. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol. Direct. 2007;2:17. doi: 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aharoni A, et al. The ‘evolvability’ of promiscuous protein functions. Nature Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 119.Thomas VL, McReynolds AC, Shoichet BK. Structural bases for stability-function tradeoffs in antibiotic resistance. J. Mol. Biol. 2010;396:47–59. doi: 10.1016/j.jmb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bloom JD, Arnold FH, Wilke CO. Breaking proteins with mutations: threads and thresholds in evolution. Mol. Syst. Biol. 2007;3:76. doi: 10.1038/msb4100119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shoichet BK, Baase WA, Kuroki R, Matthews BWA. Relationship between protein stability and protein function. Proc. Natl Acad. Sci. USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beadle BM, Shoichet BK. Structural bases of stability-function tradeoffs in enzymes. J. Mol. Biol. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 123.Peña MI, Davlieva M, Bennett MR, Olson JS, Shamoo Y. Evolutionary fates within a microbial population highlight an essential role for protein folding during natural selection. Mol. Syst. Biol. 2010;6:387. doi: 10.1038/msb.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Russell RJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 125.Ives JAL, et al. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Res. 2002;55:307–317. doi: 10.1016/s0166-3542(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 126.Gong LI, Suchard MA, Bloom JD. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife. 2013;2:e00631. doi: 10.7554/eLife.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shortle D, Lin B. Genetic analysis of staphylococcal nuclease: identification of three intragenic ‘global’ suppressors of nuclease-minus mutations. Genetics. 1985;110:539–555. doi: 10.1093/genetics/110.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shortle D, Meeker AK. Mutant forms of staphylococcal nuclease with altered patterns of guanidine hydrochloride and urea denaturation. Proteins. 1986;1:81–89. doi: 10.1002/prot.340010113. [DOI] [PubMed] [Google Scholar]
- 129.Wagner A. Neutralism and selectionism: a network-based reconciliation. Nature Rev. Genet. 2008;9:965–974. doi: 10.1038/nrg2473. This is a thoughtful Review of how the vast neutral networks accessible to evolving biological molecules shape the mode and tempo of molecular evolution.
- 130.Grutter MG, Weaver LH, Matthews BW. Goose lysozyme structure: an evolutionary link between hen and bacteriophage lysozymes? Nature. 1983;303:828–831. doi: 10.1038/303828a0. [DOI] [PubMed] [Google Scholar]
- 131.Neidhart DJ, Kenyon GL, Gerlt JA, Petsko GA. Mandelate racemase and muconate lactonizing enzyme are mechanistically distinct and structurally homologous. Nature. 1990;347:692–694. doi: 10.1038/347692a0. [DOI] [PubMed] [Google Scholar]
- 132.Nagano N, Orengo CA, Thornton JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 133.Tie J-K, Jin D-Y, Stafford DW. Mycobacterium tuberculosis vitamin K epoxide reductase homologue supports vitamin K-dependent carboxylation in mammalian cells. Antioxid. Redox Signal. 2012;16:329–338. doi: 10.1089/ars.2011.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Loeb DD, et al. Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 135.Shortle D, Stites WE, Meeker AK. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 136.Sun D, et al. Cumulative site-directed charge-change replacements in bacteriophage T4 lysozyme suggest that long-range electrostatic interactions contribute little to protein stability. J. Mol. Biol. 1991;221:873–887. doi: 10.1016/0022-2836(91)80181-s. [DOI] [PubMed] [Google Scholar]
- 137.Meeker AK, Garcia-Moreno B, Shortle D. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry. 1996;35:6443–6449. doi: 10.1021/bi960171+. [DOI] [PubMed] [Google Scholar]
- 138.Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proc. Natl Acad. Sci. USA. 2004;101:9205–9210. doi: 10.1073/pnas.0403255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li W, et al. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463:507–512. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dutton RJ, et al. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc. Natl Acad. Sci. USA. 2010;107:297–301. doi: 10.1073/pnas.0912952107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ariyoshi K, et al. Patterns of point mutations associated with antiretroviral drug treatment failure in CRF01_AE (subtype E) infection differ from subtype B infection. J. Acquir. Immune Def. Syndr. 2003;33:335–342. doi: 10.1097/00126334-200307010-00007. [DOI] [PubMed] [Google Scholar]
- 142.Bandaranayake RM, et al. The effect of clade-specific sequence polymorphisms on HIV-1 protease activity and inhibitor resistance pathways. J. Virol. 2010;84:9995–10003. doi: 10.1128/JVI.00505-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jessen TH, Weber RE, Fermi G, Tame J, Braunitzer G. Adaptation of bird hemoglobins to high altitudes: demonstration of molecular mechanism by protein engineering. Proc. Natl Acad. Sci. USA. 1991;88:6519–6522. doi: 10.1073/pnas.88.15.6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 145.Kollmann M, Løvdok L, Bartholomé K, Timmer J, Sourjik V. Design principles of a bacterial signalling network. Nature. 2005;438:504–507. doi: 10.1038/nature04228. [DOI] [PubMed] [Google Scholar]
- 146.Brzezinski P, Ädelroth P. Design principles of proton-pumping haem-copper oxidases. Curr. Opin. Struct. Biol. 2006;16:465–472. doi: 10.1016/j.sbi.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 147.Jacob F. Evolution and tinkering. Science. 1977;196:1161–1166. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- 148.Colosimo PF, et al. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science. 2005;307:1928–1933. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- 149.Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JPA. Single amino acid. mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- 150.Jeong S, et al. The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell. 2008;132:783–793. doi: 10.1016/j.cell.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 151.Manceau M, Domingues VS, Mallarino R, Hoekstra HE. The developmental role of agouti in color pattern evolution. Science. 2011;331:1062–1065. doi: 10.1126/science.1200684. [DOI] [PubMed] [Google Scholar]
- 152.Hopkins R, Rausher MD. Identification of two genes causing reinforcement in the Texas wildflower Phlox drummondii. Nature. 2011;469:411–414. doi: 10.1038/nature09641. [DOI] [PubMed] [Google Scholar]
- 153.Hoffmann FG, Storz JF, Gorr TA, Opazo JC. Lineage-specific patterns of functional diversification in the α- and β-globin gene families of tetrapod vertebrates. Mol. Biol. Evol. 2010;27:1126–1138. doi: 10.1093/molbev/msp325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Thornton JW. Resurrecting ancient genes: experimental analysis of extinct molecules. Nature Rev. Genet. 2004;5:366–375. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- 155.Bershtein S, Goldin K, Tawfik DS. Intense neutral drifts yield robust and evolvable consensus proteins. J. Mol. Biol. 2008;379:1029–1044. doi: 10.1016/j.jmb.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 156.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc. Natl Acad. Sci. USA. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. An ultra-high-throughput directed evolution study is discussed here that reveals how epistasis can lead to stochastic and irreproducible outcomes in protein evolution.
- 158.Hietpas R, Roscoe B, Jiang L, Bolon DNA. Fitness analyses of all possible point mutations for regions of genes in yeast. Nature Protoc. 2012;7:1382–1396. doi: 10.1038/nprot.2012.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.McLaughlin RN, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012 doi: 10.1038/nature11500. http://dx.doi.org/10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DNA. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J. Mol. Biol. 2013;425:1363–1377. doi: 10.1016/j.jmb.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Babajide A, Hofacker IL, Sippl MJ, Stadler PF. Neutral networks in protein space: a computational study based on knowledge-based potentials of mean force. Fold Des. 1997;2:261–269. doi: 10.1016/S1359-0278(97)00037-0. [DOI] [PubMed] [Google Scholar]
- 162.Broser M, et al. Structural basis of cyanobacterial photosystem II inhibition by the herbicide terbutryn. J. Biol. Chem. 2011;286:15964–15972. doi: 10.1074/jbc.M110.215970. [DOI] [PMC free article] [PubMed] [Google Scholar]