

Significance
The paper describes the implementation of an approach to study the chemical makeup of human skin surface and correlate it to the microbes that live in the skin. We provide the translation of molecular information in high-spatial resolution 3D to understand the body distribution of skin molecules and bacteria. In addition, we use integrative analysis to interpret, at a molecular level, the large scale of data obtained from human skin samples. Correlations between molecules and microbes can be obtained to further gain insights into the chemical milieu in which these different microbial communities live.
Keywords: human skin, 3D mapping, mass spectrometry, 16S rRNA
Abstract
The human skin is an organ with a surface area of 1.5–2 m2 that provides our interface with the environment. The molecular composition of this organ is derived from host cells, microbiota, and external molecules. The chemical makeup of the skin surface is largely undefined. Here we advance the technologies needed to explore the topographical distribution of skin molecules, using 3D mapping of mass spectrometry data and microbial 16S rRNA amplicon sequences. Our 3D maps reveal that the molecular composition of skin has diverse distributions and that the composition is defined not only by skin cells and microbes but also by our daily routines, including the application of hygiene products. The technological development of these maps lays a foundation for studying the spatial relationships of human skin with hygiene, the microbiota, and environment, with potential for developing predictive models of skin phenotypes tailored to individual health.
The skin provides the interface between our internal molecular processes and the external environment. The stratum corneum (SC), the outer layer of the epidermis, is the most exposed organ of the human body and is composed of molecules derived from our own skin cells, our microbiota, and the environment (1). In addition, it may be possible that personal hygiene products, the clothing we wear, and the food we eat can affect the chemical composition of the skin (2, 3). The chemical environment of the skin is also critical for the development of microbial communities, yet there are no systematic workflows that can be used to correlate skin chemistry and microbes. Human skin microbiome inventories are changing our views of commensal organisms and their roles in establishing and maintaining the immune system and general epithelial health (4–7), but their role in biotransformation of skin molecules is as yet poorly characterized.
Understanding the molecular topography of the surface of the SC will provide insights into the chemical environment in which microbes reside on the skin and how in turn they modify this environment. To understand the relationship between the chemical milieu and the microbial communities, we must develop workflows that map the chemistry and microbiology of the human skin and that determine the associations between them. The composition of the human skin microbiota has been correlated with skin anatomy, dividing into moist, dry, and sebaceous microenvironments, and can be influenced by beauty and hygiene products (2–9). Regions such as the face, chest, and back, areas with a high density of sebaceous glands, promote growth of lipophilic microorganisms such as Propionibacterium and Malassezia. In contrast, less exposed regions, such as the groin, axilla, and toe web, which are higher in temperature and moisture content, are colonized by Staphylococcus and Corynebacterium (7, 10, 11). Previous studies have reported mass spectrometry (MS) methods to identify compounds from human skin (12, 13). However, these studies are based on analysis performed on selected and limited areas on the skin, which do not reflect the distinct chemical microenvironments over the entire body. To date, the topographical distribution of skin molecules has not been characterized. Similarly, because the paired data were not available, skin chemistry could not be correlated to microbial diversity. Here we have developed a general approach to visualize the chemical composition and the microbial community composition of human skin surface through the creation of 3D topographical maps. We anticipate the approach outlined in this manuscript can be readily adapted to study other microbiome–chemistry associations.
Results and Discussion
General Workflow for Construction of High-Spatial Resolution 3D Models.
To demonstrate the feasibility of mapping the molecular nature of the human skin surface, two volunteers, a male and a female, were recruited for this study. We did not seek to describe general differences between men and women, which would require a larger sample size of each sex. At the same time, our individual-specific observations can test whether these two individuals are the same or different in various microbiome and chemical characteristics and provide a view of factors influencing variation within each subject from a methodological standpoint. We therefore refer to the male volunteer as person 1 and the female volunteer as person 2. Each subject was sampled twice at ∼400 sites on their skin using cotton and soft foam swabs (Materials and Methods). These swabs were subjected to MS and 16S rRNA amplicon analysis, respectively (Fig. 1). Two types of MS were applied to each sample: Ultra-performance liquid chromatography/quadrupole time-of-flight (UPLC-QTOF) data were used for metabolite and peptide capture and included data-dependent tandem MS (MS/MS), and matrix assisted laser desorption/ionization TOF (MALDI-TOF) data were used for metabolite, peptide, and protein detection. MALDI-TOF was used to detect higher molecular weight molecules from skin samples without any preliminary pretreatment and using ∼1/300th of the sample volume that would have been needed for liquid chromatography MS (LC-MS), which was advantageous due to the limited material available. Identification of ions in 3D MALDI-TOF maps was followed up by MS/MS identification using linear ion trap (IT) quadrupole Fourier transform (LTQ-FT). Such MS analysis provides richer fragmentation information when the target is known compared with postsource decay on the MALDI-TOF.
Fig. 1.

Creation of 3D topographical maps of molecules and microbes distributed on the human skin. Samples were collected at ∼400 distinct body sites from each of two volunteers. For each sample, detection of molecules was performed using MS (UPLC-QTOF and MALDI-TOF) and bacteria using 16S rRNA amplicon sequencing. After 3D modeling, a map was rendered for each molecular feature and for each taxon detected in each spot on the body sampled. The 3D maps show body distribution of sphingosine and palmitoleic acid in person 1 and person 2, respectively. The data analysis involved integrative analysis of heterogeneous big data followed up by structural biochemical analysis of molecular features of interest. See also Fig. S1.
The 3D topographical maps visualizing distributions of metabolites, peptides, and bacteria were created from UPLC-QTOF, MALDI-TOF, as well as 16S rRNA amplicon sequence data that allow visual comparative localization of metabolites, peptides, and bacteria. The 3D mapping of both molecular and bacterial inventories onto 3D topographical models was performed on computer models of people in MATLAB. The mapping of sample collection spots involved assigning each sampling location to a spot on the 3D computer graphics model (Fig. 1 and Movies S1 and S2). Then, MALDI-TOF ion masses (m/z) and LC-MS molecular features [intervals of m/z and retention time (RT)] were quantified by integrating spectral intensities and modeled onto the 3D topographical maps. For 16S rRNA amplicon data, relative abundances corresponding to each detected genus at each body site were mapped. Using a color scale, the 3D topographical maps were rendered, revealing the spatial abundances of molecules corresponding to molecular features or bacterial taxa (Fig. 1). Mapping molecular features from the LC-MS data revealed distinct spatial distributions in areas such as the head, navel, hands, axilla, groin, ears, and feet in both volunteers (Fig. S1A). Mapping the MALDI-TOF data revealed particular distributions of a complementing set of molecules of higher molecular weights from the mass range of peptides and proteins (Fig. S1B). This indicates the presence of distinct and heterogeneous chemical environments on the surface of the human skin.
Large-Scale MS Molecular Networking.
To better interpret the MS data, molecular networking (14–16) was applied to UPLC-QTOF MS/MS data. Compounds are grouped based on their MS/MS similarity serving as a proxy for structural similarity and visualized in Cytoscape (16, 17) (Fig. 1). Molecular networking allows for tractable analysis of a large number of MS/MS spectra obtained in various experiments and highlights molecular families of chemically similar molecules (14–17). Briefly, to reduce the redundancy due to many spectra potentially generated for identical molecules, MSCluster, originally designed for proteomics experiments, was adapted to merge identical and nearly identical MS/MS spectra (18), and the resulting consensus spectra were further matched between each other by using a spectral alignment algorithm that calculates a cosine score to detect pairs of spectra with highly correlated fragmentation patterns (15, 16, 19). Cosine similarity scores range from 0 to 1, where 1 indicates perfectly matched MS/MS spectra. A cosine threshold of 0.65 was used to construct the molecular network in this study. That many of the MS/MS spectra could be matched to beauty and hygiene products reflects the lasting impact of our daily regimes.
The resulting correlation scores were imported into Cytoscape (17), and the network was organized using the FM3 force-directed layout (ref. 20 and apps.cytoscape.org/apps/fm3). Because the chemistry of molecules dictates how they fragment in the gas phase, this organization of the MS/MS data reveals clusters of related MS/MS spectra that represent molecular families (14–17).
To define the origin of many of the molecules and assign the molecular features to the human, microbial, or environmental component of the skin, LC-MS/MS data were collected on beauty products that both individuals used, common raw materials used in the formulation of beauty products (cosmetic ingredients), and commercially available chemical standards. In addition, cultures of 34 different bacteria and fungi from several genera, including Propionibacteria, Corynebacteria, Acinetobacter, Pseudomonas, Streptococcus, Escherichia, Bacillus, Lactobacillus, Staphylococcus, Candida, and Malassezia, which are known to be a part of the skin microbial community, were also subjected to LC-MS/MS (12, 21). Finally, LC-MS/MS data were collected on cultured human skin cells including basal and differentiated keratinocytes and human skin tissues collected from four donors, from three locations: neck, back, and scalp. All LC-MS/MS data collected in the study together with MS/MS spectral libraries including Massbank (22), National Institute of Standards and Technology (NIST) (www.nist.gov/index.html), representative data from METLIN (23), and in-house reference spectra were then subjected to molecular network analysis. Integrative analysis of skin swab samples together with other samples and spectral libraries allowed us to identify molecules observed in 3D topographical maps and to make a hypothesis on the origins of the molecules.
Molecular networking of LC-MS/MS data from skin swab samples, cosmetic ingredients, cultured cells, human skin tissues, and chemical standards together with the MS/MS libraries resulted in 15,544 nodes representing consensus of at least three or more identical MS/MS spectra. The majority of the spectra came from the skin swab samples; 34% of them were associated with person 1 but not person 2, 25% with person 2 but not person 1, and 41% matched both subjects. Moreover, 8% of the nodes matched beauty products and/or cosmetic ingredients. Around 0.5% matched cultured keratinocytes and human skin tissues, whereas ∼1% of nodes matched to microbial cultures. A total of 0.1% of the nodes matched to cultured skin cells and human skin tissues, microbes, and human swabs. This result shows that a large portion of annotated nodes could be matched to beauty products, more than to any other origin, reflecting the lasting impact of our beauty and hygiene products on the molecular composition of the outermost layer of the skin that is exposed to the environment. Less than 3% of the nodes could be correlated to MS/MS data from MS/MS spectral libraries, suggesting that unlike skin bacterial inventories, which are now believed to be nearing complete sampling (24), the vast majority of metabolites on the skin remain uncharacterized. Even when performing high-coverage molecular networking accounting for possible molecular modifications, differences in spectra due to concentration effects (e.g., MS/MS spectra of parent ions of lower intensity may have missing lower intensity daughter ions), temperature variations of the instrumentation during the 2-wk period of continuous data collection, and other experimental considerations found in the molecular network, more than 80% of the MS/MS spectra remain uncharacterized. This is not unlike the situation in early genome sequencing projects, where less than 20% of the genes could be annotated and will require the scientific community to develop creative strategies for the analysis and annotation of the detected molecules analogous to the strategy for annotating DNA sequences that has developed over the past two decades.
The sources of these uncharacterized molecules may range from secreted dietary molecules not present in the reference database; enzymatic, light- or air-induced modifications of molecules that would not be observed in culture; and beauty products used in the past but not included in the analysis. Cultured skin cells and microbes, and excised and stored human skin tissues, may not reflect the molecular makeup of in vivo and living systems, especially if human and/or bacterial cells, produce different metabolites when they interact. Such interaction is likely, given the expanded repertoire of metabolism seen in cocultured microbes compared with individually cultured species (25, 26). Similarly, most microbial taxa lack a representative member that has been cultured, and these microbes and their interactions with each other and with the host likely contribute molecules for which we cannot yet define the origin in the network. Finally, there are likely numerous environmental contributions from sources not yet considered. Although significant challenges remain in annotating the data, the chemistry detected in this study still provides considerable insight into the different chemical environments of the skin.
Putative annotations were obtained by matching MS/MS spectra to spectral libraries and then further confirmed by follow-up manual analysis. Specific molecules, discussed in the text, were further confirmed by follow-up manual analysis. The manual analysis was necessary because, unlike in genomics or proteomics, the statistical tools for false discovery rate estimation in untargeted metabolomics are yet to be developed. Furthermore, stereochemical and region-specific modifications or isomers of related molecules are difficult to assess automatically. For the named molecular families in this article, at least some family members were matched standards based on the parent mass, MS/MS, and RT. In SI Materials and Methods, we provide the details of the level of analysis for each of the named molecules. Fig. 2 highlights representative examples of molecular families with one or more identified molecules.
Fig. 2.

Structural biochemical analysis driven by topographical maps reveals molecules from various molecular families. The molecular features were selected based on their biogeographical localization as shown on the maps. The structural analysis was performed using molecular networking of UPLC-QTOF MS/MS data. The full network is shown in the middle; a node corresponds to a consensus MS/MS spectrum, and an edge represents the similarity between MS/MS spectra. The thickness of the edges (gray lines connecting nodes) indicates the level of similarity. Network has been generated using a cosine of 0.65 and then imported and visualized in Cytoscape. Subnetworks show examples of molecular families (A–H). The network nodes were annotated with colors showing molecular origin, and larger nodes highlight molecules coming from different known origins. CAPB, cocamidopropyl betaine; PC, phosphocholine; PS, phosphoserine; SLES, sodium lauryl ether sulfate. See also Figs. S2–S5.
To highlight specific molecules, members of the molecular families of phosphatidylcholine (PC, m/z 494, 522) and phosphatidylserine (PS, m/z 526) were found in the skin swabs, cultured keratinocytes, and the fungi Malassezia furfur and Candida albicans (Fig. S2 A–D). Both PC and PS lipid families are important phospholipids in eukaryotes, having structural and signaling functions (27, 28). Examples of molecular features that matched to human origin including basal, differentiated cultured keratinocytes and human skin tissues, but not to microbial samples, are the sphingosine lipid molecular family (Fig. 2 and Fig. S2 E and F), a sterol family that includes a match to cholesterol (Fig. 2 and Fig. S2G), a PC family (m/z 496) (Fig. 2 and Fig. S3A). A molecular family that includes a match to vitamin D3 (Fig. S3B) as well as glycocholic acid (Fig. S3C) and taurocholic acid (Fig. S3D) was detected from skin samples. Cholic acids, commonly described as bile acids, are important players of the lipid metabolism in the gut and help maintain a healthy gut microbial community, thus representing a viable therapeutic intervention against pathogens (29). The function of these bile acids on the skin is unknown. Matches to phytosphingosines (Fig. S3E) were detected on the skin but not in the beauty products or cultured keratinocytes. Some molecular families were detected in both skin samples and skin bacteria cultures, such as the fatty acids oleic acid (Fig. S3F) and palmitic acid (Fig. S4A), which were found in Pseudomonas, and palmitoleic acid, which was detected in Acinetobacter, Candida, and Malassezia cultures (Fig. S4B). Tryptamine from the skin was identical to the molecular feature collected from the cultured microbe Staphylococcus (Fig. S4C). Not only did some molecular features from the human skin match molecular features from cultures, some also matched hygiene, diet, and/or beauty products and commonly used plasticizers for use in clothing and other plastics such as o-formylbenzoic acid (30) and food constituents or additives such as sinapinic acid (31, 32) (Fig. S4D) and oxidized polyethylene (33). This illustrates that our daily routines leave molecular traces on the skin that can be readily detected and are a significant component of the regiospecific chemical environment of the skin that, at the same time, can provide insight into personal habits (e.g., application of sunscreen, lotion, and so forth or not).
The molecular family that includes the C12 lauryl ether sulfate surfactant, a component of the shampoo used by person 1, was found on his head but not person 2’s (Fig. 2 and Fig. S5A). Similarly, the molecular family that includes cocoamidopropylbetaine, also known as lauroylamide propylbetaine, a surfactant present in both person 1’s and person 2’s shampoo, was found on the head of both subjects (Fig. 2 and Fig. S5B) (34). Sunscreens such as avobenzone and octocrylene (Fig. S5 C and D), ingredients in several cosmetic formulations, were observed mostly in the face and chest of the female. Finally, polymeric substances that display the characteristic 58 Da difference pattern (polypropylene glycol subunit, –CH2CH2CH2O), found in beauty products, were associated with the axilla of person 2 (Fig. S5E). Both volunteers refrained from showering and from the application of hygiene and beauty products for 3 d before sampling to increase the possibility of detecting microbially produced molecules, but despite these precautions the dominant molecular features that could be annotated, surprisingly, came from hygiene and beauty products. Consequently, the data suggest that, through the creation of topographical skin maps, we may be able to detect both past and current behaviors. The data demonstrate that the human skin is not just made up of molecules derived from human or bacterial cells. The external environment, such as polymeric materials in plastics, as found in clothing, diet, hygiene, and beauty products, contributes significantly to the skin’s chemical composition. These molecular signatures of behavioral regimens remain visible on the skin and may affect our skin microbial communities; they can now be detected and correlated with the local microbial community.
3D Mapping of 16S rRNA Data.
To identify the bacteria present on the human skin surface and relate them to molecular localizations, phylogenetic analysis of bacterial and archaeal ribosomal 16S rRNA amplicon sequences from ∼400 sites of each volunteer was performed, revealing 850 distinct microbial operational taxonomic units (OTUs) at the 97% identity level (Dataset S1). Although 36 phyla were observed, some of them at only a few locations, the most common microbial taxa observed in this study were from the phyla Actinobacteria, Firmicutes, Proteobacteria, Cyanobacteria, and Bacteroidetes, in agreement with findings in the Human Microbiome Project (4–8) (Fig. S6A, Dataset S1, and microbial distribution maps folder available at massive.ucsd.edu/ProteoSAFe/status.jsp?task=6b9dcff3899e4d5f89f0daf9489a3a5e). Topographical mapping of 16S rRNA amplicon data revealed many distinct localizations of detected taxa (microbial distribution maps are available at massive.ucsd.edu/ProteoSAFe/status.jsp?task=6b9dcff3899e4d5f89f0daf9489a3a5e). The family Staphylococcaceae was found in moist areas, such as on the foot of both volunteers, under person 2’s breast and neck, and around person 1’s nose (Fig. S6A). The genus Staphylococcus was detected mainly on the foot of both volunteers and around the nose of person 1 (Fig. 3). The genus Propionibacterium was found on the sebaceous region, including the head, face, upper back, and upper chest, of both volunteers (Fig. 3). The genus Corynebacterium was most prevalent on the head, groin, and toe regions of both volunteers (Fig. 3). In addition, organisms such as Pseudomonas, a representative Gram-negative skin commensal, which is also involved in some important infections such as cystic fibrosis, and Lactobacillus, which is generally considered a member of the skin, vaginal, oral, and gut microbiota but also previously associated with skin, was detected over the entire skin surface of both volunteers (Fig. S6B) (1). Streptococcus (an abundant genus in the oral cavity), Haemophilis, and Rothia are all part of the normal community of oral microbes (35) but are present on the skin of both individuals, primarily around the mouth of person 1 (Fig. S6C). Finally, the phylum Cyanobacteria was found in increasing amounts on the hands, knees, and lower leg of person 2 but minimally on person 1 (Fig. S6B). Our 16S rRNA maps show that chloroplasts, which group phylogenetically within the Cyanobacteria and come from the plant extracts used on the hands of the female, are localized to that subject (36). These diverse microbes and plant materials, and their specific localization, must, to some extent, define the molecules observed on the skin.
Fig. 3.

The topographical map of representative distributions of bacteria from the genera Staphylococcus, Propionibacterium, and Corynebacterium, based on their relative amounts at each body location, for the female and male individuals. Red is the highest percentage of each genus that was detected, and blue is the lowest percentage; other colors are in between. See also Fig. S6.
3D Representation of Bacterial and Molecular Diversity.
To measure and correlate the region-specific molecular diversity and bacterial diversity, we used the Shannon index (37), a widely used statistical measure of diversity that has previously been used to define high- and low-diversity regions of the skin, and the evenness of the microbial population (5, 38, 39). We generated topographical maps of both molecular and bacterial diversity, based on Shannon index measurements from n = 394 samples for person 1 and n = 362 samples for person 2. The 3D maps reveal heterogeneous regions of high and low molecular and bacterial diversity on the skin surface, separately for each volunteer. Within each subject, there was no correlation between the molecular and microbial diversity (Fig. 4). Therefore, the majority of the features that are detected by MS do not come from microbes, consistent with our observation that the features from beauty products and hygiene products are prominently detected in the LC-MS data.
Fig. 4.

Representation of the microbial and molecular diversity using the Shannon index, comparing samples based on their microbial (16S rRNA amplicon) or molecular (UPLC-QTOF-MS) profiles. The Shannon diversity maps showing values of the Shannon index calculated for each sample separately from microbial (16S) and molecular (UPLC-QTOF) collections. Both molecular and microbial diversity is displayed for each volunteer, separately, highlighting regions of high and low diversity on the skin surface. For the color scale, blue corresponds to the minimum value of the Shannon index for the individual and red corresponds to the maximum value.
Spatial Correlation Between Molecular and Bacterial Distribution.
Although the Shannon diversity revealed different spatial patterns of diversity between molecules and microbes, this does not rule out spatial correlation between localizations of individual bacterial species and metabolites. We therefore set out to find specific molecules and bacteria that shared the same biogeographical regions. The spatial correlation can be used as a hypothesis-generating methodology. However, correlation does not directly imply causation; rather, a correlation of spatial localizations of bacterial phylum and molecular species enables the formulation of testable hypotheses or provides a specific question that can then be followed up with additional experiment. Spatial correlations between all detected molecules (from LC-MS data) and microbes were computed by calculating the Pearson correlation (Colocalization Analysis of Microbial and Molecular Communities). Spatially colocalized microbial taxa and molecular species were found in both volunteers. Discovered colocalizations were represented with a network and examined. Cross-correlations (correlation > 0.5, P value < 0.05) between the genus Propionibacterium (Fig. 5A) and 491 molecular features enabled the discovery of molecular features that have similar distributions to the bacterial taxa (Fig. 5B). Approximately 73% of these molecular features belonged to the lipid molecular families as judged by their RTs (between 300 and 500 s, consistent with the hydrophobic nature of lipids) (Fig. 5 C and D). These lipids were not detected among naturally produced products of cultured Propionibacteria. Lipids such as oleic acid (Fig. S3F), palmitic acid (Fig. S4A), and monoacylated glycerols monoolein and monopalmitin (Fig. S7 A and B) were found in larger amounts on the head, face, hands, chest, and back (Fig. 5D). These are all components of acyl glycerols that make up membranes of human cells. These observations highlight molecular interrelations among the microbiota, human skin, and environment and reveal molecular microenvironments on the epidermis.
Fig. 5.
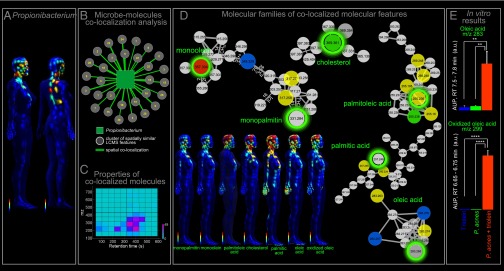
Propionibacterium genus colocalization with lipids on person 2’s body as well as in vitro study of triolein hydrolysis by P. acnes. (A) Topographical maps show the spatial distribution of this genus on the female and male individual. (B) Molecular UPLC-QTOF features spatially colocalized with the Propionibacterium bacterial taxon are displayed as a network, where a square node represents the Propionibacterium taxon, circular nodes represent clusters of tightly colocalized molecular features, and a node represents colocalization between the bacterial taxon map and a molecular cluster map. The number of tightly colocalized molecular features (having nearly identical spatial distributions) is shown inside each circle. In total, 492 molecular features were spatially colocalized with the Propionibacterium taxon. (C) The heat map represents the RT and m/z values of colocalized LC-MS features; most of them have an LC RT of 300–400 s and m/z 200–400 and a region that matches many hydrophobic molecules such as lipids. (D) Molecular networking of molecular families that have similar distribution to the Proprionibacteria and highlights the colocalized MS features and structurally related molecules of the selected molecules (green circle) found in UPLC-QTOF data. (E) In vitro analysis considering P. acnes cultures with or without triolein as well as blank growth media with triolein demonstrate the potential of microbiota to be involved in transformation of large human lipids (e.g., the triacyl glyceride triolein) into smaller lipids and fatty acids as those detected on the skin and colocalized with Propionibacterium (here oleic acid and oxidized oleic acid). Area under the peak calculation of oleic acid and oxidized oleic acid was measured for RT ranges 7.5–7.8 min for m/z 283 and 6.65–6.75 min for m/z 299. The in vitro assay was performed three times (error bars, SD) and can be interpreted as a significant (Student t test, **P < 0.01, ****P < 0.0001) increase of lipid products observed in the presence of P. acnes supplemented with triolein. See also Fig. S7.
Except for the hands of person 2, the localization of oleic acid, palmitic acid, mono-oleic, and palmitic acylated glycerols mirrors the localization of the genus Propionibacterium (Fig. 5 A and D). These correlations provided the hypothesis that some of these are products of processing of human acylated glycerols resulting from hydrolysis of triacylglycerides or diacylglycerides mediated by Propionibacterium (40). To determine whether this organism could hydrolyze acylglycerides, Propionibacterium acnes was cultured in a medium supplemented with the triglyceride triolein, and the resulting metabolic products were analyzed using UPLC-QTOF MS/MS. Products of hydrolysis to oleic acid were detected in the P. acnes cultures containing triolein but not in controls (Fig. 5E). Furthermore, an oxidized oleic acid was observed in the same cultures. Although the exact location of the oxidation is unknown, the metabolite has the same parent mass, MS/MS, and RT as the oxidized oleic acid that was detected on the skin and had a similar localization in the topographical map to the hydrolytic products such as oleic acid, palmitic acid, mono-oleoyl, and monopalmitoyl glycerol molecular families (Fig. 5 and Fig. S7C). These results strongly support the hypothesis that skin microbiota, especially Propionibacterium, not only contribute molecules to the chemical composition of the SC but also alter the chemical environment on which they live. It is anticipated that not just human skin cell molecules but also molecules produced by other microbes and from environmental origin, including diet, are altered as well.
Similarly, the automatic colocalization analysis (Colocalization Analysis of Microbial and Molecular Communities) correlated 8,122 LC-MS molecular features colocalized with seven bacteria genera (Prevotella, Butyricimonas, Clostridium, Peptoniphilus, Peptostreptococcus, Bilophila, and Rathayibacter), all localized mainly in the groin area of person 1 (Fig. 6). Among the molecules that colocalize with the groin bacterial community are the human neutrophil peptides (HNPs) (Fig. S8 A and B). Similarly, the vaginal area of person 2 contained a microbial community of six bacterial genera, including Anaerococcus, Peptoniphilus, Prevotella, Sutterella, Negativicoccus, and Peptostreptococcus, which are commonly found in oral and vaginal communities. This bacterial community is colocalized with more than 1,800 unique molecular features only detected in the groin of the female (Fig. S9A). Heme and lysophosphatidylcholine (LysoPC 16:1) were found to be localized (correlation > 0.6, P value < 0.05) in this region (Fig. S9 B–D). HNPs and LysoPC 16:1 are usually associated with inflammation (41–45). As more skin chemical maps become available in the future, they will help to gain insight into the diversity of chemistries of the skin surface and their relationship to the colocalization of microbial communities, especially how the chemistry changes over time upon environmental changes or changes of skin health such as influence of infectious agents, medications, environmental exposure, dietary, or even changes in climate. These observations demonstrate the exciting potential for human molecular topographical maps to discover relationships between these markers and specific bacterial communities.
Fig. 6.

Molecular and bacterial communities found to be colocalized in person 1. Colocalization analysis between bacteria and molecules in the male subject reveals 8,122 molecules, which are spatially colocalized with seven bacterial genera in the groin area: Prevotella, Butyricimonas, Clostridium, Peptoniphilus, Peptostreptococcus, Bilophila, and Rathayibacter. Antimicrobial peptides revealed by MALDI-TOF analysis (HNP-1 m/z 3,443 and HNP-2 m/z 3,372) colocalized with bacterial communities in the groin area of the man subject, highlighting the physiological status and unique chemical and microbial composition of this specific region of the skin. See also Figs. S8 and S9.
Conclusion
In this study, we show the technical advance of 3D molecular topographical maps to organize and visualize a large volume of data, for understanding the relationships between chemistry and microbes. This was demonstrated with creating 3D maps of the human skin surface, together with a pipeline for defining the origins of detected metabolites through molecular networking, to reveal a roadmap toward understanding the interplay between our daily routines, the molecules on our skin, and the metabolism of these molecules by our skin microbiota. At the molecular level, the skin is an enormously complex organ, in part because of the influence of the microbiota, as well as constant exposure to environmental factors. Metagenomic sequencing studies have shown that the microbiota on the skin can have greater phylogenetic diversity than in the gut (6). Our topographical maps show, at a high spatial resolution, that the complexity of the molecular distribution profiles exceeds even the complexity of the microbial profiles. This makes sense, because the chemistry that is found on the skin has many origins, and only some of this diversity comes from our microbes. Spatial analysis of these molecular components can improve our understanding of factors that drive skin microbial ecosystems. The molecular topographical maps constructed in this study represent the highest spatial resolution available to date for both human skin chemical and bacterial distributions. The approach developed here represents a starting point for future investigations into specific chemical drivers involved in the maintenance and modulation of the human skin ecosystem and how variation in this complex ecosystem impacts human health and disease. These effects are especially important for understanding the impact of molecules on the modulation of skin microbiome balance and, in particular, the influence of modern hygiene and beauty practices. Revealing the impacts of these factors on our microbes, and onto the metabolites they produce, may be essential for defining long-term skin health (46). The topographical maps thus lay the groundwork, enabling us to address such questions.
Materials and Methods
Subject Recruitment and Sample Collection.
Two healthy adults were recruited to donate samples. Both individuals signed a written informed consent in accordance with the sampling procedure approved by the University of California, San Diego Institutional Review Board (Approval 130537X). Skin preparation for sampling included avoiding bathing, shampooing, or moisturizing for 3 days before sampling and minimizing the use of personal hygiene products. The female subject did apply deodorant during this period. Two collections were performed from each skin spot. Samples were collected at roughly 400 body sites on the skin surface of each volunteer (Datasets S2 and S3), using swabs. Sampling was performed at a 2 × 2 cm area, through half of the entire human body for both volunteers, with premoistened cotton swabs at each site in 50:50 ethanol/water for MS analysis or 50 mM Tris pH 7.6, 1 mM EDTA, and 0.5% Tween 20 for nucleic acid analysis. Sample locations were noted, and swabs were placed in a deep-well 2-mL polypropylene 96-well microtiter plate. The absorbed material was then extracted in 500 μL of 50:50 ethanol/water (for mass spectrometric analysis) or Tris-EDTA buffer (50 mM Tris pH 7.6, 1 mM EDTA, and 0.5% Tween 20) for bacterial DNA extraction. After collection, samples were stored at –80 °C until further analysis. The ethanol/water extract was submitted to MS analysis including MALDI-TOF for metabolite, peptide, and protein detection and UPLC-QTOF for detection of smaller molecules, including tandem MS (MS/MS) of molecules for molecular network analysis. The second collection was subjected to prokaryotic ribosomal 16S rRNA-based sequencing to identify the bacteria present in the same location. Samples from the beauty products used by the volunteers were also subjected to the same MS analysis.
UPLC-QTOF MS Analysis.
The human swab sample, beauty product, cosmetic ingredient (raw materials), chemical standard, cultured human skin cell, bacteria strain, and human skin tissue (Datasets S2–S8) extractions were analyzed using a UltiMate 3000 UPLC system (Thermo Scientific), controlled by Chromeleon software (Thermo Scientific). UPLC conditions of analysis were 1.7 µm C18 (50 × 2.1 mm) UHPLC column (Phenomenex), column temperature of 40 °C, flow rate of 0.5 mL/min, mobile phase A of 98% water/2% acetonitrile/0.1% formic acid (vol/vol), and mobile phase B of 98% acetonitrile/2% water/0.1% formic acid (vol/vol). A linear gradient was used for the chromatographic separation: 0–0.5 min 0% B, 0.5–2 min 0–20% B, 2–8 min 20–99% B, 8–9 min 99–99% B, and 9–10 min 0% B. The UPLC-MS analysis was performed on a Maxis QTOF mass spectrometer (Bruker Daltonics), controlled by the Otof Control and Hystar software packages (Bruker Daltonics), and equipped with ESI source. MS spectra were acquired in a positive ion mode in the mass range of m/z 80–2,000. The instrument was externally calibrated before each run using ESI-L Low Concentration Tuning Mix (Agilent Technologies). Hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazene (Synquest Laboratories), m/z 922.009798, was used as an internal calibrant (lock mass) during the run. In experiments with multiple internal standards, the Maxis gets less than 0.5 ppm mass accuracy after calibration. For larger scale experiments covering several days, a mass accuracy of <10 ppm is observed for all detected molecular features. Instrument parameters were set as follows: nebulizer gas (Nitrogen) pressure, 2 Bar; Capillary voltage, 4,500 V; ion source temperature, 180 °C; dry gas flow, 9 L/min; spectra rate acquisition, 3 spectra/s. MS/MS fragmentation of the seven most intense selected ions per spectrum was performed using ramped collision-induced dissociation energy, ranging from 16 to 48 eV, to get diverse fragmentation patterns (Dataset S9). MS/MS active exclusion was set after 3 spectra and released after 30 s. An MS/MS exclusion list criterion was set for a mass range of m/z 921.5–924.5.
MALDI-TOF Analysis.
Swab samples were analyzed using a MALDI-TOF/TOF Bruker Autoflex Speed instrument (Bruker Daltonics), controlled by the flex control software (Bruker Daltonics). Samples of 1 µL were spotted directly from water/ethanol extracts using a multichannel pipette and convoluted to a 384 microtiter plate format. Plates were prespotted with matrix as noted below. Mass spectra were recorded in three distinct mass-to-charge (m/z) ranges—m/z 0–4,000, m/z 1,000–5,000, and m/z 5,000–20,000—using 2,5-dihydroxybenzoic acid (DHB), α-Cyano-4-hydroxycinnamic acid (CHCA), and 3,5-Dimethoxy-4-hydroxycinnamic acid (SA), respectively. Full-scan mass spectra were acquired in positive ion reflectron mode for mass ranges m/z 0–4,000 and 900–5,000 and in positive ion linear mode for the mass range m/z 5,000–20,000. Each mass spectrum is the result of 2,048 averaged laser shots with the laser intensity set between 55% (linear) and 65% (reflectron) of full laser intensity and a detector gain enhanced between 18× (reflectron) and 27× (linear) 4GS/s (as selected within the Bruker Flex Control software). Resulting mass spectra were analyzed using flex analysis software (Bruker Daltonics) including smoothing and baseline correction. Spectra were calibrated to PepMix internal standard solutions. For the analysis of pure compounds, with internal calibration, the mass accuracy is <25 ppm. For large-scale experiments and external calibrations, mass accuracies of 100 ppm can be expected.
DNA Extraction and Purification.
Genomic DNA extraction from swab samples previously stored in Tris-EDTA buffer (50 mM Tris pH 7.6, 1 mM EDTA, 0.5% Tween 20) was performed using the MoBio PowerSoil DNA Isolation Kit (MoBio Laboratories) in 96-well microtiter plate format as directed by the manufacturer. Extracted DNA was stored at –20 °C.
16S rRNA Amplicon Sequencing.
For each sample, PCR was completed in triplicate using the 515/806 primer pair according to the Earth Microbiome Project standard protocol, and products were pooled. Each pool was then quantified using PicoGreen (Invitrogen) and a plate reader. Once quantified, different volumes of each of the products were pooled into a single tube so that an equal amount (ng) of DNA from each sample was represented within the pool and cleaned using the UltraClean PCR Clean-Up Kit (MoBIO). Amplicons were then sequenced in a 100 bp × 13 bp × 100 bp Illumina HiSeq run using the recently described custom sequencing primers and procedures. We quality-filtered 16S rRNA amplicon sequences using the default values in QIIME version 1.60-dev (47) and grouped them into “species-level” OTUs by using the February 4, 2011 Greengenes 97% reference dataset (48), a minimum pairwise nucleotide sequence identity threshold between reads of 97%, and the UCLUST reference protocol (www.drive5.com/usearch/). Sequences that did not match reference sequences in the Greengenes database were dropped from the analysis. Taxonomy was assigned to the retained clusters (OTUs) based on the Greengenes reference sequence, and the Greengenes tree was used for all downstream phylogenetic community comparisons. Remaining samples were rarefied to 10,049 sequences (the largest number of sequences per sample, allowing retention of most of the samples). Analyses of community similarity (β-diversity) were performed by calculating pairwise distances using the phylogenetic metric UniFrac (49). The resulting distance matrices were used for principal coordinates analyses (PCoAs).
3D Modeling and Data Visualization.
To create the 3D topographical maps, we used professional triangulated 3D models of a male and a female, acquired at CGtrader.com in the STL format, which contains coordinates of vertices of triangles making up a 3D model. We performed 3D modeling manually with the help of MATLAB software separately for male and female data. The 3D modeling included finding for each sampling spot a location on the triangulated 3D model that spatially fits the best. For this, we used photos and anatomical maps with sampling spots marked and labeled. The (x, y, z) coordinates of assigned locations were recorded. The correctness of the 3D modeling was evaluated by adding spheres with the centers at the recorded (x, y, z) coordinates together with their labels, by performing the manual assignments of labels back onto photos and anatomic maps and comparing them with original photos and anatomic maps. Moreover, a radius was manually assigned to each location to improve visualization; smaller radii were used in the regions with dense distribution of locations such as face, fingers, and toes. Rendering of topographical maps corresponding to molecular features (LC-MS) (Dataset S10), m/z values (MALDI), and taxa as assessed by 16S rRNA amplicon data were performed in the same way. We considered the intensities corresponding to all spots, scaled them from 0 to 100%, and assigned colors to all intensities by using the “jet” color map, encoding colors from cold shades to hot shades (blue representing lowest intensity and red representing highest intensity). The rendering of the models was performed in MATLAB. Before rendering each model, the 3D scene was set, the default color was selected as RGB= (0.05, 0.05, 0.1), lights were added, and the views were selected; for several views of shaded regions, additional lights were added. Then, each sampling spot on the male and female models was rendered by using the color assigned from intensity. The color was made gradually, disappearing with the highest intensity at the center of the sampling location and almost no intensity at the boundary of the sphere with the selected radius. For this, a linear combination of the default color and assigned color was used, with the coefficient exponentially depending on the distance from the sampling spot center. The regions outside of the sampling neighborhoods were colored with the default color. For those sampling spots where 16S rRNA data were missing, we used the color RGB= (0.03, 0.03, 0.06), which is similar to the default color, but slightly darker. To visualize the data from MS analysis, spectral intensities were integrated over a given m/z interval for MALDI-TOF or the maximum was taken over a given molecular feature (a set of m/z × RT regions corresponding to isotopes of the same molecule eluting over a period from the LC column) for UPLC-QTOF. For MALDI-TOF, the tolerances were set as m/z –0.2, +0.3 and m/z –2, +2 for metabolic and peptide, and protein data, respectively. For LC-MS, the tolerances were –10 ppm, +10 ppm along m/z and –10, +10 s along RT.
Colocalization Analysis of Microbial and Molecular Communities.
To find communities of colocalized bacteria (those bacterial taxa whose spatial correlation values are above a threshold and correlation P values are below a threshold) as well as colocalized molecules (those molecular species whose spatial correlation values are above a threshold and correlation P values are below a threshold), we performed the following colocalization analysis separately for the female and male individuals. The overall aim of this colocalization analysis was to represent these big data as a tractable network showing spatial colocalizations between thousands of molecular and microbial maps that can be subsequently analyzed by a biochemist.
First, we performed reduction of data contained in all maps that we acquired with the aim to simplify subsequent spatial colocalization network analysis. The data reduction was applied separately to molecular and microbial data, separately for each individual, and was performed based on using spatial information only. For this, we considered all 16S amplicon spatial maps and clustered them into tight clusters (spatial correlation value > 0.5, P value < 0.05) and then considered all LC-MS maps and clustered them into tight clusters (spatial correlation value > 0.5, P value < 0.05); each cluster represents almost identical maps. No additional information other than spatial colocalization was used for clustering. The tight clustering was performed with such parameters so that (i) the clustering results can be easily interpreted and (ii) all maps inside one cluster are very similar. For clustering, we used the agglomerative clustering with the Pearson correlation distance, with complete linkage and distance threshold of 0.5 and P value < 0.05. This automatically finds clusters of nearly identical maps without specifying the number of clusters and requires all maps inside one cluster to have pairwise correlations of 0.5 or higher. We have obtained 322/319 (subject 1, female/subject 2, male) tight clusters for 16S data and 12,558/7,716 (female/male) tight clusters for LC-MS data. Then, for each subject, we calculated pairwise Pearson correlation between all maps (both 16S and LC-MS) and created networks showing a node for a map and an edge between two nodes if the correlation is above the threshold. The spatial correlation thresholds were set to 0.6 for female and 0.4 for male. Then, the networks were imported into the Cytoscape software and analyzed. To find networks of communities shown in Fig. 5, we selected the node corresponding to the Prevotella, Butyricimonas, Clostridium, Peptoniphilus, Peptostreptococcus, Bilophila, Rathayibacter, Anaerococcus, Sutterella, and Negativicoccus maps and found all nodes, which are separated by at most two edges from the Prevotella node. Then, the layout was generated, putting all found LC-MS nodes into a circle and 16S nodes aside.
Supplementary Material
Acknowledgments
We thank Elizabeth Costello, Sandy Yates, and Jeramie Watrous for assistance with sampling protocols and automation. We also thank Gary Siuzdak, TSRI (The Scripps Research Institute), for preliminary assistance of the molecular annotation of LC-MS signals. We thank Natura Innovation Brazil for encouraging the scholarship application (to C.P.) and CP Kelco, A Huber Company, for supplying the samples of raw cosmetic ingredients. This work was supported by the Keck Foundation; the San Diego Center for Systems Biology Pilot Project; National Institutes of Health (NIH) Grants GM085764 and 3-P41-GM103484; European Union 7th Framework Programme Grant 305259; the Science Without Borders Program from Conselho Nacional de Desenvolvimento Científico CNPq, Brazil; and the Howard Hughes Medical Institute. We also thank the CCMS Grant 2 P41 GM103484-06A1 and Sloan Fellowship Award. We further acknowledge Bruker and NIH Grant GMS10RR029121 for the support of the shared instrumentation infrastructure that enabled this work.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The mass spectrometry data and 3D microbial distribution maps reported in this paper have been deposited in the MassIVE database, massive.ucsd.edu/ProteoSAFe/status.jsp?task=6b9dcff3899e4d5f89f0daf9489a3a5e (identifier nos. MSV000078556 and MSV000078556, respectively). The sequence data reported in this paper have been deposited in European Molecular Biology Laboratory European Bioinformatics Institute (EMBL EBI) database, www.ebi.ac.uk/ (accession no. ERP005182). The source code for molecular networking and the molecular network documentation have been uploaded to Proteomics, proteomics.ucsd.edu/.
See Commentary on page 5261.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424409112/-/DCSupplemental.
References
- 1.Staudinger T, Pipal A, Redl B. Molecular analysis of the prevalent microbiota of human male and female forehead skin compared to forearm skin and the influence of make-up. J Appl Microbiol. 2011;110(6):1381–1389. doi: 10.1111/j.1365-2672.2011.04991.x. [DOI] [PubMed] [Google Scholar]
- 2.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stingley RL, Zou W, Heinze TM, Chen H, Cerniglia CE. Metabolism of azo dyes by human skin microbiota. J Med Microbiol. 2010;59(Pt 1):108–114. doi: 10.1099/jmm.0.012617-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, et al. The human microbiome project. Nature. 2007;449(7164):804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA, et al. NISC Comparative Sequencing Program Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science. 2014;346(6212):954–959. doi: 10.1126/science.1260144. [DOI] [PubMed] [Google Scholar]
- 8.Cogen AL, Nizet V, Gallo RL. Skin microbiota: A source of disease or defence? Br J Dermatol. 2008;158(3):442–455. doi: 10.1111/j.1365-2133.2008.08437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holland KT, Bojar RA. Cosmetics: What is their influence on the skin microflora? Am J Clin Dermatol. 2002;3(7):445–449. doi: 10.2165/00128071-200203070-00001. [DOI] [PubMed] [Google Scholar]
- 10.Findley K, et al. NIH Intramural Sequencing Center Comparative Sequencing Program Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenthal M, Goldberg D, Aiello A, Larson E, Foxman B. Skin microbiota: Microbial community structure and its potential association with health and disease. Infect Genet Evol. 2011;11(5):839–848. doi: 10.1016/j.meegid.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tseng SP, et al. Detection and distribution of endogenous steroids in human stratum corneum. Dermatologica Sinica. 2014;32(1):19–24. [Google Scholar]
- 13.Bailey MJ, et al. Chemical characterization of latent fingerprints by matrix-assisted laser desorption ionization, time-of-flight secondary ion mass spectrometry, mega electron volt secondary mass spectrometry, gas chromatography/mass spectrometry, X-ray photoelectron spectroscopy, and attenuated total reflection Fourier transform infrared spectroscopic imaging: An intercomparison. Anal Chem. 2012;84(20):8514–8523. doi: 10.1021/ac302441y. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen DD, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc Natl Acad Sci USA. 2013;110(28):E2611–E2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watrous J, et al. Mass spectral molecular networking of living microbial colonies. Proc Natl Acad Sci USA. 2012;109(26):E1743–E1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guthals A, Watrous JD, Dorrestein PC, Bandeira N. The spectral networks paradigm in high throughput mass spectrometry. Mol Biosyst. 2012;8(10):2535–2544. doi: 10.1039/c2mb25085c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics. 2011;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank AM, et al. Spectral archives: Extending spectral libraries to analyze both identified and unidentified spectra. Nat Methods. 2011;8(7):587–591. doi: 10.1038/nmeth.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JY, et al. Molecular networking as a dereplication strategy. J Nat Prod. 2013;76(9):1686–1699. doi: 10.1021/np400413s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hachul S, Junger M. Drawing large graphs with a potential-field-based multilevel algorithm. Graph Drawing. 2004;3383:285–295. [Google Scholar]
- 21.Roth RR, James WD. Microbiology of the skin: Resident flora, ecology, infection. J Am Acad Dermatol. 1989;20(3):367–390. doi: 10.1016/s0190-9622(89)70048-7. [DOI] [PubMed] [Google Scholar]
- 22.Horai H, et al. MassBank: A public repository for sharing mass spectral data for life sciences. J Mass Spectrom. 2010;45(7):703–714. doi: 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- 23.Smith CA, et al. METLIN: A metabolite mass spectral database. Ther Drug Monit. 2005;27(6):747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 24.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phelan VV, Liu WT, Pogliano K, Dorrestein PC. Microbial metabolic exchange—The chemotype-to-phenotype link. Nat Chem Biol. 2012;8(1):26–35. doi: 10.1038/nchembio.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watrous JD, et al. Microbial metabolic exchange in 3D. ISME J. 2013;7(4):770–780. doi: 10.1038/ismej.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frasch SC, Bratton DL. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog Lipid Res. 2012;51(3):199–207. doi: 10.1016/j.plipres.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Exton JH. Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta. 1994;1212(1):26–42. doi: 10.1016/0005-2760(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 29.Jones ML, Martoni CJ, Ganopolsky JG, Labbé A, Prakash S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin Biol Ther. 2014;14(4):467–482. doi: 10.1517/14712598.2014.880420. [DOI] [PubMed] [Google Scholar]
- 30.Tsuda M, Hata M, Nishida R, Oikawa S. Chemically amplified resists IV. Proton-catalyzed degradation mechanism of poly(phtaldehyde) J Photopolym Sci Technol. 1993;6(4):491–494. [Google Scholar]
- 31.Tian RR, et al. Comparison of phenolic acids and flavan-3-ols during wine fermentation of grapes with different harvest times. Molecules. 2009;14(2):827–838. doi: 10.3390/molecules14020827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galvez MC, et al. Analysis of polyphenolic compounds of different vinegar samples. Z Lebensm Unters Forsch. 1994;199(1):29–31. [Google Scholar]
- 33.US National Archives and Records Administration 2013. Code of Federal Regulations. Revised as of April 1, 2013. Title 21. Indirect Food Addictives: Polymers, Chapter 177, CITE: 21CFR177.1620.
- 34.Massey KA, Snelling AM, Nicolaou A. Quantitative analysis of surfactant deposits on human skin by liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun Mass Spectrom. 2010;24(9):1371–1376. doi: 10.1002/rcm.4528. [DOI] [PubMed] [Google Scholar]
- 35.Bik EM, et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010;4(8):962–974. doi: 10.1038/ismej.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green BR. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011;66(1):34–44. doi: 10.1111/j.1365-313X.2011.04541.x. [DOI] [PubMed] [Google Scholar]
- 37.Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27(3):379–423, 623–656. [Google Scholar]
- 38.Li K, Bihan M, Yooseph S, Methé BA. Analyses of the microbial diversity across the human microbiome. PLoS ONE. 2012;7(6):e32118. doi: 10.1371/journal.pone.0032118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lozupone CA, Knight R. Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev. 2008;32(4):557–578. doi: 10.1111/j.1574-6976.2008.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holland C, et al. Proteomic identification of secreted proteins of Propionibacterium acnes. BMC Microbiol. 2010;10:230. doi: 10.1186/1471-2180-10-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunliffe RN. Alpha-defensins in the gastrointestinal tract. Mol Immunol. 2003;40(7):463–467. doi: 10.1016/s0161-5890(03)00157-3. [DOI] [PubMed] [Google Scholar]
- 42.Braff MH, Bardan A, Nizet V, Gallo RL. Cutaneous defense mechanisms by antimicrobial peptides. J Invest Dermatol. 2005;125(1):9–13. doi: 10.1111/j.0022-202X.2004.23587.x. [DOI] [PubMed] [Google Scholar]
- 43.Philpott MP. Defensins and acne. Mol Immunol. 2003;40(7):457–462. doi: 10.1016/s0161-5890(03)00154-8. [DOI] [PubMed] [Google Scholar]
- 44.Ryborg AK, Deleuran B, Søgaard H, Kragballe K. Intracutaneous injection of lysophosphatidylcholine induces skin inflammation and accumulation of leukocytes. Acta Derm Venereol. 2000;80(4):242–246. doi: 10.1080/000155500750012090. [DOI] [PubMed] [Google Scholar]
- 45.Ryborg AK, Grøn B, Kragballe K. Increased lysophosphatidylcholine content in lesional psoriatic skin. Br J Dermatol. 1995;133(3):398–402. doi: 10.1111/j.1365-2133.1995.tb02667.x. [DOI] [PubMed] [Google Scholar]
- 46.Blaser MJ, et al. Distinct cutaneous bacterial assemblages in a sampling of South American Amerindians and US residents. ISME J. 2013;7(1):85–95. doi: 10.1038/ismej.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.