

Abstract
Hyperoxia contributes to acute lung injury (ALI) in diseases such as acute respiratory distress syndrome (ARDS). Cytochrome P450 (CYP)1A enzymes have been implicated in hyperoxic lung injury, but the mechanistic role(s) of CYP1A2 in pulmonary injury is not known. We hypothesized that mice lacking the gene for Cyp1a2 (which is predominantly expressed in the liver) will be more sensitive to lung injury and inflammation mediated by hyperoxia, and that CYP1A2 will play a protective role by attenuating lipid peroxidation and oxidative stress in the lung. Eight to ten week old WT (C57BL/6) or Cyp1a2(−/−) mice were exposed to hyperoxia (>95% O2) or maintained in room air for 24–72 h. Lung injury was assessed by determining the ratios of lung weight/body weight (LW/BW), and by histology. Extent of inflammation was determined by measuring the number of neutrophils in the lung as well as cytokine expression. The Cyp1a2(−/−) mice under hyperoxic conditions showed increased LW/BW ratios, lung injury, neutrophil infiltration, IL-6 and TNF-α levels, and augmented lipid peroxidation, as evidenced by increased formation of malondialdehyde (MDA)- and 4-hydroxynonenal (4-HNE)-protein adducts, and pulmonary isofurans compared to those of WT mice. In vitro experiments showed that the F2-isoprostane PGF2-α is metabolized by CYP1A2 to a dinor metabolite, providing evidence for a catalytic role for CYP1A2 in the metabolism of F2-isoprostanes. In summary, our results support the hypothesis that hepatic CYP1A2 plays a critical role in the attenuation against hyperoxic lung injury by decreasing lipid peroxidation and oxidative stress in vivo.
Keywords: Acute lung injury, CYP1A2, ARDS, oxidative stress
INTRODUCTION
Supplemental oxygen administration is often used in the clinical management of patients with acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) [1]. In adults, ARDS represents acute respiratory failure that affects over 200,000 patients a year in the U.S. With an estimated mortality of 30–50%, ARDS accounts for about 3 million hospital days and contributes to significant morbidity [1,2]. Hyperoxia causes lung injury in experimental animals [3–6], and exacerbates ALI/ARDS in adults [7]. Reactive oxygen species (ROS) generated during hyperoxia exposure are known to play a role in pulmonary oxygen toxicity [8–12]. Hyperoxia exposure leads to a wide array of changes in the pulmonary transcriptome [13–15] including the transcription factor Nrf2 which in turn activates many anti-oxidant genes. Hyperoxia also activates many inflammatory mediators including cytokines [16]. The resulting acute lung injury can initiate and propagate a systemic inflammatory response leading to multi-organ dysfunction [17–19].
The cytochrome P450 (CYP) are involved in the metabolism of various exogenous and endogenous compounds [20,21]. CYP1A1 and CYP1A2 are induced by polycyclic aromatic hydrocarbons (PAHs) by arylhydrocarbon receptor (AHR)-dependent mechanisms [22,23]. While CYP1A1 [24] can be induced by PAHs in liver and many other tissues [25], CYP1A2 is predominantly expressed in the liver [25–27], and is not induced in extra-hepatic tissues even after treatment with PAHs [25].
Hyperoxia induces pulmonary CYP1A1 and hepatic CYP1A1/1A2 [8,28,29]. The underlying mechanisms are not clearly understood, although the AHR pathway has been proposed to mediate these effects [28,30,31]. P450 enzymes, including CYP1A1/1A2 have been shown to modulate pulmonary oxygen toxicity [28,29,32–35] but the mechanisms by which hepatic CYP1A enzymes modulate hyperoxic lung injury are not known. In human patients, the combination of severe liver dysfunction and ARDS carries a mortality of 100% in adults [36] and liver diseases such as cirrhosis are recognized as risk factors for the development of and mortality in ARDS [37,38]. The specific role of the liver-specific enzyme; CYP1A2 in the amelioration of lung injury due to hyperoxia has not been investigated. Hyperoxia adversely affects all cellular components including DNA, protein and lipid. Isoprostanes and isofurans are formed non-enzymatically by free radical-catalyzed peroxidation of arachidonic acid [39]. These are compounds are both markers and mediators of oxidative stress [40,41].
In this study, we tested the hypothesis that mice lacking the gene for Cyp1a2, which is predominantly expressed in the liver, will be more sensitive to lung injury and inflammation mediated by hyperoxia, and that hepatic CYP1A2 protects against lung injury in part by mechanisms entailing decreases in the extent of lipid peroxidation, leading to a lowered level of oxidative injury.
MATERIALS AND METHODS
Chemicals
Tris, sucrose, NADPH, bovine serum albumin, ethoxyresorufin, methoxyresourufin, EDTA, and butylated hydroxytoluene were purchased from Sigma Chemical Co. (St. Louis, MO). Buffer components for electrophoresis and Western blotting and Goat anti-mouse IgG conjugated with horseradish peroxidase were obtained from Bio-Rad laboratories (Hercules, CA). Rat anti Mouse Ly-6B.2 monoclonal antibody, clone 7/4 (Cat no: MCA771G) was purchased from AbD Serotec (Bio-Rad laboratories, Richmond CA), the primary monoclonal antibody to CYP1A1 was obtained from Dr. P.E. Thomas, Rutgers University. All real-time RT-PCR reagents were bought from Applied Biosystems (Foster City, CA).
Animals
The animal protocol was approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine (Protocol number: AN-907) and the study was conducted in strict accordance with federal guidelines for the humane care and use of laboratory animals by. C57BL/6J wild type (WT) mice were obtained from Charles River laboratories (Wilmington, DE) and creation of the Cyp1a2(−/−) knockout mouse line, has been previously described [42]. These mice, which were on a mixed background (B6/SV129) were cross-bred into the C57BL/6J background by back-crossing for 12 generations, resulting in Cyp1a2(−/−) on > 99% B6 background.
Animals were maintained at the Feigin Center animal facility of Baylor College of Medicine, Houston, TX. A 12 h day-night cycle was maintained throughout the study. Purified tap water and food (Purina Rodent Lab Chow No. 5001 from Purina Mills, Inc. Richmond, IN) were available ad libitum. WT or Cyp1a2(−/−) mice both at eight to ten weeks old (3–6 mice per group) were maintained in room air or exposed to hyperoxia for 24, 48 or 72 h. For hyperoxia exposures, mice were placed in plexiglass chambers; oxygen was delivered through a humidified circuit at a flow rate of 5 L/min to achieve FiO2 of >95%. Oxygen concentration was monitored continuously by means of in-line analyzers at the outport of the chambers [30]. In each group (room air, 24, 48 and 72 hr of hyperoxia exposure) 3 animals were used for lung histopathology and 6 animals were used for RNA and protein extraction and other studies.
Perfusion and tissue harvesting
At the end of each experiment, 9 mice from each group were sacrificed by exsanguination while under deep pentobarbital anesthesia. Lungs (n=3 mice) were perfused in situ with phosphate buffered saline (PBS), and used for subsequent analyses of CYP1A-dependent activities and protein contents. In the remaining three animals, the left lungs were inflated through an intra-tracheal catheter and fixed at constant pressure (20 cm H2O) with zinc formalin, after which the lungs were embedded in paraffin for subsequent histological analyses of lung injury [10]. The right lungs were snap-frozen at −80° C for subsequent RNA isolation.
Liver and lung microsome isolation
Liver microsomes were prepared by calcium chloride precipitation, as reported previously [43]. Lung microsomes were isolated by differential centrifugation, as published previously [44].
CYP1A1 and CYP1A2 enzyme assays
Ethoxyresorufin O- deethylase (EROD) (CYP1A1) activity in lung and liver microsomes was assayed according to the method of Pohl and Fouts [45], as reported earlier [30]. Methoxyresorufin O-demethylase (MROD) (CYP1A2) activity was assayed as described previously [30].
Western blotting
Liver and lung microsomes (20 μg protein) prepared from individual mice were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in 7.5% acrylamide gels. The separated proteins on the gels were transferred to polyvinylidene difluoride membranes, followed by Western blotting using monoclonal antibodies to CYP1A1, which cross-reacts with CYP1A2 [10,30,31]. For loading controls, we used GRP78 (Glucose-regulated protein 78), a protein that is localized in the endoplasmic reticulum.
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) assays
Total RNA (50 ng) from liver and lung of air-breathing or hyperoxic animals was subjected to one-step quantitative TaqMan RT-PCR. ABI PRISM 7700 Sequence Detection System was used for RT-PCR reactions, as previously described [31,46]. Serial dilutions of RNA were used to optimize and validate RT-PCR conditions for the 18S gene and target genes chosen for this study. Each data point was repeated three times. Quantitative values were obtained from the threshold PCR cycle number (Ct) at which the increase in signal was associated with an exponential growth for PCR product becomes detectable. Relative mRNA levels for chosen target genes (i.e., Cyp1a1, 1a2, TNF-α, IL-6) were normalized to 18S RNA content. The primers for the target genes were obtained from Life Technologies (Carlsbad, CA); CYP1A1: Mm00487217_m1, TNF: Mm00443258_m1 and IL6: Mm00446190_m1, 18S-Hs99999901_s1. For CYP1A2, the forward primer was: TCCTGGACTGACTCCCACAAC and the reverse primer was: GAACGCCATCTGTACCACTGAA. Relative expression levels of each target gene were calculated according to the equation, 2−ΔcT, where ΔcT = Ct target gene – Ct 18S gene.
Lipid peroxidation and Inflammatory markers
Levels of F2-isoprostanes and isofurans in liver and lung homogenates were determined by GC-MS [39]. MDA- and 4-HNE-protein adducts in liver and lung tissue homogenates were analyzed by ELISA [47]. Serum and liver (tissue homogenates) were used to measure TNF-α levels by ELISA in WT and Cyp1a2(−/−) mice.
Lung injury and Inflammation
Lung weight/body weight (LW/BW) ratios were calculated as an index of lung injury in animals whose lungs were not perfused for isolation of microsomes. This ratio was used as an index of hyperoxia-induced pulmonary edema formation. Lung histology was performed by H&E staining of lung sections, as described [10,30]. Assessment of lung injury in the histopathological lung sections was performed as described in the official American thoracic society workshop report on measurement of acute lung injury in experimental animals [48]. Twenty random high-power fields (400 × total magnification) were independently scored in a blinded fashion taking care that 50% of each field was occupied by lung alveoli. Five histological findings: neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membranes, proteinaceous debris filling the airspaces and alveolar septal thickening were graded using a three tiered schema resulting in a lung injury score between zero and one. Neutrophil infiltration into lungs was determined by immunohistochemistry using the rat anti-mouse Ly-6B.2 monoclonal antibody, as reported [31]. Neutrophils were quantified by averaging the number of counts using at least 10 random high-power fields.
In vitro metabolism of PGF2-α
In order to determine the role of CYP1A2 in the metabolism of F2-isoprostanes, PGF2-α (1 μM) was incubated with recombinant human CYP1A2 and NADP(H) cytochrome P450 oxidoreductase in PBS buffer, pH. 7.4 for 30 minutes, following which the reaction was stopped with chloroform:methanol (1:3), and the product(s) were extracted using the same solvent mixture. The chloroform:methanol fraction was evaporated to dryness, and spotted on silica gel thin-layer plates. The product, which was formed in the presence of CYP1A2 and P450 oxidoreductase, but not in the absence of oxidoreductase, was scrapped off from the plate, and eluted with ethylacetate, and evaporated to dryness, dissolved in acetonitrile, and identified by LC/MS/MS (ThermoFinnigan TSQ quantum quadrupole mass spectrometer) operating in negative ion mass mode and scanning from m/z 200 to m/z 500.
Statistical analysis
Data are expressed as means ± SEM. Two-way analyses of variance (ANOVA) (effect of genotype and treatment and interaction between the two main effects), was performed followed by modified t-tests, were used to assess significant differences arising from exposure to hyperoxia and room air, and between WT and Cyp1a2(−/−) mice. P-values of < 0.05 were considered significant.
RESULTS
Cyp1a2(−/−) mice show increased LW/BW ratios under hyperoxic conditions
WT and Cyp1a2(−/−) mice exhibited significantly higher LW/BW ratios than room air controls after 48 and 72 h of hyperoxia (>95% FiO2), with the most pronounced effect occurring in the Cyp1a2(−/−) mice, which showed a doubling of LW/BW ratios compared to room air controls (Figure 1). When the LW/BW ratios were compared between Cyp1a2(−/−) and WT mice, the former had significantly higher LW/BW ratios at 72 h (~42%) than those of the latter (Figure 1).
Figure 1.

Effect of hyperoxia on LW/BW ratios in WT and Cyp1a2(−/−) mice. Representative quantitative analysis of the effects of hyperoxia exposure on LW/BW, a measure of pulmonary edema of WT and Cyp1a2(−/−) mice. Values are means ± S.E.M. from six individual animals. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05, ** P < 0.01 and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by ### P < 0.001. Main effects of genotype and treatment and interaction between the two main effects are also shown.
Cyp1a2(−/−) mice show greater histological lung injury than WT mice after hyperoxia
Signs of lung injury in the form of alveolar and perivascular edema were observed in Cyp1a2−/− mice at 48 h after hyperoxia exposure. At 72 h, both WT and Cyp1a2(−/−) mice showed increased lung injury (alveolar and perivascular edema) compared to room air controls (Figure 2A). Cyp1a2(−/−) mice showed much greater injury than WT mice after hyperoxia with severe alveolar and perivascular edema at the 72 h time point (Figure 2A). Figure 2B shows the assessment of lung injury entailing the official American Thoracic Society guidelines for the estimation of acute lung injury in experimental animals [48]. Increase in lung injury score in animals after exposure to hyperoxia for 72 h was seen in both WT and Cyp1a2(−/−) mice. Also, the pulmonary injury score was significantly greater in Cyp1a2(−/−) mice compared to WT mice after each time point of oxygen exposure (Figure 2B).
Figure 2.

H & E staining of lung histology and quantitation of lung injury. (A) Cyp1a2(−/−) mice show greater hyperoxia-induced lung edema and perivascular and alveolar injury, compared with that in WT mice. Representative hematoxylin/eosin-stained images from WT vs. Cyp1a2(−/−) lungs (n= 5 mice per group) are shown in room air and after 24, 48, and 72 h of hyperoxia exposure. Arrows and arrowheads point to perivascular and alveolar areas, respectively. (B) Lung injury scores in 20 random high-power fields (n = 3 mice per group) in hematoxylin and eosin stained lung sections obtained from WT and Cyp1a2(−/−) mice at room air and after exposure to hyperoxia (24, 48 and 72 h). Significant differences between room air and hyperoxia within genotype are indicated by ** P < 0.01 and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by # P <0.05 and ### P < 0.001. Main effects of genotype and treatment and interaction between the two main effects are also shown.
Cyp1a2(−/−) mice display increased number of neutrophils in the lungs after hyperoxia
There was increased pulmonary neutrophil infiltration after hyperoxia exposure for 72 h in both WT and Cyp1a2(−/−) mice (Figure 3A), with the Cyp1a2(−/−) mice showing greater neutrophil recruitment than WT mice. Quantitative analyses (Figure 3B) showed about a 3-fold increase in number of neutrophils in the lungs of WT mice after 72 h of hyperoxia exposure compared to room air controls. In Cyp1a2(−/−) mice, there was a marked increase in the number of neutrophils (~ 1.8-fold) after 24 h of hyperoxia over room air controls (Figure 3B). Interestingly, there was a significant decrease in neutrophil number after 48 h, followed by another increase (0.8-fold) after 72 h of hyperoxia. When neutrophil numbers were compared among Cyp1a2(−/−) and WT mice, even in room air, the former showed a 2.75-fold increase over the latter. At the 24 and 72 h time points after hyperoxic exposure, the neutrophil recruitment was augmented by 1.8- and 0.8-fold compared to the corresponding WT counterparts, respectively (Figure 3B). The decrease in neutrophils at 48 h has been observed in acute lung injury models [49,50]. This may be due to neutrophil recruitment being mediated through different pathways at 24 and 72 h. In an ALI model, it was shown that, high levels of CXCL1, CXCL2 and CXCL5 mediate the early phase of neutrophil recruitment and increases in extracellular ATP mediate the late phase [51].
Figure 3.
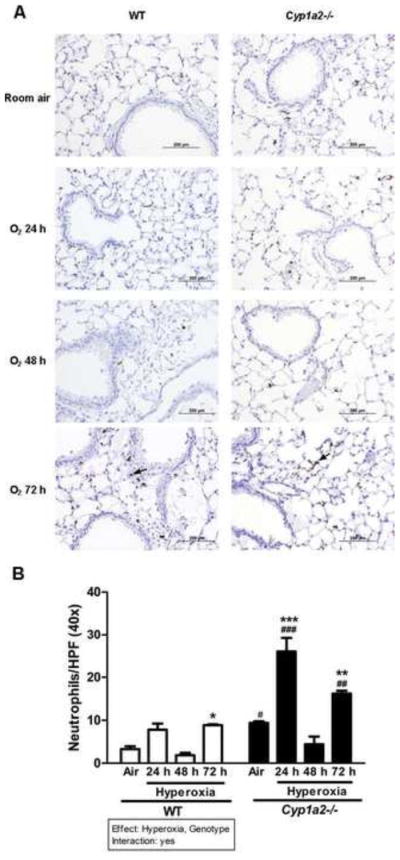
Immunohistochemistry and quantitation of pulmonary neutrophil recruitment. (A) Representative immuno-stained images for lung neutrophils. Hyperoxia-induced neutrophil recruitment was determined by immunohistochemistry with anti-neutrophil antibodies in WT and Cyp1a2(−/−) mice (n=5 mice per group) in room air and after 24, 48, and 72 h of hyperoxia. Arrows point to brown-staining neutrophils. (B) Quantitative analyses showing number of neutrophils per high-power field. Representative quantitative analysis of the hyperoxia effects on neutrophil recruitment in lungs of WT vs. Cyp1a2(−/−) mice. Values are means ± S.E.M. from at 3 individual animals. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05 and ***P <0.001. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05, ** P < 0.01and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by # P <0.05, ## P <0.01 and ### P < 0.001.
Effect of hyperoxia on pulmonary and hepatic CYP1A1 and CYP1A2 activities and apoprotein levels
There was significant induction in pulmonary ethoxyresorufin resorufin O-deethylase (EROD) CYP1A1 activity after 24 and 48 h of hyperoxia over room air levels in WT mice (Figure 4A). By 72 h, the levels returned to those seen in normoxia controls. Cyp1a2(−/−) mice showed much higher (~60%) pulmonary EROD activities than WT mice under normoxic conditions (Figure 4A). After 48 and 72 h of hyperoxia, the Cyp1a2(−/−) mice showed a significant decline in EROD activities compared to room air controls in the lung (Figure 4A). In liver, hyperoxia did not cause significant changes in EROD activities in WT mice, but there was significant decline after 72 h (Figure 4B). The Cyp1a2(−/−) mice showed repressed EROD activities in room air as well as hyperoxic conditions (24 and 48 h) compared to those of WT mice (Figure 4B). In regard to liver methoxyresorufin O-demethylase (MROD) (CYP1A2), hyperoxia caused induction of MROD activity in WT mice after 48 h, with activities declining after 72 h (Figure 4C).
Figure 4.

Representative quantitative analysis of EROD (CYP1A1 activity) in liver and lung and MROD (CYP1A2 activity) in liver of WT and Cyp1a2(−/−) mice exposed to room air or hyperoxia for 24, 48 and 72 h. Pulmonary EROD (A), hepatic EROD (B), and hepatic MROD (C) activities were measured by spectrofluorometry. Values represent means ± S.E.M. from 3 individual animals. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05, ** P < 0.01and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by ## P <0.01 and ### P < 0.001. Main effects of genotype and treatment and interaction between the two main effects are also shown.
In lungs, hyperoxia increased pulmonary CYP1A1 apoprotein contents, as determined by Western blotting, in WT mice (Figure 5A and B), with the induction declining at 72 h (Figure 5B). In Cyp1a2(−/−) mice, under normoxia and after 24 h of hyperoxia exposure, pulmonary CYP1A1 protein expression was markedly enhanced compared to that in WT mice. There was a decline in the level of CYP1A1 (Figure 5B) after 24 h of oxygen exposure. CYP1A2 was absent in the lungs of WT or Cyp1a2(−/−) mice (Figure 5A). In liver, CYP1A1 was not detectable in WT or Cyp1a2(−/−) mice (Figure 5C). An increase in CYP1A2 protein expression after 48 h of hyperoxia in WT mice was noted, and the induction declined after 72 h (Figure 5C and D). CYP1A2 was not detectable in Cyp1a2(−/−) mice as expected.
Figure 5.

Effect of hyperoxia on CYP1A1/1A2 protein expression in mice. Representative Western immunoblots and densitometric analysis showing pulmonary (A and B) CYP1A1 and hepatic (C and D) CYP1A2 protein amounts in microsomes isolated from WT and Cyp1a2(−/−) mice in room air (Air) and after 24, 48 and 72 h of hyperoxia. The positive control (labeled “PC”) was 0.5 μg of liver microsomes from 3-methylcholanthrene-treated WT mice. Under each sample lane is the corresponding GRP-78 (Glucose-regulated protein) blot to assess for protein loading. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05 and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by ### P < 0.001. Main effects of genotype and treatment and interaction between the two main effects are also shown.
Effect of hyperoxia on hepatic CYP1A1/1A2 and pulmonary CYP1A1 gene expression
Hyperoxia for 24 h led to a significant increase in pulmonary CYP1A1 mRNA after 24 h, and declined to control after 48h in WT mice (Figure 6A). In Cyp1a2(−/−) mice, CYP1A1 mRNA expression was augmented at all time points compared to the WT counterparts (Figure 6A). In the lung there was no expression of CYP1A2 in WT and Cyp1a2(−/−) mice as shown in Figure 6A. In liver, hyperoxia did not cause any significant alterations in the expression of CYP1A1 mRNA in WT mice, but in Cyp1a2(−/−) mice, there was a marked increase in the expression of CYP1A1 mRNA even in room air conditions, compared to that of WT mice (Figure 6B). In mice that were exposed to hyperoxia, there was a significant decline in CYP1A1 mRNA expression at 24 and 48 h, compared to room air controls (Figure 6B).
Figure 6.

Effect of hyperoxia on CYP1A1/1A2 mRNA expression in mice. Real time RT-PCR analysis showing effects of hyperoxia on hepatic CYP1A1 (A), pulmonary CYP1A1/A2 (B) and hepatic CYP1A2 (C) mRNA expression in WT vs. Cyp1a2(−/−) mice. Values are means ± S.E.M. from 3 individual animals. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05, ** P < 0.01and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by ## P <0.01 and ### P < 0.001. Main effects of genotype and treatment and interaction between the two main effects are also shown.
In regard to CYP1A2 gene expression in the liver, hyperoxia for 24 h resulted in a 35% increase in WT mice, but this induction declined to less than control levels (70%) by 48 h, and further declined to 10% of control by 72 h (Figure 6C). In Cyp1a2(−/−) mice, there was no detectable expression of hepatic CYP1A2, which was significantly decreased compared to WT mice.
Effect of hyperoxia on cytokine levels
Hyperoxia for 24 h led to significant increase in TNF-α mRNA levels in WT and Cyp1a2(−/−) mice, and this persisted through 72 h of hyperoxia (Figure 7A). Hyperoxia also elicited increased gene expression of IL-6 in both WT and Cyp1a2(−/−) mice, with Cyp1a2(−/−) mice displaying higher expression of IL-6 compared to WT at 72 h (Figure 7B). In serum of WT animals, TNF-α protein expression was significantly augmented after 24 h of hyperoxia, but the expression declined after 48–72 h (Figure 7C). Cyp1a2(−/−) mice showed higher TNF-α expression at 72 h compared to WT mice. In liver, exposure to hyperoxia did not elicit any induction of TNF-α at 24–48 h in WT mice, but Cyp1a2(−/−) mice showed increased TNF-α protein levels at 24 h compared to air-breathing controls (Figure 7D).
Figure 7.

Effect of hyperoxia on inflammatory markers in WT and Cyp1a2(−/−) mice. Cytokine levels in lungs (RT-PCR), serum and liver (ELISA) showing effects of hyperoxia on pulmonary (A & B) TNF-α and IL-6 mRNA levels, serum (C) and liver (D) TNF-α levels measured by ELISA in WT and Cyp1a2(−/−) mice. Values denote means ± S.E.M. from 3 individual animals. Significant differences between room air and hyperoxia within genotype are indicated by ** P < 0.01and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by # P <0.05 and ## P <0.01. Main effects of genotype and treatment and interaction between the two main effects are also shown.
Effects of hyperoxic exposure on lipid peroxidation
The effect of hyperoxia on oxidative stress was determined by assessing the extent of lipid peroxidation in liver and lung by measuring protein adducts of MDA (Figure 8A and B) and 4-HNE (Figure 8C and D). Hyperoxia exposure did not alter MDA-protein adduct levels in liver of WT mice (Figure 8A). The MDA-protein adduct levels were higher in Cyp1a2(−/−) mice compared to WT mice under normoxia, but after hyperoxia there was a decline in MDA-protein adducts in the Cyp1a2(−/−) mice (Figure 8A). In lungs, there was a statistically significant increase (doubling) of MDA-protein adducts in Cyp1a2(−/−) mice exposed to hyperoxia at 48 h (Figure 8B), and significantly higher than in WT mice at 48 and 72 h of hyperoxia exposure. In WT mice there was a modest increase in lung MDA levels, which was significant at 48 h.
Figure 8.

Assessment of lipid peroxidation. Representative quantitative analysis of hyperoxia effects on lipid peroxidation in WT and Cyp1a2(−/−) mice in liver (A and C) and lung (B and D) as measured by levels of malondialdehyde and 4-HNE- protein adducts. Also represented are levels of lung F2-isoprostanes (E), lung isofurans (F) and liver isofurans (G). Values denote means ± S.E.M. from 3 individual animals. Significant differences between room air and hyperoxia within genotype are indicated by *P <0.05, **P < 0.01 and ***P <0.001. Significant differences between corresponding WT and Cyp1a2(−/−) groups are indicated by # P <0.05, ## P <0.01 and ### P < 0.001. Main effects of genotype and treatment and interaction between the two main effects are also shown.
In regard to 4-HNE-protein adducts, in WT mice hyperoxia exposure for 48–72 h resulted in a significant decrease in hepatic 4-HNE-protein adducts compared to room air controls (Figure 8C). On the other hand, Cyp1a2(−/−) mice under hyperoxic conditions for 72 h showed augmentation of 4-HNE-protein adducts compared to room air (Figure 8C). When hepatic 4-HNE-protein adducts were compared among WT and Cyp1a2(−/−) mice, hyperoxia for 48 and 72 h showed higher levels compared to the corresponding WT counterparts. In the lungs of WT and Cyp1a2(−/−) mice, hyperoxia for 48 h augmented 4-HNE-protein adducts, compared to room air controls (Figure 8D).
In WT mice, hyperoxia for 24 h significantly increased levels of pulmonary F2-isoprostanes (Figure 8E); however, the levels of these compounds returned to room air levels at later time points. In Cyp1a2(−/−) mice, the levels of F2-isoprostanes were not altered at any time point under hyperoxic conditions (Figure 8E). Pulmonary isofurans showed increases after 24 h of hyperoxia in WT mice (Figure 8F), but the levels returned to room air levels after 48–72 h of hyperoxia. In the Cyp1a2(−/−) mice, the levels of isofurans were significantly increased at 48 h compared to the WT counterparts (Figure 8F). In liver, hyperoxia did not induce levels of isofurans at any of the time points, but Cyp1a2(−/−) mice showed increased levels of isofurans at 48 h of hyperoxia, compared to the WT counterparts (Figure 8G).
In vitro metabolism of F2-isoprostanes
In order to determine the role of CYP1A2 in the metabolism of F2-isoprostanes, PGF2-α, was incubated with purified CYP1A2 in the presence of NADPH cytochrome P450 oxidoreductase (CPR) and NADPH. The metabolites were extracted and analyzed by TLC as described in Materials and Methods in the online supplement. The major metabolite M1 was analyzed by LC-MS/MS. The unmetabolized isoprostane had a molecular ion peak at 353 (M-1) (not shown). The metabolite M1 composed exclusively of an ion with an m/z 253 (Figure 9A). The transformation of the parent compound of m/z 353 to a species of m/z 253 (loss of 100 amu) is suggestive of oxidative cleavage of the alpha (upper) side chain at the 5,6 double bond, as this would make the compound more hydrophobic (manifested as the larger Rf on TLC). Alternatively, this transformation may represent a 2,3 dinor metabolite where the carboxyl group has been converted to an aldehyde, with dehydration of the three hydroxyl groups. Incomplete dehydration would yield the m/z 271 species in negative ion mode (Figure 9B). This represents a novel P450-mediated reaction.
Figure 9.

In vitro metabolism of PGF2-α. PGF2-α (1 μM) was incubated with recombinant human CYP1A2 and NADP(H) cytochrome P450 oxidoreductase in PBS buffer, pH. 7.4 for 30 minutes, following which the reaction was stopped with chloroform:methanol (1:3), and the product(s) were extracted using the same solvent mixture. The chloroform:methanol fraction was evaporated to dryness, and spotted on silica gel thin-layer plates. The product, which was formed in the presence of CYP1A2 and NADP(H) P450 oxidoreductase, but not in the absence of oxidoreductase, was scrapped off from the plate, and eluted with ethylacetate, and evaporated to dryness, dissolved in acetonitrile, and identified by LC/MS/MS (ThermoFinnigan TSQ quantum quadrupole mass spectrometer) (A) operating in negative ion mass mode and scanning from m/z 200 to m/z 500. B. Shows the possible formation of dinor metabolite after catalysis by CYP1A2.
DISCUSSION
The primary objective of this investigation was to determine the functional role of hepatic CYP1A2 in hyperoxic lung injury using mice lacking the gene for CYP1A2, which is predominantly expressed in the liver, and is absent in the lung. We found increased LW/BW ratios, lung injury (based on histopathology), and inflammation (increased neutrophil recruitment) in Cyp1a2(−/−) mice exposed to hyperoxia, compared to similarly exposed WT mice, suggesting that liver CYP1A2 plays a protective role against lung injury and inflammation. The fact that the number of neutrophils in Cyp1a2(−/−) mice was higher compared to WT mice even under normoxia, supported the hypothesis that endogenous hepatic CYP1A2 plays a protective role against lung inflammation in WT mice.
Induction of pulmonary EROD activity after 48 h of hyperoxia in WT mice probably reflected CYP1A1 induction by hyperoxia, because EROD activity is largely catalyzed by CYP1A1 [8,30]. The decline in EROD induction after 72 h of hyperoxia was similar to previous findings in adult rat [8,30,33] and mouse models [31] showing a decline in CYP1A1 expression after prolonged hyperoxia, coinciding with respiratory distress and lung injury. The augmentation of pulmonary EROD activities in Cyp1a2(−/−) mice compared to WT mice could be explained by CYP1A2 metabolizing endogenous ligands of the AHR in the WT mice; therefore, in the Cyp1a2(−/−)mice, the AHR ligands would accumulate, resulting in CYP1A1 induction.
In liver, the decrease in EROD activity after 72 h in WT was in agreement with studies reported previously in rats [8]. The augmentation of MROD expression in the livers of WT animals exposed to hyperoxia reflected CYP1A2 induction. The observation that modulation of EROD and MROD activities by hyperoxia in liver and lung correlated overall with apoprotein expression of CYP1A1 and CYP1A2 supported the theory that the hyperoxia effects were not due to enzyme stabilization.
The augmentation of pulmonary CYP1A1 mRNA in the Cyp1a2(−/−) in room air, compared to that in WT mice may have been due to accumulation of AHR ligands that are CYP1A2 substrates. The induction of pulmonary CYP1A1 mRNA by hyperoxia for 24 h in the Cyp1a2(−/−) was also possibly mediated by AHR-mediated mechanisms, based on previous findings [31] in which hyperoxia did not elicit pulmonary CYP1A1 expression in AHR-deficient mice. The induction of hepatic CYP1A2 mRNA expression by hyperoxia for 24 h, and the subsequent decline after 48–72 h was consistent with previous findings in the rat model [8,30]. The marked induction of hepatic CYP1A1 mRNA in air-breathing Cyp1a2−/− mice was probably due to accumulation of AHR ligands that are cleared by CYP1A2 in the WT mice.
The enhancement of TNF-α and IL-6 mRNA levels in lungs of WT mice after hyperoxia for 24–72 h suggested that hyperoxia caused an inflammatory response. These results were in agreement with previous studies showing that hyperoxia leads to increased TNF-α levels in the lung [52–55]. The role of cytokines in hyperoxic acute lung injury has been extensively studied [56] and other mediators such as IL-1, IL-8, IFN-γ, MIP-2 and MCP-1 play a role in this disease process.
That hyperoxic exposures led to increased levels of serum TNFα levels, a phenomenon that was even more pronounced in Cyp1a2(−/−) mice suggested that hyperoxia elicits a systemic inflammatory response, and that in the absence of CYP1A2, this response is magnified. The present results also showed increased levels of TNF-α in livers of Cyp1a2(−/−), supporting the hypothesis that in the normal mouse, hepatic CYP1A2 plays a protective role against inflammation. Systemic inflammation in response to lung injury has been observed by other researchers. Acute lung injury impairs the integrity of the epithelial and endothelial barrier and the alveolar cytokines and other pro-inflammatory mediators generated in the lung can leak into the vascular system. This can lead to a systemic inflammatory response [19,57]. Tutor et al demonstrated increase in systemic levels of TNF-α in acute lung injury [18]. St. John et al showed that neutrophils played a major role in the systemic inflammatory effects in an acid-aspiration model of acute lung injury [58]. Jobe et al have reported increase in hepatic markers of inflammation in preterm models of lung injury [17,59].
Increased levels of hepatic MDA-protein adducts in Cyp1a2(−/−) mice under normoxia, compared to WT mice suggest that hepatic CYP1A2 metabolizes precursors of compounds that contribute to lipid peroxidation. The augmentation of pulmonary MDA-protein adducts in Cyp1a2(−/−) mice after 48 h of hyperoxia further suggests that MDA-protein adducts contributed to hyperoxic lung injury and inflammation. The enhancement of 4-HNE adducts in livers of Cyp1a2(−/−) mice exposed to hyperoxia indicated the protective role of liver CYP1A2 against oxygen-mediated lipid peroxidation.
The increased pulmonary F2-isoprostane levels after hyperoxia in WT mice after 24 h, but not after 48 or 72 h, suggests that formation of lipid hydroperoxides precedes lung injury. The elevation in the levels of pulmonary F2-isoprostanes and isofurans in rabbits following exposure to hyperoxia has been shown before [39], and has been attributed to be due to oxidative stress in vivo. The induction of CYP1A2 activity at the similar time point followed by a decrease in isofuran and F2-isoprostane levels suggest a possible role for CYP1A2 in the detoxification of these lipid peroxidation products. Cyp1a2(−/−) mice show significantly increased isofuran levels after 48h, compared with WT, suggesting that CYP1A2 metabolizes isofurans in the normal animal. Increased F2-isoprostane levels have been correlated with lung injury in neonates many of whom receive supplementary oxygen therapy during their treatment course [60]. F2-isoprostanes in tracheal aspirates were increased in the lungs of premature babies receiving oxygen and mechanical ventilation treated with the CYP inhibitor cimetidine, consistent with the hypothesis that CYP enzymes may attenuate the risk of oxidative injury [61].
In order to determine the role of CYP1A2 in the detoxification of lipid hydroperoxides, we conducted in vitro experiments with recombinant human CYP1A2, and the present results showing the formation of a dinor metabolite from PGF2-α (Figure 9B) provide direct evidence that F2-isoprostanes are endogenous substrates for CYP1A2. To our knowledge, this is the first report demonstrating a functional role for CYP1A2 in F2-isoprostane metabolism.
It is not clear as to how F2-isprostanes/isofurans would contribute to lung injury. Seto et al [62] showed that the E-ring isoprostane, 15-E2t-isOP through an EP4 receptor coupled to adenylate cyclase and soluble guanylate cyclase, activates a transepithelial Cl conductance in bovine airway epithelium. This could account for the increased mucus production observed in lung injury models. Another possibility is that F2-isoprostanes/isofurans might contribute to pulmonary vessel injury, which could in turn lead to lung injury, and this idea stems from the finding that these molecules cause constriction of isolated vessels, and could lead to pulmonary hypertension in vivo. F2-isoprostanes/isofurans may also contribute to lung injury by enhancing production of pro-inflammatory cytokines by smooth muscle, endothelial and inflammatory cells. The fact that isoprostanes are elevated in patients with acute lung injury (ALI) [63] and in experimental animals exposed to hyperoxia [39,64] lend credence to the theory that these molecules play a causative role in hyperoxic lung injury. Involvement of other reactive compounds such as eicosanoids and leukotrienes [65], or phosphatidyl serine and cardiolipin [66] in hyperoxic lung injury cannot be excluded in this study. Only a limited number of inflammatory markers were investigated in this study and sex-specific differences were not looked into. A more detailed analysis of the transcriptome between WT and knock-out animals and sex-specific differences are being currently studied in our lab.
CONCLUSIONS
In summary, in this study, we have documented the protective effects of mammalian hepatic CYP1A2 against hyperoxic lung injury (Figure 10), revealing a critical role for extra-pulmonary organs such as liver in the protection against lung injury. We postulate that hyperoxia elevates ROS-mediated lipid hydroperoxides levels in lung and liver. In the lung, these substances may cause injury via cytokine activation, leading to inflammatory cell recruitment, and eventual injury and cell death. These may also enter the systemic circulation and reach the liver where they are detoxified by CYP1A2. In the absence of CYP1A2 (in Cyp1a2(−/−) mouse), these compounds would magnify the inflammatory response to hyperoxia exposure, resulting in activation of pro-inflammatory cytokines (e.g., TNF-α and IL-6), which could result in greater lung injury. Another explanation could be that the F2-isoprostanes/isofurans that are formed in the liver directly by hyperoxia may undergo detoxification by CYP1A2 to non-toxic metabolites. In the absence of CYP1A2, these compounds would accumulate, and may be transported to the systemic circulation, and this could in turn lead to cytokine activation and increased lung injury (Figure 10). In fact, the increased lung injury in Cyp1a2(−/−) mice may entail a combination of both of these mechanisms. Thus, this study highlights the protective role of liver-specific CYP1A2 in metabolizing potential mediators of acute lung injury and provides a mechanism by which liver function could play a major role in determining survival in patients with ARDS. Further studies on the functional role of CYP1A2 in oxygen-mediated lung injury could lead to novel treatment/preventive strategies for ALI/ARDS.
Figure 10.

Schematic of the lung protective role of hepatic CYP1A2 in hyperoxic lung injury. Hyperoxia elevates ROS-mediated lipid hydroperoxides (e.g., F2-isoprostanes/isofurans) levels in lung, which then enter the systemic circulation and reach the liver where they are detoxified by CYP1A2. In the absence of CYP1A2 (in Cyp1a2(−/−) mouse), these compounds would magnify the inflammatory response to hyperoxia exposure, resulting in activation of pro-inflammatory cytokines (e.g., TNF-α and IL-6), which could result in greater lung injury. Alternatively, the F2-isoprostanes/isofurans that are formed in the liver directly by hyperoxia may undergo detoxification by CYP1A2 to non-toxic metabolites. In the absence of CYP1A2, these compounds would accumulate, and may be transported to the systemic circulation, and this could in turn lead to cytokine activation and increased lung injury.
HIGHLIGHTS.
CYP1A2 attenuates hyperoxic lung injury and inflammation.
CYP1A2 decreases lipid peroxidation and oxidative stress.
Extra-pulmonary organs such as liver may be protective against acute lung injury.
Acknowledgments
This work was in part supported by funds from RO1 grants from National Institutes of Health [ES-009132, HL-112516, HL-087174, and ES-019689] to B.M., HL-088343 to XIC, and the Center for Translational Environmental Health Research (CTEHR), P30ES023512.
ABBREVIATIONS
- 4-HNE
4-hydroxynonenal
- ARDS
Acute Respiratory Distress Syndrome
- ALI
Acute Lung Injury
- AHR
Aryl hydrocarbon receptor
- CYP
Cytochrome P450
- ELISA
Enzyme linked immunosorbent assay
- EROD
Ethoxyresorufin O-deethylase
- GC-MS
Gas Chromatography-Mass Spectrometry
- LC/MS/MS
Liquid Chromatography/Mass Spectrometry/Mass Spectrometry
- LW/BW
Lung weight/Body weight
- MDA
Malondialdehyde
- MROD
Methoxyresorufin O-demethylase
- ROS
Reactive Oxygen Species
- TLC
Thin Layer Chromatography
- WT
Wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 1301;342:1308–2000. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 2.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3.Freeman BA, Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- 4.Budinger GRS, Mutlu GM, Urich D, Soberanes S, Buccellato LJ, Hawkins K, Chiarella SE, Radigan KA, Eisenbart J, Agrawal H, Berkelhamer S, Hekimi S, Zhang J, Perlman H, Schumacker PT, Jain M, Chandel NS. Epithelial cell death is an important contributor to oxidant-mediated acute lung injury. Am J Respir Crit Care Med. 2011;183:1043–1054. doi: 10.1164/rccm.201002-0181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark JM, Lambertsen CJ. Pulmonary oxygen toxicity: a review. Pharmacol Rev. 1971;23:37–133. [PubMed] [Google Scholar]
- 6.Liu G, Feinstein SI, Wang Y, Dodia C, Fisher D, Yu K, Ho Y-S, Fisher AB. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radic Biol Med. 2010;49:1172–1181. doi: 10.1016/j.freeradbiomed.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luis M, Ayuso A, Martinez G, Souto M, Ortells J. Intraoperative respiratory failure in a patient after treatment with bleomycin: previous and current intraoperative exposure to 50% oxygen. Eur J Anaesthesiol. 1999;16:66–68. doi: 10.1046/j.1365-2346.1999.00403.x. [DOI] [PubMed] [Google Scholar]
- 8.Moorthy B, Nguyen UT, Gupta S, Stewart KD, Welty SE, Smith CV. Induction and decline of hepatic cytochromes P4501A1 and 1A2 in rats exposed to hyperoxia are not paralleled by changes in glutathione S-transferase-alpha. Toxicol Lett. 1997;90:67–75. doi: 10.1016/s0378-4274(96)03832-5. [DOI] [PubMed] [Google Scholar]
- 9.Morel Y, Barouki R. Down-regulation of cytochrome P450 1A1 gene promoter by oxidative stress. Critical contribution of nuclear factor 1. J Biol Chem. 1998;273:26969–26976. doi: 10.1074/jbc.273.41.26969. [DOI] [PubMed] [Google Scholar]
- 10.Moorthy B, Parker KM, Smith CV, Bend JR, Welty SE. Potentiation of oxygen-induced lung injury in rats by the mechanism-based cytochrome P-450 inhibitor, 1-aminobenzotriazole. J Pharmacol Exp Ther. 2000;292:553–560. [PubMed] [Google Scholar]
- 11.Frank L, Bucher JR, Roberts RJ. Oxygen toxicity in neonatal and adult animals of various species. J Appl Physiol Respir Environ Exerc Physiol. 1978;45:699–704. doi: 10.1152/jappl.1978.45.5.699. [DOI] [PubMed] [Google Scholar]
- 12.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lingappan K, Jiang W, Wang L, Couroucli XI, Moorthy B. Analysis of the transcriptome in hyperoxic lung injury and sex-specific alterations in gene expression. Lingappan, K., Jiang, W., Wang, L., Couroucli, X. I., Moorthy, B., editors. PLoS ONE. 2014;9:e101581. doi: 10.1371/journal.pone.0101581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho H-Y, Reddy SP, DeBiase A, Yamamoto M, Kleeberger SR. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic Biol Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Cho H-Y, Jedlicka AE, Reddy SPM, Kensler TW, Yamamoto M, Zhang L-Y, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. American Journal of Respiratory Cell and Molecular Biology. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 16.Bhandari V, Elias JA. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic Biol Med. 2006;41:4–18. doi: 10.1016/j.freeradbiomed.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 17.Hillman NH, Moss TJM, Kallapur SG, Bachurski C, Pillow JJ, Polglase GR, Nitsos I, Kramer BW, Jobe AH. Brief, large tidal volume ventilation initiates lung injury and a systemic response in fetal sheep. Am J Respir Crit Care Med American Thoracic Society. 2007;176:575–581. doi: 10.1164/rccm.200701-051OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tutor JD, Mason CM, Dobard E, Beckerman RC, Summer WR, Nelson S. Loss of compartmentalization of alveolar tumor necrosis factor after lung injury. Am J Respir Crit Care Med American Public Health Association. 1994;149:1107–1111. doi: 10.1164/ajrccm.149.5.8173748. [DOI] [PubMed] [Google Scholar]
- 19.Gullo A, Berlot G, Viviani M. The role of adult respiratory distress syndrome in the multiple organ dysfunction syndrome. Acta Anaesthesiol Scand Suppl. 1996;109:70–73. [PubMed] [Google Scholar]
- 20.Guengerich FP. Enzymatic oxidation of xenobiotic chemicals. Crit Rev Biochem Mol Biol. 1990;25:97–153. doi: 10.3109/10409239009090607. [DOI] [PubMed] [Google Scholar]
- 21.Guengerich FP. Cytochrome P450: what have we learned and what are the future issues? Drug Metab Rev. 2004;36:159–197. doi: 10.1081/dmr-120033996. [DOI] [PubMed] [Google Scholar]
- 22.Whitlock JP, Okino ST, Dong L, Ko HP, Clarke-Katzenberg R, Ma Q, Li H. Cytochromes P450 5: induction of cytochrome P4501A1: a model for analyzing mammalian gene transcription. FASEB J. 1996;10:809–818. doi: 10.1096/fasebj.10.8.8666157. [DOI] [PubMed] [Google Scholar]
- 23.Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 24.Badal S, Delgoda R. Role of the modulation of CYP1A1 expression and activity in chemoprevention: A mini review. J Appl Toxicol. 2014 doi: 10.1002/jat.2968. [DOI] [PubMed] [Google Scholar]
- 25.Shimada T, Inoue K, Suzuki Y, Kawai T, Azuma E, Nakajima T, Shindo M, Kurose K, Sugie A, Yamagishi Y, Fujii-Kuriyama Y, Hashimoto M. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis. 2002;23:1199–1207. doi: 10.1093/carcin/23.7.1199. [DOI] [PubMed] [Google Scholar]
- 26.Shimada T, Martin MV, Pruess-Schwartz D, Marnett LJ, Guengerich FP. Roles of individual human cytochrome P-450 enzymes in the bioactivation of benzo(a)pyrene, 7,8-dihydroxy-7,8-dihydrobenzo(a)pyrene, and other dihydrodiol derivatives of polycyclic aromatic hydrocarbons. Cancer Res. 1989;49:6304–6312. [PubMed] [Google Scholar]
- 27.Miyajima A, Furihata T, Chiba K. Functional analysis of GC Box and its CpG methylation in the regulation of CYP1A2 gene expression. Drug Metab Pharmacokinet. 2009;24:269–276. doi: 10.2133/dmpk.24.269. [DOI] [PubMed] [Google Scholar]
- 28.Okamoto T, Mitsuhashi M, Fujita I, Sindhu RK, Kikkawa Y. Induction of cytochrome P450 1A1 and 1A2 by hyperoxia. Biochem Biophys Res Commun. 1993;197:878–885. doi: 10.1006/bbrc.1993.2561. [DOI] [PubMed] [Google Scholar]
- 29.Sindhu RK, Sakai H, Kikkawa Y. Effect of hyperoxia on rat pulmonary and hepatic cytochrome P450 monooxygenases. Archives of Toxicology. 2000;73:540–546. doi: 10.1007/s002040050006. [DOI] [PubMed] [Google Scholar]
- 30.Couroucli XI, Welty SE, Geske RS, Moorthy B. Regulation of pulmonary and hepatic cytochrome P4501A expression in the rat by hyperoxia: implications for hyperoxic lung injury. Mol Pharmacol. 2002;61:507–515. doi: 10.1124/mol.61.3.507. [DOI] [PubMed] [Google Scholar]
- 31.Jiang W, Welty SE, Couroucli XI, Barrios R, Kondraganti SR, Muthiah K, Yu L, Avery SE, Moorthy B. Disruption of the Ah receptor gene alters the susceptibility of mice to oxygen-mediated regulation of pulmonary and hepatic cytochromes P4501A expression and exacerbates hyperoxic lung injury. J Pharmacol Exp Ther. 2004;310:512–519. doi: 10.1124/jpet.103.059766. [DOI] [PubMed] [Google Scholar]
- 32.Mansour H, Brun-Pascaud M, Marquetty C, Gougerot-Pocidalo MA, Hakim J, Pocidalo JJ. Protection of rat from oxygen toxicity by inducers of cytochrome P-450 system. Am Rev Respir Dis. 1988;137:688–694. doi: 10.1164/ajrccm/137.3.688. [DOI] [PubMed] [Google Scholar]
- 33.Sinha A, Muthiah K, Jiang W, Couroucli X, Barrios R, Moorthy B. Attenuation of hyperoxic lung injury by the CYP1A inducer beta-naphthoflavone. Toxicol Sci. 2005;87:204–212. doi: 10.1093/toxsci/kfi226. [DOI] [PubMed] [Google Scholar]
- 34.Couroucli XI, Liang Y-HW, Jiang W, Wang L, Barrios R, Yang P, Moorthy B. Prenatal administration of the cytochrome P4501A inducer, B-naphthoflavone (BNF), attenuates hyperoxic lung injury in newborn mice: implications for bronchopulmonary dysplasia (BPD) in premature infants. Toxicol Appl Pharmacol. 2011;256:83–94. doi: 10.1016/j.taap.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lingappan K, Jiang W, Wang L, Wang G, Couroucli XI, Shivanna B, Welty SE, Barrios R, Khan MF, Nebert DW, Roberts LJ, Moorthy B. Mice Deficient in the Gene for Cytochrome P450 (CYP)1A1 are More Susceptible than Wild Type to Hyperoxic Lung Injury: Evidence for Protective Role of CYP1A1 Against Oxidative Stress. Toxicol Sci. 2014 doi: 10.1093/toxsci/kfu106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matuschak GM. Organ interactions in critical illness: paradigms and mechanisms. New Horiz. 1994;2:413–414. [PubMed] [Google Scholar]
- 37.Goldfarb G, Nouel O, Poynard T, Rueff B. Efficiency of respiratory assistance in cirrhotic patients with liver failure. Intensive Care Med. 1983;9:271–273. doi: 10.1007/BF01691253. [DOI] [PubMed] [Google Scholar]
- 38.Shellman RG, Fulkerson WJ, DeLong E, Piantadosi CA. Prognosis of patients with cirrhosis and chronic liver disease admitted to the medical intensive care unit. Crit Care Med. 1988;16:671–678. doi: 10.1097/00003246-198807000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Fessel JP, Porter NA, Moore KP, Sheller JR, Roberts LJ. Discovery of lipid peroxidation products formed in vivo with a substituted tetrahydrofuran ring (isofurans) that are favored by increased oxygen tension. Proc Natl Acad Sci USA. 2002;99:16713–16718. doi: 10.1073/pnas.252649099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med American Thoracic Society New York, NY. 2000;162:1175–1177. doi: 10.1164/ajrccm.162.3.2001063. [DOI] [PubMed] [Google Scholar]
- 41.Montuschi P, Barnes PJ, Roberts LJ. Isoprostanes: markers and mediators of oxidative stress. FASEB J Federation of American Societies for Experimental Biology. 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 42.Dalton TP, Dieter MZ, Matlib RS, Childs NL, Shertzer HG, Genter MB, Nebert DW. Targeted knockout of Cyp1a1 gene does not alter hepatic constitutive expression of other genes in the mouse [Ah] battery. Biochem Biophys Res Commun. 2000;267:184–189. doi: 10.1006/bbrc.1999.1913. [DOI] [PubMed] [Google Scholar]
- 43.Cinti DL, Moldeus P, Schenkman JB. Kinetic parameters of drug-metabolizing enzymes in Ca 2+-sedimented microsomes from rat liver. Biochem Pharmacol. 1972;21:3249–3256. doi: 10.1016/0006-2952(72)90089-5. [DOI] [PubMed] [Google Scholar]
- 44.Matsubara T, Prough RA, Burke MD, Estabrook RW. The preparation of microsomal fractions of rodent respiratory tract and their characterization. Cancer Res. 1974;34:2196–2203. [PubMed] [Google Scholar]
- 45.Pohl RJ, Fouts JR. A rapid method for assaying the metabolism of 7-ethoxyresorufin by microsomal subcellular fractions. Anal Biochem. 1980;107:150–155. doi: 10.1016/0003-2697(80)90505-9. [DOI] [PubMed] [Google Scholar]
- 46.Bhakta KY, Jiang W, Couroucli XI, Fazili IS, Muthiah K, Moorthy B. Regulation of cytochrome P4501A1 expression by hyperoxia in human lung cell lines: Implications for hyperoxic lung injury. Toxicol Appl Pharmacol. 2008;233:169–178. doi: 10.1016/j.taap.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan MF, Wu X, Ansari GAS, Boor PJ. Malondialdehyde-protein adducts in the spleens of aniline-treated rats: immunochemical detection and localization. J Toxicol Environ Health Part A. 2003;66:93–102. doi: 10.1080/15287390306464. [DOI] [PubMed] [Google Scholar]
- 48.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. American Journal of Respiratory Cell and Molecular Biology. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perkowski S, Scherpereel A, Murciano J-C, Arguiri E, Solomides CC, Albelda SM, Muzykantov V, Christofidou-Solomidou M. Dissociation between alveolar transmigration of neutrophils and lung injury in hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1050–L1058. doi: 10.1152/ajplung.00067.2006. [DOI] [PubMed] [Google Scholar]
- 50.Reutershan J. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. AJP: Lung Cellular and Molecular Physiology. 2005;289:L807–L815. doi: 10.1152/ajplung.00477.2004. [DOI] [PubMed] [Google Scholar]
- 51.Shah D, Romero F, Stafstrom W, Duong M, Summer R. Extracellular ATP mediates the late phase of neutrophil recruitment to the lung in murine models of acute lung injury. AJP: Lung Cellular and Molecular Physiology. 2014;306:L152–L161. doi: 10.1152/ajplung.00229.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagato AC, Bezerra FS, Lanzetti M, Lopes AA, Silva MAS, Porto LC, Valença SS. Time course of inflammation, oxidative stress and tissue damage induced by hyperoxia in mouse lungs. Int J Exp Pathol. 2012;93:269–278. doi: 10.1111/j.1365-2613.2012.00823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lingappan K, Jiang W, Wang L, Couroucli XI, Barrios R, Moorthy B. Sex-specific differences in hyperoxic lung injury in mice: implications for acute and chronic lung disease in humans. Toxicol Appl Pharmacol. 2013;272:281–290. doi: 10.1016/j.taap.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Velten M, Britt RD, Heyob KM, Tipple TE, Rogers LK. Maternal dietary docosahexaenoic acid supplementation attenuates fetal growth restriction and enhances pulmonary function in a newborn mouse model of perinatal inflammation. J Nutr. 2014;144:258–266. doi: 10.3945/jn.113.179259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Britt RD, Velten M, Locy ML, Rogers LK, Tipple TE. The Thioredoxin Reductase-1 Inhibitor Aurothioglucose Attenuates Lung Injury and Improves Survival in a Murine Model of Acute Respiratory Distress Syndrome. Antioxid Redox Signal. 2014 doi: 10.1089/ars.2013.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhandari V, Elias JA. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic Biol Med. 2006;41:4–18. doi: 10.1016/j.freeradbiomed.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 57.Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:109–116. doi: 10.1164/ajrccm.160.1.9803046. [DOI] [PubMed] [Google Scholar]
- 58.St John RC, Mizer LA, Kindt GC, Weisbrode SE, Moore SA, Dorinsky PM. Acid aspiration-induced acute lung injury causes leukocyte-dependent systemic organ injury. J Appl Physiol. 1993;74:1994–2003. doi: 10.1152/jappl.1993.74.4.1994. [DOI] [PubMed] [Google Scholar]
- 59.Polglase GR, Hillman NH, Ball MK, Kramer BW, Kallapur SG, Jobe AH, Pillow JJ. Lung and systemic inflammation in preterm lambs on continuous positive airway pressure or conventional ventilation. Pediatr Res Nature Publishing Group. 2009;65:67–71. doi: 10.1203/PDR.0b013e318189487e. [DOI] [PubMed] [Google Scholar]
- 60.Comporti M, Signorini C, Leoncini S, Buonocore G, Rossi V, Ciccoli L. Plasma F2-isoprostanes are elevated in newborns and inversely correlated to gestational age. Free Radic Biol Med. 2004;37:724–732. doi: 10.1016/j.freeradbiomed.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 61.Cotton RB, Hazinski TA, Morrow JD, Roberts LJ, Zeldin DC, Lindstrom DP, Lappalainen U, Law AB, Steele S. Cimetidine does not prevent lung injury in newborn premature infants. Pediatr Res. 2006;59:795–800. doi: 10.1203/01.pdr.0000219397.35473.5f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seto V, Hirota C, Hirota S, Janssen LJ. E-Ring Isoprostanes Stimulate a Cl Conductance in Airway Epithelium via Prostaglandin E2-Selective Prostanoid Receptors. American Journal of Respiratory Cell and Molecular Biology. 2008;38:88–94. doi: 10.1165/rcmb.2007-0117OC. [DOI] [PubMed] [Google Scholar]
- 63.Carpenter CT, Price PV, Christman BW. Exhaled breath condensate isoprostanes are elevated in patients with acute lung injury or ARDS. Chest. 1998;114:1653–1659. doi: 10.1378/chest.114.6.1653. [DOI] [PubMed] [Google Scholar]
- 64.Jansson A-H, Eriksson C, Wang X. Effects of budesonide and N-acetylcysteine on acute lung hyperinflation, inflammation and injury in rats. Vascul Pharmacol. 2005;43:101–111. doi: 10.1016/j.vph.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 65.Rogers LK, Tipple TE, Britt RD, Welty SE. Hyperoxia exposure alters hepatic eicosanoid metabolism in newborn mice. Pediatr Res. 2010;67:144–149. doi: 10.1203/PDR.0b013e3181c2df4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyurina YY, Tyurin VA, Kaynar AM, Kapralova VI, Wasserloos K, Li J, Mosher M, Wright L, Wipf P, Watkins S, Pitt BR, Kagan VE. Oxidative lipidomics of hyperoxic acute lung injury: mass spectrometric characterization of cardiolipin and phosphatidylserine peroxidation. AJP: Lung Cellular and Molecular Physiology. 2010;299:L73–L85. doi: 10.1152/ajplung.00035.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]