

Abstract
Herein, we demonstrate that nanotopographical cues can be utilized to enable biologics >66 kDa to be transported across epithelial monolayers. When placed in contact with epithelial monolayers, nanostructured thin films loosen the epithelial barrier and allow for significantly increased transport of FITC-albumin, FITC-IgG, and a model therapeutic, etanercept. Our work highlights the potential to use drug delivery systems which incorporate nanotopography to increase the transport of biologics across epithelial tissue.
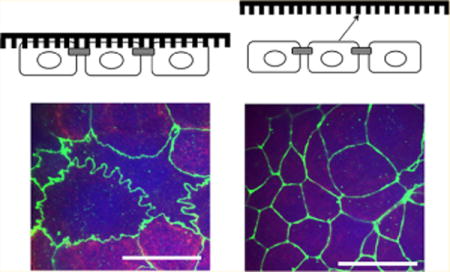
Keywords: Nanotopography, drug delivery, epithelial permeability, tight junctions, transport
Although injections have been the mainstay technology for delivering macromolecular therapeutics, bolus injections are limited by the patient discomfort and the need for physician oversight which can lead to a lack of patient compliance. Alternative drug delivery systems have been developed to utilize routes through more convenient and accessible tissues, such as the epithelia of the nose, mouth, eye, skin, and gastrointestinal tract.1–6 However, these approaches are limited to low molecular weight therapeutics. Because the physiological function of the epithelia is to prevent the entry of toxins into the body, numerous barriers prevent absorption of large molecular weight therapeutics. One significant obstacle to drug delivery across the epithelium is the tight junctional complex that links adjacent cells together and occludes the paracellular space. In fact, it has been shown that tight junctions alone prevent the entry of molecules larger than 20 kDa in the gastrointestinal tract and only 0.5 kDa in the stratum corneum of the skin.
To improve the absorption of biologics without the use of hypodermic needle injections, new approaches need to be developed that would have specific control over drug transport mechanisms in epithelial tissue. Recent studies have shown that nano- and microstructures can induce cellular restructuring via mechanotransduction pathways by interacting with cells at previously unattainable length scales.7–11 In the context of drug delivery systems, it has been shown that structure and geometry play a large role in determining the efficiency of particle uptake by cells.12,13 More recently, Fischer et al. and Uskokovic et al. demonstrated considerable cytoadhesive properties for drug delivery applications of nanoengineered microparticles with nanowires having dimensions comparable to the size of cellular microvilli.10,11 Inspired by this structure-mediated approach to direct specific cellular behavior, we report that nanotopography increases the transport of FITC-BSA, FITC-IgG, and commercially available etanercept (MW = 150 kDa) across the epithelial barrier without the use of chemical permeation enhancers. The ability to increase epithelial transport via nanotopography may have dramatic implications for drug delivery applications where the epithelial barrier presents an obstacle to high molecular weight therapeutics.
In this work, we demonstrate that nanostructured thin films placed in contact with epithelial monolayers enhance protein transport across the barrier. Fabrication of a mold for the nanostructured thin films was performed using electron beam lithography (JEOL JBX-9300FS EBL). A nanostructured pattern was generated on a polymethylmethacrylate (PMMA) resist that had been spin-casted onto an underlying silicon substrate. After developing away the PMMA resist, anisotropic reactive ion-etching was employed to precisely etch the underlying silicon substrate, resulting in the nanofeatured mold shown in Figure 1. The mold was stamped into FDA-approved polypropylene (Premier Lab Supply Inc., 25.4 mm) through nanoimprint lithography (NIL) techniques, which is a facile and versatile process that allows for rapid patterning of large areas. This method allows for virtually any pattern to be generated at a resolution as small as 10 nm in a reproducible manner.14,15 Therefore, virtually any kind of pattern can be fabricated to systematically investigate particular structural parameters such as pitch, diameter, height, aspect ratio, and so forth. Briefly, the polypropylene thin film was placed in contact with the silicon mold and exposed to T = 170 °C and P = 2 GPa using an Obducat 6-in. nanoimprint lithography system. Afterward, the mold was removed to reveal well-defined nanofeatures on a polypropylene thin film. Through scanning electron microscopy inspection, it was apparent that the features consisted of an array of nanopillars, each with an average height (H) of 300 nm, an average diameter (D) of 200 nm (aspect ratio (AR) = 1.5), and a pitch spacing of 300 nm. The root-mean-square (rms) surface roughness of the nanostructured thin film was 47 nm, as measured utilizing atomic force microscopy. Another nanostructured thin film was fabricated that had higher AR nanopillars (H = 16 μm, D = 800 nm, AR = 20, surface roughness = 850 nm). These two nanostructured thin films were tested to investigate the effect of two different aspect ratios on drug transport. The two nanostructured thin films will be referred to as P(1.5) and P(20) where P represents polypropylene and the number inside the parentheses represents the AR of the nanopillars.
Figure 1.
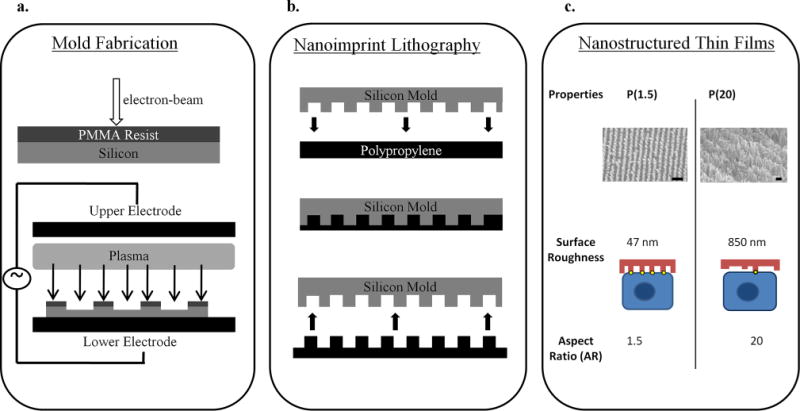
Nanostructured thin film fabrication. Molds for NIL were fabricated using (a) electron beam lithography followed by anisotropic reactive ion etching to generate small features on the nanometer length scale. (b) Next, nanoimprint lithography was employed to imprint the nanofeatures from the nanofeatured mold into a polypropylene thin film through a stamping process. (c) A scanning electron microscopy image of the low AR nanostructured film, P(1.5), shows nanopillar features with an average height of 300 nm and an average diameter of 200 nm. The scale bar is 400 nm. The high AR nanostructured film, P(20), has features with an average height of 16 μm and an average diameter of 800 nm. The scale bar is 3 μm. The schematic demonstrates how P(1.5) with the lower surface roughness is capable of more focal contact points with the cell (yellow dots) compared to the fewer contact points between the cell and P(20).
Once fabricated, the nanostructured thin films were used to investigate their effect on increasing the permeability of epithelial tissue. To this end, human intestinal Caco-2 cells (American Type Culture Collection, Manassas, VA) were used as an in vitro epithelium model. These cells differentiate and polarize such that their phenotype resembles the enterocytes of the small intestine, both morphologically and functionally. Most importantly for this application, Caco-2 cells express tight junctions and form an effective barrier to large molecular weight compounds. The cells were grown to confluency on Transwell permeable inserts (Corning) until they polarized and expressed tight junctions, as indicated by transepithelial electrical resistance (TEER) values greater than 350 Ω·cm2 (World Precision Instruments).16 When a tight barrier was formed, the nanostructured thin films were placed directly in contact with the Caco-2 monolayer as shown in Figure 2. The two controls consisted of an untreated monolayer and an unimprinted polypropylene thin film to control for the same film weight and material chemistry. Next, a phosphate buffered saline solution (Invitrogen) containing either fluorescein isothiocyanate conjugated to bovine serum albumin (FITC-BSA, Sigma-Aldrich, 66 kDa), fluorescein isothiocyanate conjugated to Immunoglobulin G (FITC-IgG, Sigma-Aldrich, 150 kDa), or etanercept (Immunex Corp., Thousand Oaks, CA, 150 kDa), all at the concentration of 0.1 mg/mL, was introduced to the apical side of the transwell insert. The concentration on the basal side of the insert was sampled at various time points for measurement.
Figure 2.
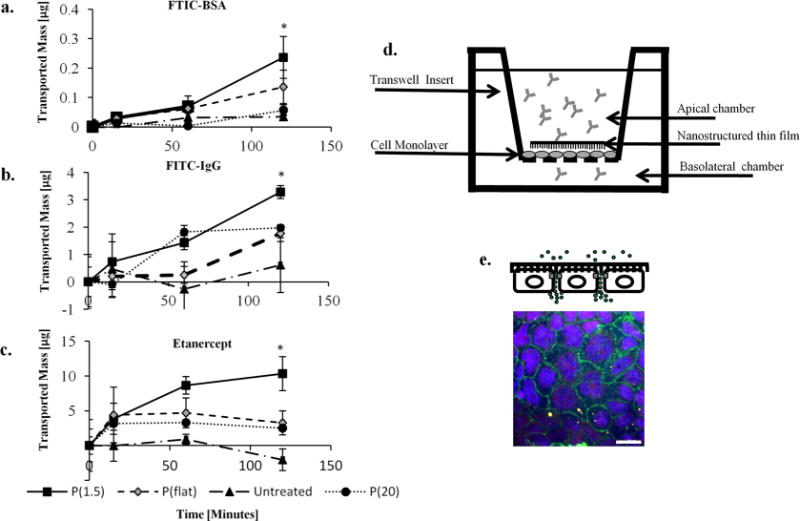
In vitro transport studies. Transport studies show that P(1.5) significantly enhances the transport of high MW species across the Caco-2 cell monolayers over 2 h. Data are displayed as the mean mass in micrograms (± standard deviation). Parts (a), (b), and (c) are the transport of FITC-BSA, FITC-IgG, and etanercept, respectively. Part (d) is a schematic of the transport study setup. The nanostructured thin film is placed directly in contact with the Caco-2 monolayer. The drug solution is placed in the apical chamber and is sampled (with PBS replacement) periodically from the basal camber. (e) It appears that the IgG-FITC (green) is located around the Caco-2 cells (blue Hoechst) directly in the paracellular space. The scale bars represent 20 μm.
As presented in Figure 2, the P(1.5) nanostructured thin film induced substantially higher transport of FITC-BSA, FITC-IgG, and etanercept compared to the two control groups and P(20). Interestingly, the transported mass of FITC-BSA, FITC-IgG, and etanercept does not follow the predicted trend for molecular weight. For example, FITC-BSA has a much lower molecular weight than FITC-IgG and etanercept; yet, its transport across the barrier was smallest among the three compounds (refer to Figure 2). Previous studies have described how drug permeability decreases as the molecular weight increases due to larger radii of gyration and the related diffusivity effects. However, it is important to note that size is only one of many physicochemical properties that contribute to drug permeability. Other properties such as lipophilicity and electrophilicity are critical parameters for influencing transport across the epithelial barrier. Therefore, although FITC-IgG and etanercept were transported across the monolayer in greater amounts than FITC-BSA, these results could be due to the differences in other physicochemical properties besides molecular weight. Future studies will explore how nanotopographical cues influence the transport of drugs with varying partition coefficients and electrophilicities.
With regards to the nanofeature AR, previous work has demonstrated that silicon nanowires with AR’s greater than 16 (length = 3 m, diameter = 60 nm) tend to collapse over on themselves and mat together, reducing the overall available surface area for cytoadhesion.11 This theory may be implicated for the P(20) nanostructured surface which induced lower drug transport. The high AR nanopillars of P(20) are more compliant and have a lower critical buckling load, which reduces the ability to impart the necessary mechanical stimulus on the cells. Studies have shown throughout the literature that mechanical stimuli applied to cells via nanotopographical cues invoke numerous cytoskeletal rearrangements through force-induced mechanotransduction pathways.8,9,17,18 Therefore, the AR of the nanofeatures may be a critical parameter to influence the degree of mechanical stimuli on the cells, which ultimately affects the drug transport across the epithelium. Future studies will consist of optimizing high molecular weight drug transport by tuning the AR’s of the features around 1.5 with NIL fabrication.
Additionally, it is well-established that surface roughness on the nanoscale influences the interface between nanomaterials and different epithelial cell types such as keratinocytes and mammary epithelial cells.19–21 For example, studies have demonstrated that surface roughness on the nanoscale alters cytoskeletal rearrangements and cellular focal adhesions as observed through morphological changes.19–21 These cytoskeletal components, such as F-actin, are tethered directly to tight junction proteins (zonula occludens, claudins, occludin, and junctional adhesion molecules), which are the elements responsible for epithelial barrier function.22–25 Therefore, the surface roughness on the nanoscale may influence junction restructuring through physical cues mediated by integrin-ligand engagement and cytoskeletal rearrangements. These mechanotransduction cues may be stronger when the surface roughness lies between an optimal range which is less than 850 nm but higher than the surface roughness of the flat control film as we observed. The high AR nanostructured thin film, with its high surface roughness close to 1 μm, provides fewer contact points and thus a weaker physical interaction with the cells to influence tight junction rearrangement as shown in the schematic of Figure 1. Similarly, the unimprinted polypropylene control film provides minimal surface interaction with the cells and consequently induces a lower amount of transport across the epithelial monolayer. Although there is a nonzero amount of drug that is transported in the presence of the control film, this may be due to the weight effects of the film. Taken together, these result suggest that there may be an optimal aspect ratio and surface roughness for sufficient cell–material interactions to impart epithelial permeabilization for high molecular weight drug transport.
Additionally, as shown in Figure 4, the TEER values decreased significantly when P(1.5) and the flat unimprinted films were brought into contact with the cells. This result indicates that charged ions are more easily able to penetrate the barrier in the presence of these two treatments. However, after the nanostructured films were removed and incubated at 37 °C for 24 h, the TEER values returned to the original resistance levels, suggesting that the effect is reversible and nontoxic. These results are significant because the three proteins that transported across the epithelium were up to 1 order of magnitude larger than the macromolecular sizes previously demonstrated as able to penetrate the gastrointestinal epithelium (20 kDa). Additionally, this enhancement in permeability is not cytotoxic as evidenced in the Supporting Information. Currently, chemical agents, ultrasound techniques, electrophoresis methods, and combinations of both have been extensively explored throughout the literature as approaches to permeabilizing the epithelium. For example, a recent report demonstrated that dual-frequency ultrasound pretreatments of the skin resulted in enhanced delivery of glucose (0.180 kDa) and inulin (5 kDa), drugs that are 2 to 3 orders of magnitude smaller in molecular weight than the proteins that we investigated. The study demonstrated a cumulative mass delivery across epithelial tissue of 50 and 3.5 μg of glucose and inulin, respectively, from a 1 mg/mL drug reservoir solution.26 Although their approach achieves similar mass transport as our method while using a 10 times higher drug concentration gradient, the long-term tissue viability from such an aggressive approach is uncertain, and the simultaneous application of dual-frequency ultrasound is a comparatively complicated setup. Additionally, other approaches utilize chemical permeability enhancers such as sodium deoxycholate. This chemical was reported to significantly increase mucosal epithelial permeability to salicylic acid in the paracellular route by causing uncoiling and extension of protein helices, which oftentimes permanently damages cellular membranes.27 Other agents such as ionic surfactants have demonstrated increases in epithelial permeability, but they have been shown to illicit toxic and irritative potential at relatively low concentrations.28 Although these chemical permeability enhancements have shown enhanced delivery effects, there are permanent toxicity risks that must be addressed. Therefore, the use of nanostructured thin films to induce large molecule transport across the epithelial barrier presents a significant finding for translational drug delivery applications.
Figure 4.
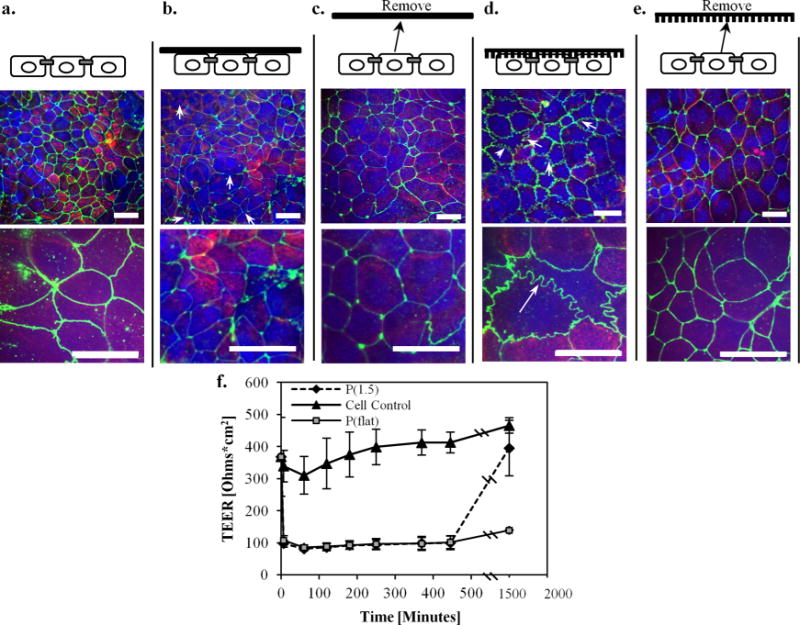
Tight junction morphological changes. Immunofluorescence staining of the tight junction protein, zonula occluden (ZO-1), was performed. (a) Staining of the untreated caco-2 monolayer shows a normal cobblestone morphology. (b) When the flat unimprinted polypropylene control film is placed on the monolayer, minimal disruptions in ZO-1 are observed as indicated by the discontinuous lines (pointed out by the white arrows). (c) When the flat film is removed, the relatively low staining intensity of the ZO-1 remains the same after 24 h. (d) However, it is apparent that the nanostructured thin film induces a dramatic ruffled morphology after 2 h (see arrows), indicating tight junction remodeling and a loosening of the epithelial barrier to allow for paracellular transport. (e) After the nanostructured thin film was removed from the monolayer and incubated for 24 h, the ZO-1 morphology reverted back to the normal cobblestone architecture, indicating a reversible rearrangement. (f) TEER measurements before and after the nanostructured surface is placed in contact with the cells. TEER measurements decreased in the presence of both the nanostructured and the flat films. However, the monolayer that had been in contact with the nanostructured film eventually increased after 24 h which demonstrates the reversible and nondeleterious effects of the nanostructures on the cells. In contrast, the monolayer that had been in contact with the flat film does not recover to a higher TEER value after 24 h. Scale bars for the three top images and three bottom images are 10 and 20 μm, respectively.
Next, we investigated whether the nanotopography induces etanercept transport by passive or active cellular processes. It is hypothesized that nanostructured thin films could: mechanically disrupt the tight junction proteins, trigger active tight junction remodeling to allow for paracellular transport, and/or induce transcellular pathways requiring energy-dependent processes. To investigate these mechanisms and elucidate whether active or passive processes were at play, the transport studies were performed at 4 and 37 °C in the presence of the nanostructured thin films. As displayed in Figure 3, when the monolayers were maintained at 37 °C, transport was significantly enhanced. However, when the monolayers were maintained at 4 °C compared to the physiological temperature (37 °C), etanercept transport remained relatively unchanged over 4 h. These results implicate the active transport mechanism and not the one due to passive, mechanical puncturing of the epithelial barrier. To investigate these mechanisms related to nanotopographical cues, we tested the contributions of both the active transcellular route and active tight junction remodeling in the paracellular pathway.
Figure 3.
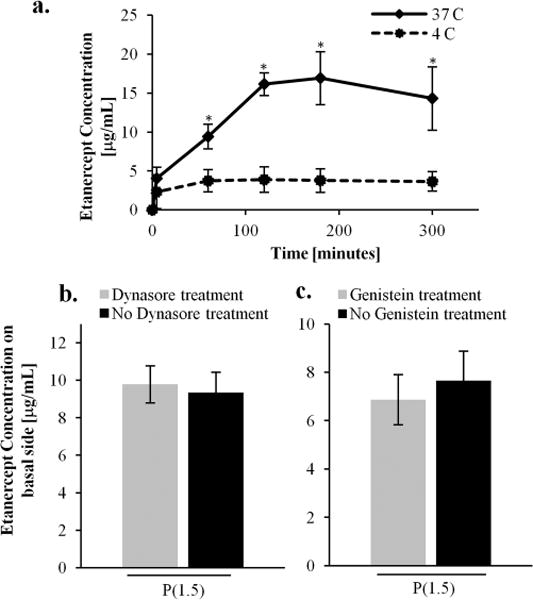
Active transport processes. (a) Etanercept transport studies performed at 4 and 37°C show significantly higher drug concentration in the basal chamber at physiological temperature. The transport is retarded at 4°C. These results indicate that the enhanced transport is due to active transport instead of passive mechanisms. (b) Dynasore was used to inhibit dynamin-mediated endocytosis and shows no significant effect on the transport of etanercept across the epithelial monolayer. (c) Similarly, genistein was used to inhibit caveolae-mediated endocytosis and also does not affect the transport of the etanercept.
Because the three proteins have radii of gyration that are approximately twice as large as the tight junction pores in the paracellular space (1–3 nm), we suspected that they permeate the epithelium via the transcellular pathway.29,30 Therefore, we explored this route by testing two small molecule endocytosis inhibitors that block transcellular transport and have been used extensively for this application in other reported studies.31 Dynasore is a small molecule inhibitor that specifically blocks the GTPase activity of dynamin and has been shown to not influence other GTPase activity.32 This molecule was used to block vesicular dynamin-mediated endocytosis, which is involved in both clathrin- and caveolin-mediated. Genistein was used to inhibit protein tyrosine kinases that block internalization by caveolae.31 To this end, etanercept transport studies were performed in the presence of these two chemical inhibitors in separate experiments to block clathrin-, caveolin-, and dynamin-mediated endocytosis across the Caco-2 monolayer. To determine the sufficient dose of dynasore and genistein, two control experiments were performed with transferrin and albumin, which are known molecules to transport via the clathrin and caveolae pathways, respectively. Dynasore (BioVision) was reconstituted in DMSO at 80 mM and then diluted in MEM-α medium to 80 μM. After washing the Caco-2 monolayers with PBS, the cells were incubated at 37 °C in the presence of the Dynasore solution for 1 h. Transport studies were performed as described above in the presence of the nanostructured films. To block caveolae endocytosis, Caco-2 monolayers were incubated with genistein (200 μM) for 1 h at 37 °C before performing the transport studies in the presence of the nanostructured thin films as previously reported.15 The results in Figure 3 show that the inhibitors did not significantly decrease the etanercept transport across the epithelial barrier. This suggests that the nanotopography does not induce transcellular transport related to clathrin-, dynamin-, or caveolae-mediated endocytosis. However, further studies will be performed to elucidate other contributions to transcellular transport.
As shown in Figure 4d, the TEER values were reduced when the nanostructured thin films were placed in contact with the cells. This result is a direct indication that the tight junctions in the paracellular space were affected. Therefore, we investigated the paracellular pathway by performing immunofluorescence imaging of the tight junctions to morphologically examine whether the nanotopography induced active remodeling of these proteins. Briefly, cell monolayers from the transport studies were stained for zonula occludens-1 (ZO-1) to visualize the morphology of the tight junctions after contacting the nanostructured thin films for 2 h and then at 24 h, after the nanostructured surfaces were removed. The cells were fixed and permeabilized with cold methanol at 4 °C and then incubated with a primary antibody (ZO-1 rabbit polyclonal antibody, Invitrogen) solution diluted 1:100 at 25 °C for 1 h. After washing with PBS, a 1:100 secondary antibody solution (Abcam) was added for 1 h at 25 °C. The samples were then mounted for confocal microscopy imaging. As shown in Figure 4, the cell monolayer that had been in contact with P(1.5) for 2 h exhibited remarkable ZO-1 membrane ruffling. This result is in contrast to the untreated control monolayer which displays an intact cobblestone morphology and to the P(flat) control which shows low intensity ZO-1 staining. The presence of the nanostructured thin films in contact with the monolayer did not affect cell viability (Supporting Information). After 2 h, the nanostructured surfaces were removed, and the monolayers were incubated at 37 °C for 24 h. Within the 24 h, the ruffled morphology reverted back to the intact tight junction configuration, and the TEER values recovered to their original values, which suggests the reversibility of this remodeling process. It is interesting to note that the TEER values also decreased for the monolayers in contact with the P(flat) control films. However, when the control films were removed, the resistance did not return to the original value. Therefore, the control films do not exhibit the reversible effect that was demonstrated by the nanostructured films. The weight of the control films themselves may be disrupting the monolayer in a nonspecific way. This general disruption from the weight may contribute to the enhanced permeability to small ions and electrolytes as measured by the decrease in TEER. However, the tight junction proteins do not remodel and respond in the same way they do to the nanostructured thin films which exhibit ZO-1 morphological ruffling.
The dramatically ruffled morphology of ZO-1 indicates that the nanotopography remodels the tight junction proteins and loosens the paracellular pathway for etanercept transport. Studies by Teo et. al and others demonstrated that nanotopography can alter cellular morphology when the cells were grown directly on the nanostructured surface.33 Although their setup was subtly different from ours, they also demonstrated that nanotopography induced membrane ruffling that was coordinated by the actin cytoskeleton. In the present study, we hypothesize that the membrane curvature induced by the nanopillars may contribute to the loosening of the tight junction barrier through mechanotransduction pathways. In addition, as a direct indication that paracellular transport is at play, we imaged the monolayer immediately after performing the transport studies to visualize the location of FITC-IgG relative to the cells. From the maximum intensity projection image in Figure 2, it is apparent that the FITC-IgG is located in the paracellular space between the epithelial cells.
To further examine the paracellular transport route, different signaling molecules were investigated through gene expression studies. The results from the real time PCR were analyzed using the ΔΔCt method and normalized to GAPDH transcript levels. From Figure 5, it is apparent that the nanostructured thin film has a dramatic effect on the overall mRNA transcription levels. For example, myosin light chain kinase (MLCK), an important regulator of paracellular permeability, increased by 5.6-fold and 19-fold in cells that contacted the nanostructured thin films compared to the unimprinted polypropylene and untreated controls, respectively. MLCK is a protein kinase associated with tight junctions through the contraction of the cortical actin cytoskeleton and has been previously reported to enhance paracellular permeability.34 These results suggest that the nanotopography may lead to increased paracellular transport by activating mechanotransduction pathways. Additionally, focal adhesion kinase (FAK) transcript expression was examined to measure the formation of focal adhesions as an indication of the level of signaling through integrin binding and clustering. Extensive work in the literature has demonstrated the coordinated cross talk between integrin-binding and the junctional complex for the maintenance of epithelial integrity.35 To this end, we demonstrated that the FAK mRNA expression was increased over 16-fold in cells that contacted the nanostructured thin film. This data supports the observation that tight junction loosening may be triggered by integrin engagement mediated by FAK. Although our strategy utilizes a physical cue and not a biological peptide sequence, the mechanical stimulus from the nanostructures may imitate receptor–ligand interactions on the nano scale. Therefore, our approach offers a novel platform to investigate interactions that may be related to both physical and biological mechanisms.
Figure 5.
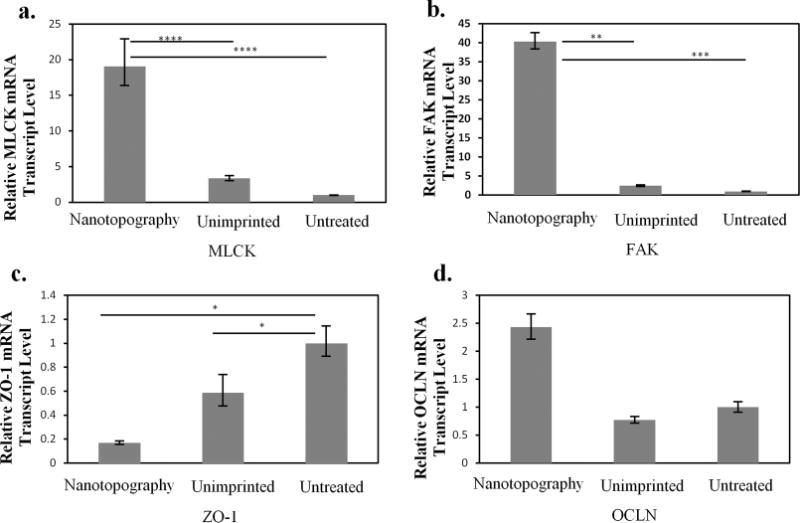
Quantitative PCR studies. The gene expression levels of the signaling molecules, myosin light chain kinase (MLCK) and focal adhesion kinase (FAK), are displayed in (a) and (b). The gene expression levels of tight junction proteins zonula occludin-1 (ZO-1) and occludin (ocln) are also displayed in (c) and (d). Data are normalized by expression levels of each gene by the controls (Untreated) and presented as an average ± standard deviation. *p < 0.05, **p < 0.005, ***p < 0.002, and ****P < 0.001, n = 3.
This restructuring of the tight junctions to allow for paracellular drug transport is further supported by the gene expression results for tight junction proteins. There is a significant decrease in ZO-1 mRNA expression levels compared to the controls, as shown in Figure 5. Many studies have reported that a correlation exists between a decrease in ZO-1 protein expression and enhanced paracellular permeability. For example, Tian et. al demonstrated that stimulation of HK-2 cells with TGF-β resulted in a decrease in ZO-1 protein expression, indicating tight junction disassembly and a subsequent increase in paracellular permeability.36 Similarly, hepatic growth factor (HGF) stimulated RPE monolayers were observed to lose barrier function resulting from a decrease in ZO-1 protein expression in the presence of HGF.37 Therefore, a decrease in ZO-1 gene expression is consistent with tight junction disassembly and enhanced paracellular permeability.
An increase in the expression of occludin (ocln) due to the nanostructured surface was also observed. Ocln is a transmembrane component of tight junctions that regulates paracellular permeability. Consistent with these results, Wang et. al also observed an increase in protein expression of ocln and enhanced paracellular permeability when BMECs were treated with VEGF.38 Our results suggest that the nanostructures are influencing mechanotransduction pathways to actively remodel the tight junctions and facilitate the transport of etanercept. Furthermore, this phenomenon appears to be a reversible process as indicated by the TEER values returning to their original values when the nanostructured surfaces are removed after 24 h. This result suggests that the tight junctions recover and that the nanostructure does not illicit cytotoxic effects. The nanostructures seem to dramatically affect the paracellular pathway by directly modulating the tight junction proteins.
In this work, nanostructured thin films were fabricated using NIL and were utilized as an epithelial permeability enhancer for the transport of high molecular weight proteins. The transport of three high molecular weight biologics was significantly improved by leveraging the interaction of nanoscale features with cellular processes related to tight junction remodeling. This interaction allowed for increased transport across an epithelial monolayer. By taking advantage of nanostructure-mediated transport, it may be possible to safely deliver high molecular weight biologics through epithelial tissue without the use of hypodermic needle injections.
Supplementary Material
Acknowledgments
We would like to thank Dr. Lily Peng, Dr. Vuk Uskoković Dr. Daniel Bernards, Professor Ronald Fearing, and the UCSF Nikon center for their valuable insight and advice, Devin Brown and Nicole Devlin at the Georgia Tech Microelectronics Research Center for generating the NIL molds and Sunland Biotechnology for ELISA testing. Funding for this work was kindly provided by Kimberly-Clark Corporation and the National Institutes of Health (NIH).
Footnotes
Methods that were used for the gene expression studies as well as a figure that verifies cell viability in the presence of the nanostructures. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Xia H, Gao X, Gu G, Liu Z, Zeng N, Hu Q, Song Q, et al. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Oh DH, Chun KH, Jeon SO, Kang JW, Lee S. Eur J Pharm Biopharm. 2011 doi: 10.1016/j.ejpb.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Gratieri T, Gelfuso GM, de Freitas O, MelaniRocha E, Lopez RFV. Eur J Pharm Biopharm. 2011 doi: 10.1016/j.ejpb.2011.05.006. [DOI] [Google Scholar]
- 4.Polat BE, Hart D, Langer R, Blankschtein D. J Controlled Release. 2011 doi: 10.1016/j.jconrel.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonaje K, Lin KJ, Wang JJ, Mi FL, Chen CT, Juang JH, Sung HW. Adv Funct Mater. 2010 doi: 10.1002/adfm.201001014. [DOI] [Google Scholar]
- 6.Shofner JP, Phillips MA, Peppas NA. Macromol Biosci. 2010 doi: 10.1002/mabi.200900223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci. 2008 doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalby MJ. Med Eng Phys. 2005 doi: 10.1016/j.medengphy.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Dalby MJ, Riehle MO, Sutherland DS, Agheli H, Curtis ASG. Eur J Cell Biol. 2004 doi: 10.1078/0171-9335-00369. [DOI] [Google Scholar]
- 10.Uskoković V, Lee PP, Walsh LA, Fischer KE, Desai T. A Biomaterials. 2012 doi: 10.1016/j.biomaterials.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer KE, Alemán BJ, Tao SL, Daniels RH, Li EM, Bünger MD, Nagaraj G, Singh P, Zettl A, Desai TA. Nano Lett. 2009 doi: 10.1021/nl803219f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doshi N, Mitragotri S. J Roy Soc Interfaces. 2010 doi: 10.1098/rsif.2010.0134.focus. [DOI] [Google Scholar]
- 13.Champion JA, Katare YK, Mitragotri S. J Controlled Release. 2007 doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou SY, Krauss PR, Renstrom PJ. Science. 1996 doi: 10.1126/science.272.5258.85. [DOI] [Google Scholar]
- 15.Chou SY, Krauss PR. Microelectron Eng. 1997 doi: 10.1016/S0167-9317(96)00097-4. [DOI] [Google Scholar]
- 16.Moyes SM, Morris JF, Carr KE. Int J Pharm. 2011 doi: 10.1016/j.ijpharm.2010.12.033.17. [DOI] [PubMed] [Google Scholar]
- 17.Yu D, et al. Proc Natl Acad Sci USA. 2010;107:8237–8241. doi: 10.1073/pnas.0908869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magjarevic R, Goldberg DS, Swaan PW, Ghandehari H. In: Herold KE, Vossoughi J, Bentley WE, editors. 26th Southern Biomedical Engineering Conference SBEC 2010; College Park, Maryland, USA. Berlin: Springer; 2010. pp. 236–239. [Google Scholar]
- 19.Puckett S, Pareta R, Webster TJ. Int J Nanomed. 2008;3:229–241. doi: 10.2147/ijn.s2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puckett SD, Lee PP, Ciombor DM, Aaron RK, Webster TJ. Acta Biomater. 2010 doi: 10.1016/j.actbio.2009.12.016. doi:16/j.actbio.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Khang D, Lu J, Yao C, Haberstroh KM, Webster T. J Biomaterials. 2008 doi: 10.1016/j.biomaterials.2007.11.009. doi:16/j.biomaterials.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg DS, Swaan PW, Ghandehari H. IFMBE Proc 26th Southern Biomedical Engineering Conference, SBEC; 2010. pp. 236–239. [Google Scholar]
- 23.Wang N, Butler JP, Ingber DE. Science. 1993 doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 24.McNeil E, Capaldo CT, Macara IG. Mol Biol Cell. 2006 doi: 10.1091/mbc.E05-07-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalby MJ, Biggs MJP, Gadegaard N, Kalna G, Wilkinson CDW, Curtis ASG. J Cell Biochem. 2007 doi: 10.1002/jcb.21058. [DOI] [PubMed] [Google Scholar]
- 26.Schoellhammer CM, Polat BE, Mendenhall J, Maa R, Jones B, Hart DP, Langer R, Blankschtein D. J Controlled Release. 2012 doi: 10.1016/j.jconrel.2012.08.019.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gandhi R, Robinson JR. Int J Pharm. 1992 doi: 10.1016/0378-5173(92)90142-O. [DOI] [Google Scholar]
- 28.Sohi H, Ahuja A, Ahmad FJ, Khar RK. Drug Dev Ind Pharm. 2010 doi: 10.3109/03639040903117348. [DOI] [PubMed] [Google Scholar]
- 29.Kilár F, Simon I, Lakatos S, Vonderviszt F, Medgyesi GA, Závodszky P. Eur J Biochem. 1985 doi: 10.1111/j.1432-1033.1985.tb08712.x. [DOI] [PubMed] [Google Scholar]
- 30.Kitchens K, Kolhatkar R, Swaan P, Eddington N, Ghandehari H. Pharm Res. 2006 doi: 10.1007/s11095-006-9122-2. [DOI] [PubMed] [Google Scholar]
- 31.Sadekar S, Ghandehari H. Adv Drug Delivery Rev. 2012 doi: 10.1016/j.addr.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dev Cell. 2006 doi: 10.1016/j.devcel.2006.04.002. doi:16/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 33.Teo BKK, Goh SH, Kustandi TS, Loh WW, Low HY, Yim EKF. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.08.088. [DOI] [PubMed] [Google Scholar]
- 34.Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. J Cell Sci. 2006 doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- 35.Ojakian GK, Ratcliffe DR, Schwimmer R. J Cell Sci. 2001;114:941–952. doi: 10.1242/jcs.114.5.941. [DOI] [PubMed] [Google Scholar]
- 36.Tian YC, Phillips AO. Am J Pathol. 2002 doi: 10.1016/S0002-9440(10)61109-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin M, Chen Y, He S, Ryan SJ, Hinton DR. Invest Ophthalmol Vis Sci. 2004 doi: 10.1167/iovs.03-0355. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Dentler WL, Borchardt RT. Am J Physiol Heart Cir Physiol. 2001;280:434–440. doi: 10.1152/ajpheart.2001.280.1.H434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.