

Significance
Protein disulfide isomerase (PDI) is a chaperone protein in the endoplasmic reticulum. It is up-regulated in mouse models of, and brains of patients with, neurological protein folding diseases. Irreversible inhibition of PDI activity by the small molecule 16F16 results in protection in cell and organotypic brain slice culture models of Huntington disease. Here, we identified lead optimized compound (LOC)14 as a nanomolar, reversible inhibitor of PDI that protects PC12 cells and medium spiny neurons from the toxic mutant huntingtin protein. LOC14 has improved potency compared with 16F16 and displays favorable pharmaceutical properties, making it a suitable compound to evaluate the therapeutic potential of inhibiting PDI in multiple disease models.
Keywords: small molecule, protein disulfide isomerase, drug, inhibitor, neuroprotection
Abstract
Protein disulfide isomerase (PDI) is a chaperone protein in the endoplasmic reticulum that is up-regulated in mouse models of, and brains of patients with, neurodegenerative diseases involving protein misfolding. PDI’s role in these diseases, however, is not fully understood. Here, we report the discovery of a reversible, neuroprotective lead optimized compound (LOC)14, that acts as a modulator of PDI. LOC14 was identified using a high-throughput screen of ∼10,000 lead-optimized compounds for potent rescue of viability of PC12 cells expressing mutant huntingtin protein, followed by an evaluation of compounds on PDI reductase activity in an in vitro screen. Isothermal titration calorimetry and fluorescence experiments revealed that binding to PDI was reversible with a Kd of 62 nM, suggesting LOC14 to be the most potent PDI inhibitor reported to date. Using 2D heteronuclear single quantum correlation NMR experiments, we were able to map the binding site of LOC14 as being adjacent to the active site and to observe that binding of LOC14 forces PDI to adopt an oxidized conformation. Furthermore, we found that LOC14-induced oxidation of PDI has a neuroprotective effect not only in cell culture, but also in corticostriatal brain slice cultures. LOC14 exhibited high stability in mouse liver microsomes and blood plasma, low intrinsic microsome clearance, and low plasma-protein binding. These results suggest that LOC14 is a promising lead compound to evaluate the potential therapeutic effects of modulating PDI in animal models of disease.
Neurodegenerative disorders constitute a class of diseases that express characteristic misfolded proteins that aggregate and induce neuronal toxicity and death. Huntington disease (HD) is one such fatal protein misfolding disease that afflicts primarily medium spiny neurons in the striatum. HD is caused by expansion to more than 36 CAG trinucleotide repeats in the huntingtin gene. These CAG repeats translate into an expanded polyglutamine tract in the huntingtin protein, causing it to aggregate, and drive neuronal dysfunction and progressive neuronal loss. Currently, no therapeutic avenue can delay or stop the progression of the disease. In this context, there is a need to develop therapeutics and drug targets that can prevent or delay pathogenesis in neurodegenerative diseases, such as HD, involving protein misfolding.
Previously, it was reported that modulation of protein disulfide isomerase (PDI) by small molecules is beneficial in cell and brain slice models of HD (1). PDI is a thiol-oxidoreductase chaperone protein that is responsible for the isomerization, reduction, and oxidation of nonnative disulfide bonds in unfolded proteins entering the endoplasmic reticulum (ER). Structurally, PDI consists of four domains with a thioredoxin fold: a, b, bʹ, and aʹ, an extended C terminus with KDEL ER retention sequence, and an interdomain linker x between the bʹ and aʹ domains. The a and aʹ domains are catalytically active, contain the WCGHC active site and independently can perform oxidation and reduction reactions (2). However, all four domains are needed to achieve the isomerization and chaperone activity of PDI. Besides its catalytic role involving thiols and disulfides, PDI also serves an essential structural role as the β subunit of prolyl-4-hydroxylase (3) and as a microsomal triglyceride transfer protein (4).
PDI is up-regulated in mouse models of, and in brains of patients with, neurological protein folding diseases (5–7). In addition, PDI has also been implicated in a number of cancers (8–10), HIV-1 pathogenesis (11), and blood clot formation (12), suggesting the growing importance of understanding this enzyme. One challenge has been the lack of available drug-like inhibitors, especially for in vivo evaluation in neurodegenerative disease models. Reported inhibitors of PDI are (i) irreversible binders to the catalytic site cysteines (1, 8, 13), (ii) not cell permeable, because they were designed for the inhibition of extracellular PDI (14, 15), or (iii) nonselective hormones and antibiotics, such as estrone and bacitracin, that act broadly on multiple target proteins (15, 16). Irreversible inhibitors, although having promise in ovarian cancer, have mechanism-based toxicity that is not likely well tolerated in neurons. PDI is an essential protein, whose irreversible genetic silencing is cytotoxic to cells and probably in animal models as well, because no genetic PDI null has been generated. The related PDI A3 (ERp57) protein KO resulted in embryonic lethality in mice (17). Thus, irreversible inhibitors of PDI may exhibit the same level of cytotoxicity in vivo. We hypothesized that reversible, noncovalent inhibitors of PDI might exhibit a therapeutic window on PDI inhibition and would have improved pharmaceutical properties. Here, we report the discovery of a neuroprotective, reversible modulator of PDI that has nanomolar potency, has high in vitro stability in liver microsomes and blood plasma, and is protective for medium spiny neurons in a brain slice model for HD. This scaffold represents a class of reversible modulators of PDI that can probe its potential as a drug target for neurological diseases with misfolded proteins.
Results
High-Throughput Screen Identifies Small Molecule Inhibitors of PDI.
To identify neuroprotective PDI modulators with attractive pharmaceutical properties, we assembled a library with lead-optimized compounds (18, 19). Initially, a database of 3,372,615 commercially available small molecules (from Asinex, Life Chemicals, Enamine, TimTec, InterBioScreen, and Chembridge suppliers) was compiled and stringently filtered computationally. The compounds were filtered to adhere to the Lipinski rule of five (20) and a molecular weight minimum of 235; compounds with noxious or reactive properties and water solubility less than 0.5 mM were eliminated. Strong emphasis was placed on compounds suitable for lead development; therefore, potential nonspecific binders that might be acting as cationic and nonionic detergents were removed. Scaffolds with more than five rotatable bonds and topological polar surface area larger than 70 Å2 were eliminated to improve the likelihood of blood–brain barrier penetration. As a final step, the compounds were clustered based on their Tanimoto coefficient of 0.7 and only a diverse subset was purchased. The final lead optimized compound (LOC) library contained 9,719 unique small molecules.
We used a cascade of two assays, involving both a phenotypic high-throughput screening (HTS) assay and an in vitro PDI reductase assay, to identify neuroprotective PDI-inhibiting compounds. PC12 cells stably transfected with an inducible plasmid for mutant huntingtin protein (21) (mHTTQ103) were used for the screen, because they previously showed reliance on PDI inhibition for survival from misfolded mHTTQ103-induced cell death (1). Each compound in the LOC library was screened in triplicate at three different concentrations, 4, 1, and 0.25 µg/mL, resulting in nine data points per compound, to maximize the probability of identifying effective compounds. Alamar blue was used as a fluorescent readout for viability after 48 h of compound treatment and mHTTQ103 induction. The overall Zʹ factor for the screen was 0.78 with a signal-to-noise ratio of 165 and coefficient of variation of 5.8%, indicating a robust assay for hit identification (22). Of 9,719 compounds, 9 compounds rescued PC12 mHTTQ103 cells to at least 45% viability in the primary screen. All of the candidate hit compounds were retested in a twofold dilution series. The viability curves of the eight compounds that reproducibly exhibited >50% viability are shown in Fig. 1A and Fig. S1A. Of these, three compounds had EC50 values in the nanomolar range; two compounds, LOC14 (EC50 = 500 nM) and LOC9 (EC50 = 600 nM) (Fig. 1A) were more potent than the previously identified irreversible neuroprotective PDI inhibitor 16F16 (EC50 = 1,500 nM) (1).
Fig. 1.

High-throughput screen identifies neuroprotective PDI inhibitors. (A) Dose–response curves of three top hits that rescued PC12 cells from mHTTQ103-induced cell death as measured by Alamar blue fluorescence after 48-h treatment. Data from cells induced to express mHTTQ103 (blue) and cells not expressing mHTTQ103 (red) are plotted as mean percent of DMSO-treated uninduced cells ± SD. Experiments were performed in triplicate. (B) Secondary screen of the top three hits (75 μM) for their ability to inhibit the enzymatic activity of PDIa (5 μM) in an insulin aggregation assay. Experiments were performed in duplicate with data plotted as mean ± SEM.
Because neuroprotection of PC12 mHTTQ103 cells can occur via additional pathways other than PDI modulation, e.g., caspase inhibition, the hits from the cell-based assay were screened for inhibition of PDI’s reductase activity using insulin and the recombinant catalytic a domain of human PDI A1 (referred to as PDIa), which can perform the same catalytic oxidation and reduction reactions as full-length PDI with one inactive domain (2). In this insulin aggregation assay (8, 14, 15, 23), PDIa reduced the two disulfide bonds between the α- and β-chains of insulin, causing the β-chain to aggregate and precipitate, resulting in an increase in absorbance at 650 nm. Of eight hit compounds from the cell culture screen, two, LOC14 and LOC6, were able to almost completely inhibit PDIa enzymatic activity (Fig. 1B and Fig. S1B).
At this stage, LOC14 emerged as the most potent small molecule that could both rescue PC12 mHTTQ103 cells and inhibit PDIa reductase activity; we therefore selected LOC14 as a lead compound for further analyses.
LOC14 Binds with Nanomolar Affinity to PDI.
To confirm the compound’s identity, we resynthesized LOC14 (SI Materials and Methods and Fig. S2). The biochemical activity of the resynthesized LOC14 was identical to the commercially obtained compound.
We next investigated the binding mode of LOC14 to PDIa using isothermal titration calorimetry (ITC). ITC measures the heat released or absorbed during a biomolecular interaction. It is a direct analytical method for determining binding and thermodynamic parameters, such as reaction stoichiometry (n), binding constants (Ka and Kd), enthalpy (∆H), entropy (∆S), and free energy (∆G), of an interaction.
Calorimetric titration of LOC14 against PDIa showed exothermic binding (Fig. 2A) with a dissociation constant (Kd) of 61.7 ± 5.6 nM. The compound titration into buffer alone was subtracted from the raw binding data to account for the heat of dilution. The thermodynamic parameters plot (Fig. 2C, Left) showed that the overall favorable affinity of LOC14 to PDIa is driven by both favorable (negative) enthalpic and entropic contributions. This finding indicates that likely both polar and hydrophobic interactions are contributing to LOC14 binding affinity to PDIa.
Fig. 2.

Sulfur in LOC14 is important for tight binding to PDIa. Calorimetric titration of (A) 400 µM LOC14 into 30 µM PDIa or (B) 400 µM Oxy-LOC14 into 40 µM PDIa. (Upper) Raw data of the heat released. (Lower) Binding isotherm of the reaction. Data are fitted to a one-site binding model after subtracting the heat released from titrating the compound alone into buffer. One of three representative experiments is shown. (C) Summary of thermodynamic parameters for binding. Data are plotted as mean ± SD (n = 3).
To test whether the sulfur atom on LOC14 is important for interaction with the protein, we synthesized an isoxazolone analog of LOC14, termed Oxy-LOC14 (SI Materials and Methods) and tested its binding affinity to PDIa. Oxy-LOC14 had 39-fold loss in binding affinity compared with LOC14 and a Kd of 2,430 ± 760 nM by ITC (Fig. 2B). The thermodynamic parameters plot showed that Oxy-LOC14 had almost complete loss of its enthalpic binding component (Fig. 2C, Right). This difference in the thermodynamic signatures due to a single atom, sulfur to oxygen, substitution indicated that the sulfur atom on LOC14 can form favorable interactions with the protein.
LOC14 Is a Reversible Modulator of PDI.
The importance of the sulfur atom on LOC14 suggested that it might be binding covalently to the active site of PDIa, which contains two cysteines with reactive thiol groups. To investigate the mode of binding further and to determine whether the binding was reversible or irreversible, we used two separate methods to assess reversibility. First, we analyzed the fluorescence of the LOC14-PDIa complex before and after buffer dialysis. LOC14 has a distinct emission spectrum when excited at 280 nm that is different from the emission of PDIa alone or from the LOC14-PDIa complex (Fig. S3A). If LOC14 binds irreversibly to the protein, then we would expect to see the same fluorescence spectrum of the LOC14-PDIa complex before and after dialysis in the dialysis chamber and none in the buffer compartment, assuming the fluorescence of LOC14 is not dramatically altered on binding. LOC14 and PDIa were incubated together overnight (to allow for the maximum binding to occur in the case that LOC14 was a time-dependent irreversible binder) and the next day were dialyzed with buffer four times using an Amicon Ultra 10-kDa cutoff size exclusion filtration device. As a control, samples that contained only PDIa or LOC14 were also used. The emission spectrum was recorded of samples from the flow-through and dialysis chamber. In the control samples, the fluorescence of LOC14 alone was observed only in the flow-through fraction, whereas the fluorescence of PDIa alone was only observed in the dialysis compartment (Fig. S3 B and C) as was expected. Furthermore, their respective emission spectra were identical before and after the buffer exchange. The PDIa-treated LOC14 sample, however, showed different fluorescence profile before and after dialysis (Fig. S3 B and C). The emission spectrum in the dialysis chamber resembled the fluorescence spectrum of PDIa and the flow-through fraction resembled the fluorescence profile of LOC14. These results illustrated that LOC14 binding to PDIa is reversible.
In the second approach, we analyzed the recovery of enzymatic activity of PDIa after dilution of preformed concentrated PDIa-LOC14 complexes. LOC14 (750 μM) was incubated with a concentrated solution of PDIa (500 μM). The mixture was then diluted 100-fold, and PDI’s enzymatic activity was measured in the insulin reduction assay. As a control, the same procedure was also performed on the sample containing an irreversible inhibitor 16F16 (16F16 at 750 μM and PDIa at 500 μM). Without dilution, concentrated 16F16 (750 μM; Fig. 3A, green) or concentrated LOC14 (750 μM; Fig. 3B, gray) were both able to inhibit PDI’s reduction of insulin. However, after dilution, the samples containing LOC14-PDIa diluted complexes showed complete recovery of PDI’s enzymatic activity that lead to insulin reduction and precipitation (Fig. 3B, blue). PDI’s enzymatic activity was still inhibited, after dilution, in samples that contained 16F16-PDIa diluted complexes (Fig. 3A, purple). These results illustrated that, unlike 16F16, LOC14 binding to PDIa is reversible.
Fig. 3.

Recovery of enzymatic activity of PDIa shows LOC14 reversibly binds to PDIa. PDIa (500 μM) was incubated with either (A) irreversible inhibitor 16F16 (750 μM) or (B) LOC14 (750 μM) for 3 h at room temperature and then diluted 100-fold into assay buffer and analyzed for its ability to inhibit the enzymatic insulin aggregation. Diluted complexes were compared with samples containing 5 μM PDIa only (red) or 5 μM PDIa with either 7.5 or 750 μM compound LOC14 or 16F16. Experiments were performed in triplicate with data plotted as mean ± SEM.
LOC14 Binds Adjacent to the Active Site and Oxidizes PDI.
To identify residues on PDIa involved with the LOC14 interaction, we performed 1H-15N heteronuclear single quantum correlation (HSQC) binding studies on the uniformly 15N-labeled PDIa, with and without LOC14. 1H-15N HSQC spectra displays directly bonded N-H resonance peaks from each amino acid in a protein. On ligand binding, the perturbations in the local environment induce a change in the chemical shift of the resonance peaks. Only the residues that are either involved in the binding site or in the conformational change of the protein on compound binding will be perturbed (24). Knowing the resonance assignments is beneficial when mapping the binding site from chemical shift perturbation data.
The resonance assignments of oxidized PDIa have been previously determined (25); however, the resonances for the reduced form of the protein are not known. By matching the conditions reported by Kemmink et al. (25), we were able to efficiently transfer the assignments of unaltered residues in the oxidized protein to the peaks of 1H-15N HSQC spectrum of reduced PDI protein. To validate the assignments, 15N total correlated spectroscopy-HSQC and 15N-nuclear Overhauser effect spectroscopy-HSQC were performed (Fig. S4). These 3D-NMR data were then used to identify the individual spin systems and sequentially assign the reduced PDIa protein (Table S1).
The resulting HSQC spectrum with one-to-one molar equivalence ratio (PDIa to LOC14) on LOC14 binding is shown in Fig. 4A. Titrating LOC14 beyond one-to-one (protein:compound) molar ratio resulted in no additional shift changes (Fig. 4B), indicating that by one molar equivalent, the protein is fully saturated with ligand. This result is consistent with the calculated Kd data from the ITC experiments that determined nanomolar affinity of LOC14 for PDIa. The lowest concentration of reduced PDIa that we could use for optimal sensitivity in HSQC experiments was 50 μM and, as this is above the nanomolar Kd, we would expect to see one-to-one stoichiometric binding.
Fig. 4.
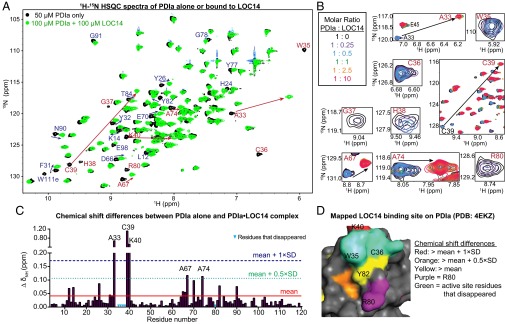
Chemical shift changes in PDIa on binding LOC14. (A) Superimposed HSQC spectra of PDIa alone (black) and PDIa treated with 1 mol. equiv. of LOC14 (green). Resonances with largest chemical shifts (mean shift change + 1 × SD) are labeled in red. (B) Zoom-in on the most shifted peaks. The abrupt progression of PDIa peaks as LOC14 is titrated at 0- (black), 0.25- (purple), 0.5- (blue), 1- (green), 2.5- (orange), and 10- (red) fold molar excess. After the saturation point with 1 mol. equiv. of LOC14 (green), no further shifts are observed. (C) Chemical shift differences (∆ δNH) for each residue in the PDIa sequence on 1:1 PDIa:LOC14 binding. Weighted mean of 1H and 15N chemical shift changes is plotted as a red line; the mean shift change + 1 × SD is plotted as a dotted blue line. (D) Chemical shift perturbations were used to map the LOC14 binding site onto the molecular surface of reduced PDIa (Protein Data Bank ID code 4EKZ).
Furthermore, the majority of the perturbed peaks initially decreased in peak intensity and then either appeared in a new location or completely disappeared (Fig. 4B). This behavior suggests slow exchange on the NMR timescale and is indicative of tight binding and conformational change in the protein.
Closer examination of the HSQC spectrum of reduced PDIa liganded to LOC14 revealed that, on compound binding, PDIa adopts an oxidized conformation. This result is evident by the perfect overlay between the HSQC spectrum of oxidized protein alone and the HSQC spectrum of reduced PDIa liganded with LOC14 (Fig. S5A). One major difference between these two, however, is seen in residue R80. The resonance peak for R80 is present in the oxidized PDIa HSQC spectrum, but disappears when the reduced PDIa is treated with LOC14. These data are indicative of a protein-ligand interaction. All other residues that disappear on compound treatment such as W35, C36, G37, and H38 also are absent in the oxidized protein HSQC spectrum. [The residue numbering in this report is based on the sequence of the mature PDI protein, i.e., residue 1 of the mature PDI corresponds to residue 18 in the full-length PDI. The first 17 amino acids in full-length PDI are the signal sequence that is processed out to generate the mature PDI.]
Mapping the residues with the most significant chemical shift change (Fig. 4C) and residues that disappear on compound titrations to the structure of PDIa, illuminated a small area between the active site and R80 (Fig. 4D). The few residues not localized to this pocket, e.g., L12, K14, Y26, and N90, are involved in the conformational change of the protein on ligand binding.
Calorimetric titration of LOC14 against oxidized PDIa showed a loss in binding affinity (compared with LOC14 titration into reduced PDIa; Fig. S5B). These data indicate that the presence of free cysteine thiols on PDIa is important for the tight interaction with LOC14. Additionally, this reinforces that the observed HSQC shifts that reflect oxidative conformational change in PDIa are due to LOC14 binding.
LOC14 Binds to a Different Site Than 16F16.
Based on the mapped binding site from the NMR studies and the fact that sulfur atom was essential to retain tight binding of LOC14 to the protein, we next examined whether LOC14 was binding to the two cysteines in the protein, C36 and C39, both in the active site.
Previously, 16F16 was reported to function as an irreversible inhibitor of PDI A1 and PDI A3 proteins (1). The compound 16F16 contains a chloroacetyl group that covalently modifies free cysteine thiols. To confirm the likely covalent nature of the interaction and identify the cysteines on PDIa modified by 16F16, LC-MS/MS fragmentation was performed. Compound 16F16 selectively bound to the only cysteines in the PDIa protein, and it was able to covalently modify both C36 and C39 (Fig. 5 A–C). To test whether LOC14 bound to the same active site cysteines as 16F16, PDIa was preincubated with 16F16 overnight and then analyzed by ITC on LOC14 titrations. After pretreatment, the two thiols in the active site would be irreversibly bound to 16F16 and not be available for LOC14 interaction. ITC data showed that the binding affinity was reduced with 16F16 pretreatment but not completely obliterated (Fig. 5D).
Fig. 5.
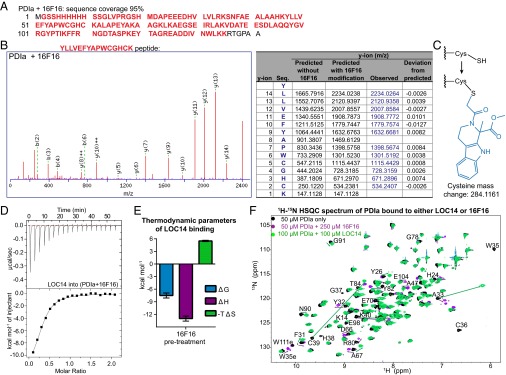
LOC14 has a different mode of binding to PDIa than irreversible inhibitor 16F16. (A) LC/MS showed 95% sequence coverage of PDIa (red bold) when treated with 16F16. (B) Predicted and observed fragment ion (ms/ms) mass spectrum and table of the YLLVEFYAPWCGHCK peptide from the trypsin digested PDIa (100 μM) treated with 16F16 (500 μM) overnight. Ion score was 58, precursor RMS error was 3 ppm, and product RMS error was 5 ppm. The predicted and observed y-ion masses are different because of a 284.1161 m/z (monoisotopic) modification at each cysteine. Blue are observed y-ion masses. (C) Schematic showing the modification at each cysteine on 16F16 binding to PDIa, which causes a 284.1161 mass increase. (D) ITC titration of 400 µM LOC14 against 40 µM PDIa that has been pretreated overnight with irreversible inhibitor 16F16 (200 µM). (Upper) Raw data of the heat released. (Lower) Binding isotherm of the reaction, fit to one-site binding model after subtracting the heat released from titrating LOC14 into buffer with 16F16. One of three representative experiments is shown. (E) Summary of thermodynamic parameters for binding. Data are plotted as mean ± SD (n = 3). (F) Superimposed HSQC spectra of 50 µM PDIa alone (black), 100 µM PDIa treated with 100 µM LOC14 (green), and 50 µM PDIa treated with 250 µM 16F16 (purple). The arrows indicate the direction of the shift.
Additionally, the thermodynamic parameters plot (Fig. 5E) showed a different mode of binding than when the protein was treated with LOC14 alone (Fig. 2C). Even though the overall ∆G of binding was favorable (negative), there was a large entropic penalty (positive ∆S) when LOC14 bound, most likely due to the conformational change in the protein. This result suggests that the protein adopts one conformation when 16F16 is bound to the active site cysteines, most likely one that minimizes the steric clash of having such a bulky group. Then, on LOC14 binding, the protein is forced into another conformation, one that resembles its oxidation state, but paying the cost of unfavorable entropy.
The different mode of binding to PDIa between these small-molecule modulators is also supported by NMR 1H-15N HSQC data (Fig. 5F). PDIa treated with 16F16 displays a different protein conformation, seen by the different chemical shift changes, than when LOC14 is bound to PDIa.
LOC14 Can Protect Medium Spiny Neurons from Neurotoxicity Induced by Mutant Huntingtin Protein.
Having elucidated aspects of the biophysical mechanism of action of LOC14 binding to PDI, we explored whether LOC14 would be a good candidate for in vivo studies. We first examined its activity in an organotypic postnatal brain slice model for HD focusing on the medium spiny neurons (MSNs) of the striatum (26). MSNs are the first population of neurons to degenerate in patients with HD and are the most vulnerable to the toxicity associated with mutant huntingtin dysfunction. Rat corticostriatal brain slice explants were cotransfected with yellow fluorescent protein (YFP) and the first exon of mutant HTT (mHTT-Q73) to induce neurodegeneration and then treated with LOC14. In the absence of LOC14, very few healthy MSNs remained, as assessed by the lack of normal sized and shaped cell bodies, absence of long primary dendrites, and lack of continuous expression of YFP throughout the cell (Fig. 6). Compound LOC14 rescued MSNs in a concentration-dependent manner, even at low micromolar concentrations (Fig. 6). This result indicated that LOC14 oxidation of PDI is neuroprotective in both cell culture and brain tissues.
Fig. 6.

LOC14 rescues striatal MSNs from mutant huntingtin-induced neurodegeneration in brain slice explants. Rat corticostriatal brain slice explants cotransfected with YFP and the first exon of mutant HTT (mHTT-Q73) were treated with LOC14, a positive control compound mixture of 50 µM KW-6002 and 30 µM SP600125, or DMSO only for 4 d. Data are plotted as means ± SEM from one of two representative experiments. *Significant by ANOVA followed by Dunnett's post hoc comparison test at P < 0.05.
LOC14 Is Metabolically Stable Compound for In Vivo Studies.
Next, metabolic in vitro stability studies were performed with LOC14 to determine its suitability for in vivo studies. LOC14 showed high stability in mouse liver microsomes, had a low intrinsic clearance value of less than 0.5 mL/min/g, and had a half-life of more than 90 min (Table S2). This result indicates that LOC14 is not metabolically reactive with liver enzymes such as cytochrome P450s and may have a suitably long half-life in vivo. LOC14 was also relatively stable in mouse plasma with a half-life of 2.4 h (Table S3). Furthermore, low binding was observed between LOC14 and the plasma proteins (Table S4), indicating that in vivo, the bulk of LOC14 is free to be distributed to tissues to exert pharmacological effects.
In Vivo Pharmacokinetic Study with LOC14.
Showing promising in vitro metabolic properties, LOC14 was tested in a single-dose pharmacokinetic (PK) study. This was a pilot study to evaluate the ability of LOC14 to traverse the blood–brain barrier (BBB). In this study, LOC14 was administered via two routes, i.v. (Fig. 7A) or orally (Fig. 7B), at a single-dose of 20 mg/kg to WT C57BL/6j mice. Data from the PK study showed that LOC14 was well tolerated at high dose of 20 mg/kg, penetrated the BBB, and accumulated at reasonable concentrations in the brain regardless of the administration route (Fig. 7 A and B).
Fig. 7.

LOC14 can traverse the BBB in vivo. LOC14 administered via (A) i.v. or (B) oral route to wild-type C57BL/6j mice at 20 mg/kg. The concentration of compound in the brain tissue and plasma is shown for each individual mouse (represented by dots). The horizontal lines represent the mean and SD (n = 3).
Discussion
In this study, we identified and characterized LOC14 as the first reversible, neuroprotective, nanomolar inhibitor of PDI. We found that LOC14 reversibly binds to a region adjacent to the active site of PDI, induces the protein to adopt an oxidized conformation, and inhibits its reductase activity. A possible mechanism of inhibition is shown in Fig. S6. We found that the oxidation of PDI by LOC14 is protective in PC12 cells and in MSNs that degenerate from transfected mutant huntingtin protein expression. Furthermore, LOC14 displayed high in vitro metabolic stability in mouse liver microsomes and blood plasma and penetrated the BBB in vivo, making it a promising candidate for in vivo mouse studies of PDI’s role in protein misfolding diseases.
This study is the first report, to our knowledge, that oxidation of PDI is neuroprotective. One possible explanation has to do with PDI’s binding protein ER oxidoreductin 1 (Ero1). During protein folding, PDI cycles between oxidized and reduced disulfide states. When it forms a disulfide bond with a substrate protein, its own catalytic site becomes reduced. In vitro, PDI can be oxidized by GSSG, but in vivo the protein Ero1 is needed to accomplish this task. Ero1 is a flavin-adenine-dinucleotide-(FAD)-bound protein that takes electrons from reoxidized PDI and passes them onto molecular oxygen as a terminal acceptor, in the process creating hydrogen peroxide and thus generating reactive oxygen species (ROS). By oxidizing PDI with LOC14, Ero1 can be bypassed, reducing the generation of ROS and hence providing neuroprotection. This conclusion is consistent with previous findings that reported that overexpression of PDI is toxic in neuron-like PC12 cells, but can be protected from this overexpression toxicity with the irreversible PDI inhibitor 16F16 (1). By increasing the level of PDI production, one would increase Ero1 oxidation of PDI, leading to increased ROS generation. It is possible that irreversible PDI inhibitors covalently bind to the active site cysteines, prevent the Ero1-PDI interaction, and thus prevent oxidation.
To our knowledge, this is the first compound reported to date that binds reversibly to PDI with low nanomolar affinity and causes protection in neuronal cells and tissue. Inhibition of PDI activity causing neuroprotection has not been validated yet in in vivo mouse models of neurodegeneration, due to the lack of drug-like inhibitors with low cytotoxic properties. Previously reported PDI inhibitors that are cell permeable bind covalently and irreversibly to PDI. Ultimately, this type of binding completely inactivates the protein, can be nonselective, and can result in haptenization, causing unpredicted idiosyncratic toxicity from the immune system in vivo, leading to liver failure and blood disorders. The reversible modulation of PDI with LOC14 overcomes these challenges. LOC14 forms covalent, but reversible, bonds with the protein, ultimately acting like a noncovalent inhibitor (because of its potent, but reversible, effects on the protein). Thus, in vivo, LOC14 may not result in idiosyncratic toxicities. Furthermore, LOC14 showed high stability in liver microsomes and blood plasma, was tolerated at a high dose of 20 mg/kg in mice, and penetrated the BBB in vivo, making it a promising candidate for future in vivo work.
We used the catalytic a domain of PDI A1 as a prototype of the redox reactions that the PDI family of proteins catalyze. The N-terminal cysteine of the a domain in PDI A1 is less reactive than the N-terminal cysteine of the aʹ domain of PDI A1, and both have lower hyperactivity than the catalytic cysteines in PDI A3 (ERp57). Thus, it is very likely that LOC14 will react and oxidize both catalytic domains of PDI A1 and PDI A3. Previously, it was reported that both PDI A3 and PDI A1 proteins (1) (and possibly PDI A4 and PDI A6) (13) were the target of 16F16. Because numerous PDI family members reside in the ER and their distinct roles are still unclear, it is possible that modulation of the whole family by LOC14 is neuroprotective.
In summary, we identified a previously unidentified scaffold, LOC14, for reversible inhibition of PDI’s reductase activity. This compound, although targeting similar residues of PDI as the irreversible inhibitor 16F16, forces the protein to adopt a different conformation that resembles the native oxidized form. LOC14 has improved solubility, potency, and in vitro metabolism properties compared with other reported PDI inhibitors, and it protects neuron-like PC12 cells as well as bona fide striatal MSNs from mutant huntingtin toxicity. Validating PDI as target for neurodegenerative disorders may open new therapeutic strategies to treat and understand these diseases.
Materials and Methods
LOC Library.
The LOC library was assembled as described previously (19). Briefly, structures of 3,372,615 commercially available compounds from six suppliers (Asinex, Life Chemicals, Enamine, TimTec, InterBioScreen, and Chembridge) were compiled into one database and stringently filtered computationally for optimized lead-like properties. The final library totaling 9,719 diverse compounds was purchased, dissolved in DMSO at 4 mg/mL, formatted into 384-well mother plates, and stored frozen at −80 °C.
Enzymatic Insulin Reduction Assay.
The assay was carried out in a 384-well black, clear bottom plate. Each well contained 80 μL of the reaction mixture in buffer A (10 mM Tris⋅HCl, pH 8, 150 mM NaCl, and 2 mM EDTA) with 5 μM PDIa, 100 μM bovine insulin, 350 μM DTT, and 75 μM test compound. All experiments were done in duplicate. The assay plate was incubated at 25 °C for 1 h, and then the absorbance at 650 nm was read on a Tecan Infinite 200 microplate reader for each sample consecutively at 5-min intervals for 1 h. Increase in absorbance is indicative of insulin’s β-chain aggregation and precipitation out of solution.
NMR, ITC, LC/MS, Biochemical, and Cellular Assays.
See SI Materials and Methods for details.
Supplementary Material
Acknowledgments
We thank Dr. Mike Goger from New York Structural Biology Center (NYSBC) for assistance with 3D-NMR data acquisition, Dr. Andras Bauer and Alexandra Cantley for compiling and formatting the LOC library, and Dr. Gisun Park for synthesizing 16F16. We also thank Dr. John Decatur for the use of the Columbia Chemistry NMR core facility instruments provided by National Institutes of Health (NIH) Grant 1S10RR25431-1A1 and National Science Foundation Grant CHE-0840451. A.G.P. and B.R.S. are members of the New York Structural Biology Center. The data collected at NYSBC was made possible by a grant from NYSTAR (Empire State Development Division of Science, Technology and Innovation). This research was funded by the Howard Hughes Medical Institute; NIH Grants 5R01CA097061, 5R01GM085081, and R01CA161061; and New York Stem Cell Science Grant C026715 (to B.R.S.), as well as the Alzheimer’s Drug Discovery Foundation (B.R.S. and D.C.L.), NIH Grant GM50291 (to A.G.P.), and Training Program in Molecular Biophysics Grant T32GM008281 (to A.K. and M.M.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1500439112/-/DCSupplemental.
References
- 1.Hoffstrom BG, et al. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6(12):900–906. doi: 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darby NJ, Creighton TE. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry. 1995;34(37):11725–11735. doi: 10.1021/bi00037a009. [DOI] [PubMed] [Google Scholar]
- 3.Koivu J, et al. A single polypeptide acts both as the beta subunit of prolyl 4-hydroxylase and as a protein disulfide-isomerase. J Biol Chem. 1987;262(14):6447–6449. [PubMed] [Google Scholar]
- 4.Wetterau JR, Combs KA, Spinner SN, Joiner BJ. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J Biol Chem. 1990;265(17):9800–9807. [PubMed] [Google Scholar]
- 5.Yoo BC, et al. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 2002;334(3):196–200. doi: 10.1016/s0304-3940(02)01071-6. [DOI] [PubMed] [Google Scholar]
- 6.Colla E, et al. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci. 2012;32(10):3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkin JD, et al. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30(3):400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Xu S, et al. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc Natl Acad Sci USA. 2012;109(40):16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashida T, Kotake Y, Ohta S. Protein disulfide isomerase knockdown-induced cell death is cell-line-dependent and involves apoptosis in MCF-7 cells. J Toxicol Sci. 2011;36(1):1–7. doi: 10.2131/jts.36.1. [DOI] [PubMed] [Google Scholar]
- 10.Lovat PE, et al. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008;68(13):5363–5369. doi: 10.1158/0008-5472.CAN-08-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbouche R, Miquelis R, Jones IM, Fenouillet E. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J Biol Chem. 2003;278(5):3131–3136. doi: 10.1074/jbc.M205467200. [DOI] [PubMed] [Google Scholar]
- 12.Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118(3):1123–1131. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge J, et al. Small molecule probe suitable for in situ profiling and inhibition of protein disulfide isomerase. ACS Chem Biol. 2013;8(11):2577–2585. doi: 10.1021/cb4002602. [DOI] [PubMed] [Google Scholar]
- 14.Jasuja R, et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122(6):2104–2113. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan MM, et al. Discovery of a small molecule PDI inhibitor that inhibits reduction of HIV-1 envelope glycoprotein gp120. ACS Chem Biol. 2011;6(3):245–251. doi: 10.1021/cb100387r. [DOI] [PubMed] [Google Scholar]
- 16.Karala AR, Ruddock LW. Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 2010;277(11):2454–2462. doi: 10.1111/j.1742-4658.2010.07660.x. [DOI] [PubMed] [Google Scholar]
- 17.Garbi N, Tanaka S, Momburg F, Hämmerling GJ. Impaired assembly of the major histocompatibility complex class I peptide-loading complex in mice deficient in the oxidoreductase ERp57. Nat Immunol. 2006;7(1):93–102. doi: 10.1038/ni1288. [DOI] [PubMed] [Google Scholar]
- 18.Cantley AM, et al. Small molecule that reverses dexamethasone resistance in T-cell acute lymphoblastic leukemia (T-ALL) ACS Med Chem Lett. 2014;5(7):754–759. doi: 10.1021/ml500044g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon SJ, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 21.Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16(3):546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 23.Smith AM, et al. A high-throughput turbidometric assay for screening inhibitors of protein disulfide isomerase activity. J Biomol Screen. 2004;9(7):614–620. doi: 10.1177/1087057104265292. [DOI] [PubMed] [Google Scholar]
- 24.Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Kemmink J, Darby NJ, Dijkstra K, Scheek RM, Creighton TE. Nuclear magnetic resonance characterization of the N-terminal thioredoxin-like domain of protein disulfide isomerase. Protein Sci. 1995;4(12):2587–2593. doi: 10.1002/pro.5560041216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinhart PH, et al. Identification of anti-inflammatory targets for Huntington’s disease using a brain slice-based screening assay. Neurobiol Dis. 2011;43(1):248–256. doi: 10.1016/j.nbd.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.