

Significance
Enterohemorrhagic Escherichia coli (EHEC) is a foodborne pathogen that can cause bloody diarrhea and hemolytic uremic syndrome, which can lead to severe clinical complications such as kidney failure. The main factors triggering disease are well known and include type 3 secreted effectors, adhesins, and Shiga toxins. Much less is known about how these factors are induced in response to the environmental transition that bacteria experience during transfer into and passage through the host. We show here that although positive regulators of virulence are induced during passage through the host, they are only activated to increase virulence as a result of force generated by host cell contact. Thus, mechanosensation is a way of integrating multifactorial environmental cues to fine-tune virulence regulation.
Keywords: enterohemorrhagic Escherichia coli, locus of enterocyte effacement, attaching/effacing pathogens, gastrointestinal infection, mechanosensing
Abstract
Enterohemorrhagic Escherichia coli (EHEC) is a foodborne pathogen causing hemorrhagic colitis and hemolytic uremic syndrome. EHEC colonizes the intestinal tract through a range of virulence factors encoded by the locus of enterocyte effacement (LEE), as well as Shiga toxin. Although the factors involved in colonization and disease are well characterized, how EHEC regulates its expression in response to a host encounter is not well understood. Here, we report that EHEC perceives attachment to host cells as a mechanical cue that leads to expression of LEE-encoded virulence genes. This signal is transduced via the LEE-encoded global regulator of LEE-encoded regulator (Ler) and global regulator of Ler and is further enhanced by levels of shear force similar to peristaltic forces in the intestinal tract. Our data suggest that, in addition to a range of chemical environmental signals, EHEC is capable of sensing and responding to mechanical cues to adapt to its host’s physiology.
Pathogens frequently undergo drastic environmental transitions as a direct result of their transmission between different environmental and host niches. In doing so, their gene expression patterns dramatically change to achieve niche adaptation and ensure the energy efficiency necessary for survival. Individual cues causing such environmental switches are generally well understood across a range of pathogenic organisms. How integration of such multifactorial cues and, as a result, robust regulation of virulence in response to a range of different hosts is achieved and has evolved is much less understood.
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 is a foodborne pathogen and an important cause of bloody diarrhea worldwide (1). In some cases, EHEC infection can lead to hemolytic uremic syndrome and severe clinical complications, including kidney failure. EHEC can persist in environmental niches, as well as colonize the gastrointestinal tract of ruminants and human hosts. Virulence factors contributing to intestinal colonization and establishment of disease in humans are well characterized and include type 3 secreted effector proteins, factors mediating intimate adhesion (Tir/Intimin), and Shiga toxins. Factors implicated in the formation of attaching and effacing lesions, which leads to the loss of microvilli from the intestinal brush border and, as a result, severe diarrhea, include the type 3 secretion system, as well as Tir (translocated intimin receptor) and Intimin (2, 3). These are encoded by a pathogenicity island called the locus of enterocyte effacement (LEE), consisting of five major transcriptional units, LEE1–LEE5 (4). All five units are subject to shared regulation by Ler (LEE-encoded regulator), the master regulator of LEE and of other, non-LEE-encoded virulence factors (5). This genetic organization is conserved across other attaching and effacing pathogens, including enteropathogenic E. coli and Citrobacter rodentium (6, 7). Ler is encoded in the first transcriptional unit of LEE (LEE1) and works mainly by antagonizing global gene repression imposed by the histone-like nucleoid-structuring (H-NS) protein (8). Regulation of Ler is responsive to many environmental cues, reflective of the transition in lifestyle, as a result of uptake by and passage through the host. These include changes in metabolites, CO2 concentration, and the presence of host immune effectors and adrenal hormones, among others (9–12). Many of these cues directly converge on Ler, whereas others require the global regulator of Ler (GrlA), a LEE-encoded positive regulator of Ler expression, but all result in global regulation of LEE-encoded genes, and thus virulence (13–15). However, it is not known how these multifactorial environmental cues are integrated to achieve a spatially and temporally coordinated response to the presence of the host tissue. Here, we describe how initial attachment to host cells generates a mechanical cue, which is further enhanced by fluid shear levels present in the host intestinal tract and is required to fully activate Ler and, thus, LEE-encoded virulence mechanisms, in a GrlA-dependent manner. Our data suggest that, in addition to a range of chemical signals, EHEC is capable of directly sensing and responding to mechanical cues to adapt to its host’s physiology and to fine-tune virulence activation. In light of recently published data demonstrating mechanosensation as a regulatory cue inducing Pseudomonas aeruginosa virulence, this study highlights a remarkable case of parallel evolution in which functionally distinct pathogens have integrated mechanosensation as a basic physical mechanism into their regulatory circuitry to achieve control of virulence pathways (16).
Results
Attachment to Host Cells Triggers LEE Induction in a GrlA-Dependent Manner.
LEE1 is the first transcriptional unit within the LEE region and encodes Ler, the master regulator of EHEC virulence gene expression. Previous reports show only a moderate induction of LEE1 promoter activity on exposure to individual environmental cues, but many of these studies were done using E. coli K12 as a surrogate strain, thus eliminating many EHEC-specific factors relevant to virulence regulation (15, 17). Others were done in EHEC strains, but not in the context of host cells (18). In this study, we set out to investigate the direct effects of host cell attachment on LEE-encoded virulence gene regulation in the EHEC strain Sakai 813, a Shiga-toxin-negative derivative of the original Sakai isolate. We analyzed LEE1 promoter (PLEE1) activity, using EHEC reporter strains transformed with either PLEE1-lacZ or PLEE1-gfp transcriptional fusions, on contact with host cells. We infected HeLa epithelial cells with EHEC for 4 h and first analyzed LEE1 promoter induction and infection phenotype in situ, using fluorescence microscopy of PLEE1-gfp reporter strains. Wild-type bacteria efficiently attached to HeLa cells and formed actin pedestals, apparent from the fluorescent-actin staining test, as previously described (Fig. 1A) (19). Most host-attached bacteria also showed strong LEE1 promoter activation. Strikingly, bacteria adsorbed to the glass slide, rather than attached to host cells, showed no or low GFP fluorescence, indicating that ler induction is enhanced on attachment to host cells compared with exposure to DMEM alone, which has previously been described as a cue for ler activation (Fig. 1C) (17). Because GrlA is a LEE-encoded activator of ler, and thus the entire LEE region, we also tested LEE1 promoter activation in a ΔgrlA background. In contrast to wild-type bacteria, LEE1 promoter activity remained low in a ΔgrlA background, even in bacteria attached to host cells (Fig. 1 B–D). Lower LEE1 promoter induction, and thus lower activation of the entire LEE region in the ΔgrlA background, was also apparent from the infection phenotype: both the number of attached bacteria per host cell and the bacteria’s ability to form actin pedestals was significantly decreased (Fig. 1 E and F). Introduction of the different extrachromosomal transcriptional reporters did not, in itself, alter the bacteria’s ability to attach or form pedestals: both EHEC wild-type and wild-type containing a previously described, constitutively active PLEE1-gfp fusion (PLEE199T-gfp) showed similar levels of attachment and pedestal formation (Fig. S1) (17).
Fig. 1.

Attachment to host cells triggers LEE1 promoter activation in a GrlA-dependent manner. EHEC wild-type (A) or EHEC ΔgrlA (B) harboring a PLEE1-gfp transcriptional fusion as reporter were used to infect HeLa cells [multiplicity of infection (MOI) 10, 4 h]. Samples were fixed and DNA (Hoechst), reporter activation (GFP), and F-actin (rhodamine-phalloidin) were visualized by fluorescence microscopy. Several actin pedestals caused by EHEC attachment are marked by arrows. Example of an EHEC bacterium adsorbed to the glass slide, rather than attached to host cells, is marked by an asterisk. (Scale bar, 10 μm.) Percentage GFP-positive bacteria (C), average GFP intensity per bacterium (for GFP-positive cells) (D), number of attached bacteria per host cell (E), and number of pedestals per host cell (F) were determined from these experiments. Data are representative of three independent experiments (>100 HeLa cells each). The asterisk denotes significant differences between samples based on Student’s t test (P < 0.05). ns, not significant (P ≥ 0.05).
We also tested LEE1 promoter induction in EHEC strains transformed with PLEE1-lacZ transcription fusions. β-galactosidase activity was measured in host-attached or nonattached reporter strains isolated from infected host cell cultures and normalized to bacterial counts determined from these samples (Fig. S2A). Exposure to DMEM (the cue experienced by nonadherent bacteria isolated from infected cultures) resulted in a moderate increase in ler induction, which is in agreement with previous findings (17). Host-adherent bacteria, in contrast, showed strongly increased LEE1 promoter activity (∼14-fold compared with EHEC grown in LB and approximately sevenfold compared with DMEM-induced, nonadherent bacteria). Similar to what we observed with the PLEE1-gfp reporter strain, induction of PLEE1-lacZ was GrlA-dependent (Fig. S2B). LEE1 induction was also observed using PLEE1-gfp and PLEE1-lacZ transcription reporters in wild-type, but not ΔgrlA, strains on bacterial attachment to Caco-2 intestinal epithelial cells, similar to what was observed in HeLa cells (∼10-fold induction compared with DMEM-induced, nonadherent bacteria; Fig. S3). The ΔgrlA strain showed significantly lower levels of attachment and pedestal formation compared with the wild-type strain. However, the overall level of bacterial attachment was lower in Caco-2 cells compared with in HeLa cells.
Attachment-Dependent LEE1 Promoter Activation Is Bacteria-Driven and Is Independent of the Host Response to Infection.
Stable attachment of EHEC to host cells is a multifactorial process and is the result of a complex interplay between bacterial and host cell signaling. This raises the question of whether GrlA-dependent LEE1 induction is driven by bacterial signaling alone or whether host-derived signals, which form part of the host response to infection, are required as well. First, we tested whether de novo protein synthesis in the host cells was required for attachment-dependent LEE1 induction. Pretreatment of HeLa cells with cycloheximide before infection did not change the overall infection phenotype, nor did it alter LEE1 induction levels (Fig. 2). Next, we asked whether host cytoskeletal rearrangements leading to pedestal formation were required for LEE1 induction. We analyzed infection phenotype and LEE1 promoter activity in EHEC wild-type-infected HeLa cells after pretreatment with cytochalasin D, which inhibits actin polymerization, and thus pedestal formation. Although cytochalasin D treatment abolished pedestal formation, neither overall bacterial attachment nor LEE1 activation was affected by the drug treatment (Fig. 2 C–G). We conclude that LEE1 promoter activation is likely bacteria-driven, as it does not require cues based on de novo protein synthesis or actin rearrangements derived from the host cells as a result of infection.
Fig. 2.

Induction of LEE1 is bacteria-driven, and a host response to infection is not required for signal transmission to GrlA. HeLa cells were infected with EHEC harboring a PLEE1-gfp transcriptional fusion (MOI 10, 4 h), following pretreatment with DMSO as control (A), 10 μg/mL cycloheximide (B), or 1 μg/mL cytochalasin D (C) for 1 h. Samples were fixed and DNA (Hoechst), reporter activation (GFP), and F-actin (rhodamine-phalloidin) were visualized by fluorescence microscopy. (Scale bar, 10 μm.) Percentage GFP-positive bacteria (D), average GFP intensity per bacterium (for GFP-positive cells) (E), number of attached bacteria per host cell (F), and number of pedestals per host cell (G) were determined for untreated (U), cycloheximide-treated (CHX), and cytochalasin d-treated (CD) cells. Data are representative of three independent experiments (>100 HeLa cells each). The asterisk denotes significant differences between samples based on Student’s t test (P < 0.05). ns, not significant (P ≥ 0.05); NA, not analyzed (no pedestals formed in CD-treated cells).
LEE1 Activation Results Directly from Host Attachment and Is not the Result of Positive Selection for Stochastic LEE1 Activation through Adhesion.
Arguably, the selective induction of LEE1 we observe in host-adherent cells could be brought about by at least two different mechanisms: LEE1 induction could be a result of host attachment, and thus adhesion would act as a cue for induction, or it could a result of stochastic LEE1 activation in nonadherent cells, and then positive selection of bacteria with high LEE activation levels for host attachment, through their enhanced capability to engage with the host cell surface. To distinguish between these two mechanisms, we measured LEE1 induction, using a fluorescence plate assay. EHEC wild-type strains containing promoterless gfp, inducible PLEE1-gfp, or constitutively active PLEE199T-gfp were incubated in a plate either in the presence or absence of host cells, and total fluorescence per well was measured over time. In the presence of host cells, fluorescence of the constitutively active reporter was initially high and slightly increased during the 4-h course of the experiment, reflecting bacterial proliferation (Fig. 3A). Fluorescence of the promoterless reporter (background fluorescence) remained low over the same time course. Fluorescence from the inducible LEE1 promoter (PLEE1-gfp) was initially low, but increased significantly over the course of the experiment to reach levels to match those of the constitutive reporter at 4 h. The rate of fluorescence increase over time was thus much higher for the PLEE1-gfp than the PLEE199T-gfp reporter strain, indicating LEE1 induction, rather than an increase resulting from cell proliferation alone. In the absence of host cells, both rates matched, indicating that LEE1 induction was a result of host attachment, rather than selective attachment to host cells resulting from adhesion-independent stochastic activation (Fig. 3B). No significant increase in the fluorescence rate of the PLEE1-gfp reporter was observed in a ΔgrlA background, even in the presence of host cells (Fig. 3C). Because the growth rates of both wild-type and mutant strains are similar (Fig. S4), this confirms the GrlA-dependence of adhesion-dependent LEE1 induction. We further tested EHEC deletion strains deficient for either Tir (Δtir) or Intimin (Δeae), two factors involved in the stable attachment of EHEC to host cells. Neither of these two mutants showed an increased rate of fluorescence (and thus LEE1 induction) compared with PLEE199T-gfp (Fig. 3 D and E). Growth rates were unaffected by either tir or eae deletion (Fig. S4). Taken together, these data better align with a scenario in which host attachment precedes and acts as a cue for LEE1 induction.
Fig. 3.

Population-level analysis of LEE1 induction rates in EHEC wild-type and mutant strains. Fluorescence intensity (AFU) was measured as a read-out for promoter activation, using promoterless gfp (blue), PLEE1-gfp (red), or PLEE199T-gfp (green) reporter constructs in EHEC wild-type cells grown in the presence (A) or absence (B) of host cells. Fluorescence was also measured in EHEC ΔgrlA (C), Δtir (D), and Δeae (E) strains incubated in the presence of HeLa cells for 1, 2, 3, or 4 h. Data are representative of three independent experiments done in triplicate. Asterisks denote significant differences between samples based on Student’s t test (P < 0.05). ns, not significant (P ≥ 0.05).
Attachment-Dependent Activation via GrlA Underlies Positive Feedback Regulation.
EHEC produces several adhesins that facilitate its interaction with host cells, including fimbriae and Tir/Intimin (20). Because both Intimin and its type 3-secreted receptor, Tir, are part of the LEE regulon, we investigated whether attachment underlies positive feedback regulation. Deletion of either tir or eae, encoding Tir and Intimin, respectively, decreased host adhesion significantly, both at early (1 h) and later (4 h) points (Fig. 4). The grlA deletion mutant showed no significant difference in its initial attachment to host cells. However, after 4 h of infection, the number of host-adherent bacteria was significantly decreased (approximately fourfold) compared with wild-type bacteria. This coincides with the time frame for full LEE1 induction (Fig. 3A).
Fig. 4.

Bacterial attachment over time in EHEC wild-type and deletion strains. HeLa cells were infected with EHEC wild-type or deletion strains (MOI of 10), and bacterial attachment to host cells was determined after 1 h (gray bars) or 4 h (black bars) of infection by dilution plating. Data are representative of three independent experiments done in triplicate. The asterisk denotes significant differences between wild type and deletion strains at the respective time, based on Student’s t test (P < 0.05). ns, not significant (P ≥ 0.05).
The LEE1 Promoter Is Mechanoresponsive, and Its Induction Is Independent of the Mode of Attachment.
In a bid to identify whether a specific host receptor is required for attachment-dependent LEE1 induction, we immobilized EHEC on a range of pure substrates, each mimicking a different type of interaction between bacteria and the host cell surface. These included electrostatic interactions between the negatively charged bacterial cell wall and positively charged poly-l-lysine, Tir–Intimin interaction, and immobilization using an antibody recognizing the O-antigen moiety of EHEC lipopolysaccharide. Immobilization on all three types of substrates induced LEE1 in a GrlA-dependent manner, albeit to different degrees (Fig. 5). In contrast, treatment of bacteria with these adhesion substrates in solution had no significant effect on LEE1 induction (Fig. S5). However, in each case, exposure of substrate-immobilized bacteria to increasing levels of fluid shear (0.1–10 dynes/cm2) caused a further increase in LEE1 promoter activity compared with the activity observed under static conditions. Although this behavior was independent of the mechanism of bacteria–substrate interaction, the rate of induction with increasing fluid shear varied, depending on the substrate used for immobilization, but saturated at ∼17,000 AFU (arbitrary fluorescence units) per cell (corresponding to sevenfold induction compared with static conditions; Fig. 5 D, H, and L). The number of immobilized bacteria per field did not change significantly with increasing fluid shear, meaning bacteria could withstand the increasing shear force and remained stably attached to the substrate in each case. The level of substrate attachment did not generally alter between wild-type and grlA deletion mutant, with the exception of bacteria immobilized on Tir peptide, in which case attachment was lower for the ΔgrlA strain but also remained stable with increased shear force (Fig. S6).
Fig. 5.

LEE1 induction is independent of the mode of attachment, but the shape of the force response curve is substrate-dependent. EHEC wild-type (A, E, I) or ΔgrlA (B, F, J) strains containing a PLEE1-gfp reporter were introduced into substrate-coated flow cells and incubated for 1 h under static conditions, followed by 3 h of flow to give a defined fluid shear force ranging from 0 to 10 dynes/cm2. Substrates included poly-l-lysine (A–D), Tir-peptide (E–H), and α-LPS antibody (I–L) and were chosen to represent different modes of bacterial attachment. Images are representative of bacteria incubated under static conditions (0 dynes/cm2). (Scale bar, 5 μm.) After the experiment, average fluorescence intensity (AFU) per bacterium was determined from image analysis, and values were blotted as fold-change compared with wild-type EHEC on poly-K under static conditions (D, H, L). Data are representative of three independent experiments (>100 cells each). The asterisk denotes significant differences between samples based on Student’s t test (P < 0.05).
To analyze LEE1 induction and phenotypic changes during infection, host-adherent EHEC strains were exposed to increasing levels of fluid shear (Fig. 6). Using imaging analysis of gfp-reporter strains attached to HeLa cells, we observed gradual LEE1 induction in a GrlA-dependent manner under increasing levels of fluid shear (0.1–10 dynes/cm2). The level of LEE1 induction increased under fluid shear compared with static conditions, but saturated at ∼19,000 AFU per cell (corresponding to 3.5-fold induction compared with static conditions) and did not further increase under shear flows of up to 10 dynes/cm2 (Fig. 6B). This increase in LEE1 induction in response to fluid shear was partially mirrored by a change in infection phenotype, with more attached bacteria progressing to stable attachment (i.e., pedestal formation) under flow compared with static conditions (Fig. 6 C and D). Nonadherent bacteria exposed to flow conditions did not show increased levels of LEE1 induction (Fig. 6E).
Fig. 6.
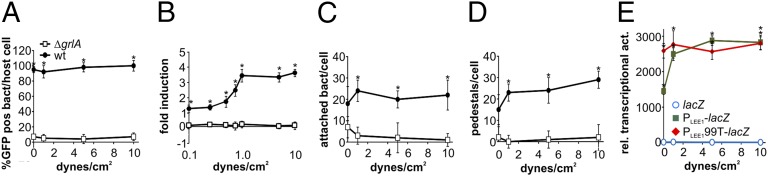
Fluid shear exacerbates LEE1 activation in host-attached bacteria. EHEC wild-type (black circles) or ΔgrlA strains (white squares) containing a PLEE1-gfp reporter were used to infect HeLa cells grown in glass flow cells and incubated for 1 h under static conditions, followed by 3 h of flow to give a defined fluid shear force ranging from 0 to 10 dynes/cm2. After the experiment, percentage GFP-positive bacteria per cell (A), fold-change in average GFP intensity per bacterium compared with static conditions (B), attached bacteria per cell (C), and pedestals per cell (D) were determined from image analysis. Data are representative of three independent experiments (>100 HeLa cells each). HeLa cells grown in glass flow cells were also infected with EHEC wild-type strain containing promoter-less lacZ (blue), PLEE1-lacZ (green), or PLEE199T-lacZ (red) reporters, as described earlier. After the experiment, cells were detached from the flow cells, using Triton-X100, and samples were used to determine relative transcriptional activities (E). Data are representative of three independent experiments performed in triplicate. The asterisk denotes significant differences between samples based on Student’s t test (P < 0.05).
Only Free, not GrlR-Bound, GrlA Is Mechanoresponsive.
It is well documented that GrlR acts as a repressor of GrlA-mediated LEE1 promoter induction, and thus LEE activation, by sequestering a portion of the cell’s GrlA in a (GrlR)2–GrlA complex (21). We therefore tested whether both free and GrlR-bound pools of GrlA are mechanoresponsive. If host attachment acts on the GrlRA complex to relieve GrlR-mediated repression, deletion of grlR should mimic the effect of host attachment. We thus compared LEE1 induction in EHEC wild-type and ΔgrlR strains containing PLEE1-lacZ transcriptional fusions. Deletion of grlR enhanced LEE1 induction by approximately twofold, but did not mimic the strong induction seen in host-adherent bacteria (Fig. S7). This suggests that attachment-mediated activation of GrlA is not achieved merely by relieving GrlR-mediated suppression of GrlA, and that other, GrlR-independent modes of regulating GrlA activity exist.
We also analyzed LEE1 promoter activity and infection phenotype in EHEC wild-type cells overexpressing GrlR, both GrlR and GrlA, or GrlA alone. Cells infected with EHEC expressing additional GrlR showed a very similar phenotype to cells infected with the ΔgrlA strain: PLEE1-gfp activity, number of attached bacteria, and pedestal formation were significantly decreased compared with in cells infected with EHEC wild-type bacteria (Fig. 7A). GrlA overexpression, in contrast, led to a hyperinfective phenotype, with an approximately twofold increase in both the number of attached bacteria and pedestals formed (Fig. 7C), but this phenotype was not recapitulated with the GrlRA overexpressing strain (Fig. 7B), which behaved similar to the EHEC wild-type strain. These results were recapitulated using PLEE1-lacZ reporter strains overexpressing GrlR, GrlRA, or GrlA (Fig. 7H). LEE1 induction was slightly enhanced in both the GrlRA and GrlA overexpressing wild-type cells harvested from the supernatant during infection, or from cells grown in planktonic cultures. This slight enhancement in LEE1 induction was exaggerated by host attachment, where GrlA overexpression caused a ∼13-fold induction of LEE1 over wild-type cells (which, themselves, show a 14-fold induction compared with planktonic cells). These data confirm that only free GrlA is mechanoresponsive and can induce LEE1, whereas GrlRA complex remains unaffected by this stimulus. Our data also suggest that the cellular pool of free GrlA is not, in itself, competent to fully induce LEE1, but becomes activated as a result of host attachment via an as-yet-unidentified mechanism.
Fig. 7.

Only free, but not GrlR-bound, GrlA is competent for attachment-mediated LEE1 induction; attachment does not relieve GrlR-mediated repression of GrlA. EHEC wild-type strain harboring a PLEE1-gfp transcriptional fusion as reporter and GrlR (A), GrlRA (B), or GrlA (C) expression vectors were used to infect HeLa cells (MOI 10, 4 h). Samples were fixed and DNA (Hoechst), reporter activation (GFP), and F-actin (rhodamine-phalloidin) were visualized by fluorescence microscopy. (Scale bar, 10 μm.) Percentage GFP-positive bacteria (D), average GFP intensity per bacterium (for GFP-positive cells) (E), number of attached bacteria per host cell (F), and number of pedestals per host cell (G) were determined from these experiments. Data are representative of three independent experiments (>100 HeLa cells each). HeLa cells were also infected (MOI 10, 4 h) with EHEC wild-type strain harboring a PLEE1-lacZ transcriptional fusion as reporter and empty vector (cont), GrlR, GrlRA, or GrlA expression constructs (H). Nonadherent bacteria (red) were recovered from the supernatant. Host cells were then washed and Triton-X100 lysed to recover adherent bacteria (green). Both fractions were used to determine β-galactosidase activity, and results were normalized to cfu/mL and are shown as relative transcriptional activity. Relative transcriptional activity was also determined for bacteria grown in planktonic LB cultures (blue). The asterisk denotes significant differences between bacteria harboring empty vector and expression constructs, based on Student’s t test (P < 0.05; n = 3). ns, not significant (P ≥ 0.05).
Discussion
Human disease caused by EHEC infection is usually the result of foodborne transmission. Thus, bacteria exit the ruminant gastrointestinal tract and persist on contaminated food matter before being taken up into a human host, where they colonize and cause diarrheal disease. After human uptake, bacteria are exposed to a range of host-specific cues, including a shift in temperature, passage through the acidic stomach environment, neutralization through bicarbonate exposure, and finally, the intestinal environment. It has always been assumed that sequential exposure to these host-specific triggers is sufficient to induce virulence exclusively within the human host niche, the intestine. Previous studies have indeed demonstrated induction of Ler, and thus LEE, in response to environmental stimuli. For example, GrlA is expressed in response to bicarbonate released by the pancreas, which partially induces LEE, and thus virulence (22, 23). Here, we show that although the levels of GrlA have a subtle effect on Ler activation, full virulence induction is only achieved through host attachment. This departs from our previous understanding of GrlA-based regulation, which was thought to require GrlR for inhibition and release of GrlA from the GrlR complex to achieve activation. In contrast to this, our data give strong evidence supporting the hypothesis that full induction by GrlA relies on mechanically stimulated activation of free GrlA, whereas the same cue does not activate GrlR-bound GrlA. How exactly GrlA becomes competent to bind to or activate the LEE1 promoter is clearly more complex than a transition from GrlR-bound to unbound states. It could be a result of a change in subcellular localization, posttranslational modification, or additional binding partners; these possibilities will be addressed in future work. This mechanism of virulence induction underlies positive feedback regulation, as the LEE includes both Tir and Intimin, factors required for intimate host attachment. Although EHEC adhesion is mediated by multiple components, and thus LEE induction does not strictly require Tir/Intimin, their presence reinforces existing bacterial attachment, and thus optimizes mechanotransduction.
Taken together, our data suggest that although exposure to early host environmental triggers may cause basal activation of the LEE, and thus poise the system to respond, full activation of virulence requires two components of mechanosensation: First, direct contact with and attachment to the host cell surface, which contributed to an approximately sevenfold induction over host exposed but nonattached bacteria, and second, enhancement of the thus-generated force in response to fluid shear levels comparable to those in the intestinal lumen, which leads to a further three- to fourfold activation of LEE1 in bacteria experiencing fluid shear compared with static conditions. Levels of fluid shear in the intestinal tract vary, depending on the exact physical location. According to hydrodynamic calculations, shear forces can approach 5 dynes/cm2 on the exposed brush border surface and can decrease to 2–3 dynes/cm2 between microvilli, depending on the flow rate (24). This highlights the physiological relevance of the LEE1 induction observed in our experiments, which reaches its maximum around 1 dyne/cm2. The basic physical sensation of mechanical forces thus acts to integrate a variety of host-specific chemical signals and ensures the complex arsenal of virulence factors is only fully expressed once the pathogen has reached its dedicated niche. Although such chemical stimuli may vary between different environments, and even different host organisms, these physical parameters are a conserved signal indicating the presence of a host surface.
Further work will be needed to understand what bacterial envelope components are involved in transduction of the mechanical signal sensed at the outer membrane in response to attachment to GrlA, the cytoplasmic regulator of virulence genes. The plate-based fluorescence assay used here to measure promoter activation in response to attachment (Fig. 3) can be easily adopted to conduct high-throughput screens to identify further bacterial components involved in signal perception and transduction across the bacterial cell envelope. The EHEC surface contains multiple mechanoresponsive elements and factors, which could have a putative role in signaling attachment, including flagella (during the early stages of attachment), fimbrial adhesins, or, as recently reported, the tip-associated adhesin PilY (16, 25–27). Recently, Siryaporn et al. described mechanosensing as the inducing signal for virulence in P. aeruginosa and implicated PilY as the outer membrane component of the signal transduction pathway, although further components of the transduction mechanism remained elusive (16). In comparison with attaching/effacing pathogens such as EHEC, P. aeruginosa colonizes different niches within the host and comprises a different arsenal of virulence mechanisms. However, surface attachment equally acts as a general and evolutionary conserved signal for the presence of a host cell. This opens up the exciting perspective that mechanoperception is an evolutionary robust and widely used principle used by microbial pathogens to integrate a large and divergent set of specific environmental cues.
Materials and Methods
The wild-type strain used in this study was an EHEC O157:H7 Sakai shiga-toxin-negative derivative strain (Sakai 813), a derivative of RIMD 0509952 (28). The gene-doctoring procedure was used to introduce gene deletions in this background, as previously described (29). All described strains and plasmids are listed in Table S1. Details of growth conditions, infection experiments under static and flow conditions, surface coating, imaging, and measurements of transcriptional activity are described in the SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank S. Sasakawa for the kind gift of EHEC strain Sakai 813. We thank Dave Lee, Jack Bryant, and Laura Sellars for technical assistance with the construction of transcriptional fusions and EHEC deletion strains. We thank Mark Webber for providing equipment access and technical assistance with flow cell experiments. We thank members of the A.M.K. and S.J.B. laboratories for critical reading and comments on the manuscript. This work was supported by grants from the Biotechnology and Biological Sciences Research Council (to A.M.K. and S.J.B.), the Leverhulme Trust (to S.J.B.), a King Abdulaziz University Scholarship (to G.A.), and a Commonwealth Academic Fellowship (to M.S.I.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1422986112/-/DCSupplemental.
References
- 1.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11(1):142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDaniel TK, Kaper JB. A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol Microbiol. 1997;23(2):399–407. doi: 10.1046/j.1365-2958.1997.2311591.x. [DOI] [PubMed] [Google Scholar]
- 3.Jerse AE, Yu J, Tall BD, Kaper JB. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc Natl Acad Sci USA. 1990;87(20):7839–7843. doi: 10.1073/pnas.87.20.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci USA. 1995;92(5):1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elliott SJ, et al. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 2000;68(11):6115–6126. doi: 10.1128/iai.68.11.6115-6126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott SJ, et al. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol Microbiol. 1998;28(1):1–4. doi: 10.1046/j.1365-2958.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- 7.Deng W, et al. Dissecting virulence: Systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci USA. 2004;101(10):3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bustamante VH, Santana FJ, Calva E, Puente JL. Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: Ler antagonizes H-NS-dependent repression. Mol Microbiol. 2001;39(3):664–678. doi: 10.1046/j.1365-2958.2001.02209.x. [DOI] [PubMed] [Google Scholar]
- 9.Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. Bacteria-host communication: The language of hormones. Proc Natl Acad Sci USA. 2003;100(15):8951–8956. doi: 10.1073/pnas.1537100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pacheco AR, et al. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492(7427):113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoh M, Bi Z, Matsuyama J, Nagayama K, Honda T. Effect of environmental conditions on proteins secreted by enterohemorrhagic Escherichia coli O26:H11. Microbiol Immunol. 2003;47(1):1–6. doi: 10.1111/j.1348-0421.2003.tb02779.x. [DOI] [PubMed] [Google Scholar]
- 12.Branchu P, et al. NsrR, GadE, and GadX interplay in repressing expression of the Escherichia coli O157:H7 LEE pathogenicity island in response to nitric oxide. PLoS Pathog. 2014;10(1):e1003874. doi: 10.1371/journal.ppat.1003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barba J, et al. A positive regulatory loop controls expression of the locus of enterocyte effacement-encoded regulators Ler and GrlA. J Bacteriol. 2005;187(23):7918–7930. doi: 10.1128/JB.187.23.7918-7930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyoda S, et al. The GrlR-GrlA regulatory system coordinately controls the expression of flagellar and LEE-encoded type III protein secretion systems in enterohemorrhagic Escherichia coli. J Bacteriol. 2006;188(16):5682–5692. doi: 10.1128/JB.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell RM, Sharp FC, Rasko DA, Sperandio V. QseA and GrlR/GrlA regulation of the locus of enterocyte effacement genes in enterohemorrhagic Escherichia coli. J Bacteriol. 2007;189(14):5387–5392. doi: 10.1128/JB.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siryaporn A, Kuchma SL, O’Toole GA, Gitai Z. Surface attachment induces Pseudomonas aeruginosa virulence. Proc Natl Acad Sci USA. 2014;111(47):16860–16865. doi: 10.1073/pnas.1415712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Islam MS, Bingle LE, Pallen MJ, Busby SJ. Organization of the LEE1 operon regulatory region of enterohaemorrhagic Escherichia coli O157:H7 and activation by GrlA. Mol Microbiol. 2011;79(2):468–483. doi: 10.1111/j.1365-2958.2010.07460.x. [DOI] [PubMed] [Google Scholar]
- 18.Tobe T, et al. Dual regulatory pathways integrating the RcsC-RcsD-RcsB signalling system control enterohaemorrhagic Escherichia coli pathogenicity. Mol Microbiol. 2005;58(1):320–333. doi: 10.1111/j.1365-2958.2005.04828.x. [DOI] [PubMed] [Google Scholar]
- 19.Knutton S, Baldwin T, Williams PH, McNeish AS. Actin accumulation at sites of bacterial adhesion to tissue culture cells: Basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 1989;57(4):1290–1298. doi: 10.1128/iai.57.4.1290-1298.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farfan MJ, Cantero L, Vidal R, Botkin DJ, Torres AG. Long polar fimbriae of enterohemorrhagic Escherichia coli O157:H7 bind to extracellular matrix proteins. Infect Immun. 2011;79(9):3744–3750. doi: 10.1128/IAI.05317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padavannil A, et al. Structure of GrlR-GrlA complex that prevents GrlA activation of virulence genes. Nat Commun. 2013;4:2546. doi: 10.1038/ncomms3546. [DOI] [PubMed] [Google Scholar]
- 22.Abe H, Tatsuno I, Tobe T, Okutani A, Sasakawa C. Bicarbonate ion stimulates the expression of locus of enterocyte effacement-encoded genes in enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 2002;70(7):3500–3509. doi: 10.1128/IAI.70.7.3500-3509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tauschek M, et al. Transcriptional analysis of the grlRA virulence operon from Citrobacter rodentium. J Bacteriol. 2010;192(14):3722–3734. doi: 10.1128/JB.01540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo P, Weinstein AM, Weinbaum S. A hydrodynamic mechanosensory hypothesis for brush border microvilli. Am J Physiol Renal Physiol. 2000;279(4):F698–F712. doi: 10.1152/ajprenal.2000.279.4.F698. [DOI] [PubMed] [Google Scholar]
- 25.Cairns LS, Marlow VL, Bissett E, Ostrowski A, Stanley-Wall NR. A mechanical signal transmitted by the flagellum controls signalling in Bacillus subtilis. Mol Microbiol. 2013;90(1):6–21. doi: 10.1111/mmi.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. Bacterial adhesion to target cells enhanced by shear force. Cell. 2002;109(7):913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 27.Tchesnokova V, et al. Shear-enhanced binding of intestinal colonization factor antigen I of enterotoxigenic Escherichia coli. Mol Microbiol. 2010;76(2):489–502. doi: 10.1111/j.1365-2958.2010.07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makino K, et al. (1998) Complete nucleotide sequences of 93-kb and 3.3-kb plasmids of an enterohemorrhagic Escherichia coli O157:H7 derived from Sakai outbreak. DNA Res 5(1):1–9. [DOI] [PubMed]
- 29.Lee DJ, et al. Gene doctoring: A method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol. 2009;9:252. doi: 10.1186/1471-2180-9-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.