

Abstract
Local release of drugs may have many advantages for tissue repair but also presents major challenges. Bioengineering approaches allow microstructures to be fabricated that contain bioactive peptides for sustained local delivery. Heart tissue damage is associated with local increases in mechano growth factor (MGF), a member of the IGF-1 family. The E domain of MGF peptide is anti-apoptotic and a stem cell homing factor. The objectives of this study were to fabricate a microrod delivery device of poly (ethylene glycol) dimethacrylate (PEGDMA) hydrogel loaded with MGF peptide and to determine the elution profile and bioactivity of MGF. The injectable microrods are 30 kPa stiffness and 15 μm widths by 100 μm lengths, chosen to match heart stiffness and myocyte size. Successful encapsulation of native MGF peptide within microrods was achieved with delivery of MGF for 2 weeks, as measured by HPLC. Migration of human mesenchymal stem cells (hMSCs) increased with MGF microrod treatment (1.72±0.23, p<0.05). Inhibition of the apoptotic pathway in neonatal rat ventricular myocytes was induced by 8 h of hypoxia (1 % O2). Protection from apoptosis by MGF microrod treatment was shown by the TUNEL assay and increased Bcl-2 expression (2±0.19, p<0.05). Microrods without MGF regulated the cytoskeleton, adhesion, and proliferation of hMSCs, and MGF had no effect on these properties. Therefore, the combination microdevice provided both the mechanical cues and 2-week MGF bioactivity to reduce apoptosis and recruit stem cells, suggesting potential use of MGF microrods for cardiac regeneration therapy in vivo.
Keywords: Microdevice, Microenvironmental niche, Tissue regeneration, Drug delivery
1 Introduction
Cells are regulated by both mechanical and chemical stimuli arising from the surrounding extracellular matrix (ECM). They respond by subcellular reorganization, proliferation, differentiation, migration and other functions (Scadden 2006; Discher, et al. 2009). One goal of bioengineering is to mimic this physico-chemical microenvironment in order to control the behavior of cells for tissue regeneration. Our group has shown that microtopography affects cellular structure and function (Motlagh et al. 2003; Norman and Desai 2005; Norman et al. 2008; Collins et al. 2010; Biehl et al. 2009; Doroudian et al. 2013). Furthermore, the stiffness of the surface on which stem cells are grown is sufficient to alter lineage commitment (Engler et al. 2006). The greatest effect on fibroblast cell function was induced by microrods with a combination of stiffness and microtopography in a three dimensions (3D) gel culture system (Ayala et al. 2010). The range of stiffness for normal cardiac ventricular tissue is 20–30 kPa (Berry et al. 2006) and the logical range for microstructures to be relevant for the study of cardiac cell regulation. The micron size scale of cells in cardiac tissue is also used.
Equally important to the physical parameters are the many chemical signals, which provide complex cues for the functional control of different cell types. For cardiac repair, some critical chemicals from the cellular microenvironment are those that recruit stem cells or prevent myocyte death. It is interesting that most studies now show that functional improvements associated with the direct delivery of mesenchymal stem cells (MSCs) to the heart are due to their secretion of soluble factors rather than the engraftment of stem cells per se (Fazel et al. 2006; Paul et al. 2009). The universal stem cell homing factor, SDF-1, is an important chemokine attracting stem cells to the heart. SDF-1 is produced by ischemic tissue and affects migration and mobilization of proangiogenic cells, however, it undergoes rapid proteolysis in blood, limiting its therapeutic potential (Hattori et al. 2001).
Cell homing to an injury site is also a property of insulin-like growth factor 1 (IGF-1). Local IGF-1 produced by the muscle acts to increase myocyte growth and preserve the injured myocardium (Napier et al. 1999; Donath et al. 1998; Stavropoulou et al. 2009). Rapid binding of IGF-1 to proteins in the circulation severely limits the bioavailability of IGF-1. An alternative splicing of IGF-1 yields a special E domain and has a distinct function from IGF-1 and does not bind to IGF-1 binding proteins. IGF-1Ec in humans (IGF-1Eb in rodents) is also known as the mechano growth factor (MGF). Additionally, native MGF blocks apoptosis of injured myocytes as well as attracting stem cells (Ates et al. 2007; Carpenter et al. 2008; Musaro et al. 2004). The E-domain of the MGF peptide, which consists of 24 amino acids, caused increased migration of human mesenchymal stem cells (hMSCs) and human myogenic precursor cells (Collins et al. 2010; Mills et al. 2007). MGF may affect proliferation of some cell types, such as myoblast C2C12, myocardial H9C2, and osteoblasts (Yang and Goldspink 2002; Kandalla et al. 2011; Li et al. 2013) but not others such as MSCs and chondrocytes (Collins et al. 2010; Schlegel et al. 2013).
Clinical regenerative therapy would benefit by enhancing tissue repair without the need for exogenous stem cells. Therefore, it is attractive to consider what steps are necessary to engineer the crucial features in an acellular microdevice. A rational physical design that might foster natural cardiac repair processes would be a combination of microtopographic features, like the myocyte-shaped microrod, with stiffness in the cardiac range (30 kPa). Additionally, MGF is an attractive choice of a peptide for incorporation into microrods for cardiac regeneration and repair given its chemokine and anti-apoptotic properties. Therefore, this study takes the initial steps to manufacture an MGF-eluting microrod that can be used as an injectable microdevice for the localized delivery of bioactive MGF over sufficient time for potential regenerative therapy of the injured heart in vivo.
2 Materials and methods
2.1 MGF peptide
The native form of MGF E-domain (peptide sequence: YQPPSTNKNTKSQRRKGSTFEERK, [Fig. 1a] was custom-synthesized with a C-terminal cysteine cap at a purity of >90 % and delivered in lyophilized aliquots (Genescript Corp, NJ). Peptides were dissolved in 80 % acetonitrile (stock 4 mg/mL) and diluted in molecular grade water to 1,000 ng/mL yielding a final concentration (30–70 ng/mL) in media. For the control without MGF, equal volumes of molecular grade water were added to media.
Fig. 1.
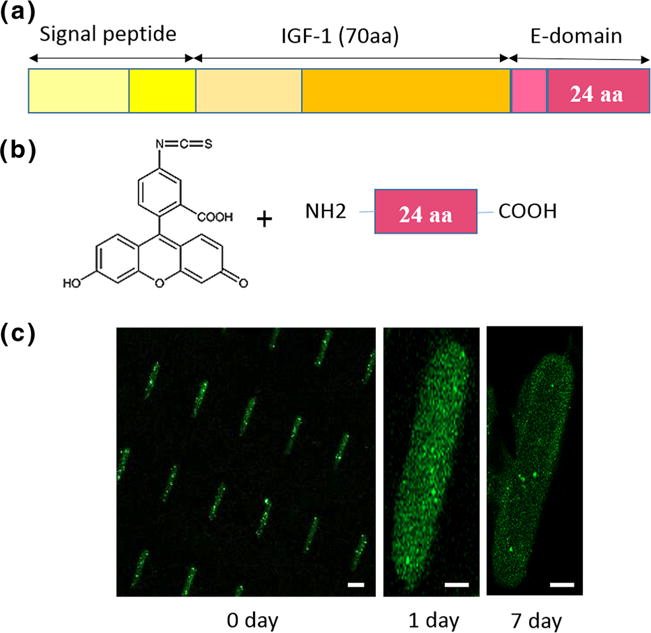
FITC-MGF encapsulation into PEG-DMA microrods. a MGF is an isoform of IGF-1, which includes 24 amino acids in the C terminal of the E-domain; b FITC is conjugated to the N terminal of the MGF peptide; c MGF-FITC encapsulated in the microrods still attached to the wafer (0 day), and at 1 day and 7 days in PBS, as seen by fluorescence microscopy (green, FITC). Scale bar, 20 μm
2.2 Microfabrication and encapsulation of microrods with MGF
Microrods were fabricated photolithographically using commercially available materials by processes developed by us (Ayala et al. 2010). Briefly, the precursor solution was made by mixing poly(ethylene glycol) dimethacrylate (PEG-DMA) (MN 750, Sigma Aldrich) with 1× phosphate buffered saline (PBS) and adding the photo-initiator 2, 2-dimethoxy-2-phenylacetophenone (Sigma Aldrich) solubilized in 1-vinyl-2-pyrrolidone (Sigma Aldrich) at a concentration of 150 mg/mL.
Lyophilized E-domain native MGF (Genescript Corp, NJ) was resuspended at a concentration of 4.0 mg/mL in a solution of 80 % acetonitrile (HPLC Grade, Sigma Aldrich) and 20 % PBS and added to the hydrogel precursor solution. Based on the desired specifications of sufficient mechanical stiffness, high hydrogel porosity for drug loading, and sufficient viscosity of precursor solution for thin layer formation, the ratio of components chosen was 4 parts PEG-DMA : 3 parts PBS : parts photo-initiator solution : 1 part MGF solution. The final MGF concentration was 0.458 mg/mL. For control microrods not containing peptide, the 1 part MGF solution was replaced with a solution of 80 % acetonitrile and 20 % PBS with no MGF to maintain consistency between controls and peptide-loaded samples.
MGF-tagged FITC was incorporated [Fig. 1b] to evaluate the encapsulation and distribution of MGF in microrods. Green fluorescence of FITC shows MGF still bound to the microrods after initial washing and after one to 7 days in PBS [Fig. 1c].
2.3 Microrod size and stiffness
The PEGDMA hydrogel microrods were designed to be the size scale and shape of cardiac myocytes, namely microrods with approximately 15×15 μm2 cross-section and length 100 μm. Light microscopy measured length and width [Fig. 2a]. The microstructure height was accurately measured using an Ambios Technology XP-2 profilometer [Fig. 2b]. Scanning electron microscopy (SEM) images show the structure of empty and MGF microrods [Fig. 2c, d]. By cross-interpolating from previous work (Ayala et al. 2010), the stiffness was designed to be approximately 30 kPa chosen to mimic adult heart stiffness and based on the PEGDMA concentration and cross-linker ratio used in the precursor solution during photolithographic processing. The small size (<3 kDa) and low volume of the MGF incorporated into each microrod, along with its non-reactivity during the photopolymerization process makes it unlikely that the inclusion of this peptide will result in appreciable changes in stiffness and will still allow us to achieve the minimum stiffness threshold of 10–20 kPa in order to properly mimic native heart tissue stiffness.
Fig. 2.
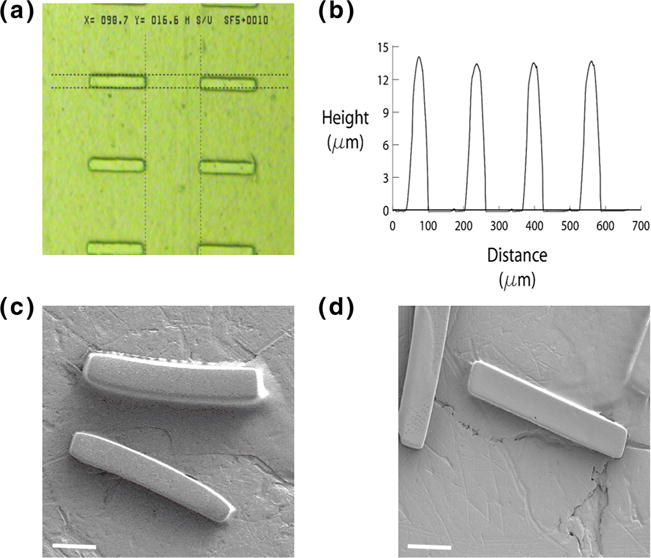
Microrod dimension and structure. a A phase microscopy shows microrods with approximately 15 μm width and 100 μm length; b The microstructure height was measured approximately 15 μm using an Ambios Technology XP-2 profilometer (note: the x axis distance value in this figure does not represent the actual width of the microrods); SEM images represent the structure of empty microrods c and MGF microrods d. Scale bar, 20 μm
2.4 Microrod degradation
Microrods were resuspended in sterile, warm saline and shaken in an incubator at 37 °C. Microscopic images recorded over a period of 2 months were used to determine the rate of degradation by width measurement using ImageJ software. The mean width of 20 isolated microrods was measured at each time point.
2.5 MGF elution from the microrods
Elution of MGF from the hydrogel matrix of the microrods was assessed by harvesting the MGF microrods into 450,000 microrod aliquots immediately after fabrication and suspending in 125 μL of PBS. The microrods were agitated gently at 37 °C. At day 1, 2, 4, 7, 14, and 21, the samples were removed from the incubator, agitated again to mix the solution thoroughly, then gently centrifuged to collect the MGF microrods at the bottom. A 110 μL sample was then drawn from the vial and replaced with fresh PBS. The collected sample was stored at −20 °C until analysis.
Prior to beginning the experiment, a standard curve for the detection of MGF was established using a 1260 Infinity HPLC system (Agilent Technologies, CA) and samples of MGF prepared in a 20 % acetonitrile solution at concentrations of 0.5, 1, 5, 10, 20, and 50 μg/mL in triplicate with a PBS blank sample run between each standard curve sample. Samples were run through a Luna 5 μm C18 (2) 100A, 250 × 4.6 mm column (Phenomenex, Inc, CA) equilibrated to room temperature using a process adapted from previously published protocols (Tucker et al. 2012). Solvents were made from HPLC grade water, acetonitrile, and trifluoroacetic acid (TFA) (VWR, PA). Solvent A was 100 % water and solvent B was 100 % acetonitrile. TFA was added to both solvents at a concentration of 0.1 % (v/v). The mobile phase was eluted from the column at 1 mL/min beginning with 95 % solvent A and 5 % solvent B and increasing to 80 % solvent A and 20 % solvent B from 0 to 10 min, which was then maintained from 10 to 15 min. From 15 to 30 min, the mobile phase was transitioned to 10 % solvent A and 90 % solvent B. From 30 to 30.5 min, the solvent mixture was returned to the starting mixture and the column was flushed with this solution and re-equilibrated from 30.5 to 35 min. Samples were analyzed by a UV detector at 205 nm. The MGF peak eluted at approximately 11.7 min and the area under the curve of the peak at this time was calculated to establish the linear range of the standard curve.
To ensure that small chain PEG molecules that were potentially eluted over the 14 days did not interfere with MGF detection, additional standards with 10 μg/mL MGF were doped with 0.1, 1, and 10 % (v/v) un-crosslinked PEGDMA. No interference with the MGF peak was noted at these concentrations of PEG-DMA inclusion.
Experimental samples were run with the HPLC protocol described above including a PBS blank between each sample and interpolated on the standard curve to back-calculate the mass of MGF in solution at each time point. The elution experiment was repeated with n=5 aliquots of 450,000 MGF-containing microrods and the results averaged. Control aliquots (n=2) of empty microrods were also incubated over the 14 days, sampled at each time point for comparison, and subtracted from the readings on MGF for normalization.
2.6 Cell culture for human mesenchymal stem cells and neonatal rat ventricular myocytes (NRVM)
Institutional approval was received to obtain and use mesenchymal stem cells (hMSCs) isolated from human bone marrow aspirates supplied by Texas A&M Health Science Center College of Medicine Temple, TX. Microarray analyses indicate that gene expression was consistent for hMSCs from different donors, isolated and expanded as described previously. Experiments were performed on passage three or lower from hMSCs obtained from 3 separate donors. hMSCs were cultured in complete culture media (CCM) consisting of MEM-α supplemented with 16.5 % fetal bovine serum (FBS), 2 mM L-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin, and incubated at 37 °C as used by us (Doroudian et al. 2013).
Primary heart cultures were obtained from neonatal rats according to Institutional Animal Care and Use Committee and National Institutes of Health guidelines for the care and use of laboratory animals. Hearts were removed and cells were isolated from 1- to 2-day-old neonatal Sprague–Dawley rats with collagenase (Worthington), as previously described by our group (Boateng et al. 2003). The cells were re-suspended, filtered through a metal sieve to remove large material, and plated in PC-1 medium (Biowhittaker/Cam- brex) on fibronectin coated plates (25 μg/ml). Cells were left undisturbed for 24 h in a 5 % CO2 incubator. Unattached cells were removed by aspiration, and PC-1 media was replenished. Cells were allowed to establish beating over at least 1 day prior to experimental use.
2.7 MGF bioactivity assessed by hMSC migration
Migration was used to test the bioactivity of MGF microrods (MGF-rods). Migration of hMSCs was determined with a 10-μm-thick polycarbonate porous membrane insert with 8-μm pores (FluoroBlok, BD Biosciences). hMSCs were cultured to approximately 80 % confluence in CCM. Plates were washed with PBS and cells starved with non-serum, low glucose (0.1 mM) DMEM media (LGM) for 4 h. Cells were then detached, counted and 50,000 cells in LGM were added to the upper compartment of the porous insert in a Boyden chamber. The bottom of each well contained LGM with or without growth factors. Groups were no MGF in media (control), empty rods (negative control), 30 ng MGF in media (positive control), and 100 K MGF-rods which were added below the chamber. Cells migrated and attached for 22 h at 37 °C, 5 % CO2. Then the inserts were removed, washed with PBS and stained with Calcein-AM. Plates were scanned using a FlexStation II (Molecular Devices) bench top scanning fluorometer afterward. To confirm hMSC migration, images were collected of Calcein-AM labeled hMSCs, which migrated through the membrane. Fluorescence was observed on a Nikon Microphot-FXA/SA epifluorescent microscope.
2.8 Microrod and MGF effects on subcellular structure
In order to analyze subcellular features of the actin cytoskeleton and focal adhesion, hMSCs were cultured with the microrods still adhered to the silicon wafers with or without MGF for 48 h. Cells were fixed with 4 % paraformaldehyde in phosphate buffered saline (PBS) for 10 min at room temperature, rinsed three times with PBS and permeabilized by 0.1 % Triton X-100 in PBS for 10 min, and washed 3 times with PBS. Cells were pre-incubated in blocking solution (PBS, 1 % bovine serum albumin (BSA)) for 15 min and then incubated with rhodamine conjugated phalloidin (Molecular Probes) at a dilution of 1:400 to stain actin, or paxillin antibody (Abcam) at a dilution of 1:250 for 1.5 h followed by another incubation with secondary antibody Alexa Fluor 488 (Invitrogen) at a dilution of 1:1,000 for 45 min to stain the focal adhesions of the cells. 4′, 6-Diamidino-2-phenylindole (DAPI) (Sigma) was used for nuclear staining, which artificially stained the microrods as well. Confocal images of actin and focal adhesions were obtained with Zeiss LSM 510 META and LSM 710 microscopes.
2.9 Microrod and MGF effects on proliferation
To assess cell proliferation of hMSCs after 48 h of culture on microrods with or without MGF, cells underwent a 1-h incorporation of 5-ethynyl-2′-deoxyuridine (EdU, 10 mM, Invitrogen Corp.). Once incorporation was complete, the wafers were cut and mounted onto glass slides with DAPI for nuclear staining. The microrods were artificially stained with both DAPI (blue) and Alexa Fluor 594 (red) used in the EdU kit.
2.10 MGF bioactivity to reduce apoptosis of NRVM induced by hypoxia
In order to induce physiological apoptosis, neonatal rat ventricular myocytes (NRVM) were cultured for 4 days after isolation and then placed in a humidified hypoxic chamber (Sanyo, Inc) with 5 % CO2, 1 % O2 and the remainder balanced with N2 for 8 h at 37 °C with MGF in media, E-rods, or MGF-rods. Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end-labeling (TUNEL) assay was used to assess cell apoptosis. TUNEL reaction preferentially labels DNA strand breaks generated during apoptosis. The negative control was normoxia, and the positive control was induced by a DNase I recombinant (Roche Applied Science).
Changes in gene expression of Bcl-2 were assessed as an index of apoptosis protection from hypoxia. Total RNA was isolated from NRVMs from the experimental conditions of control, MGF in media, E-rods, and MGF-rods. The RNA Mini Kit (QIAGEN) was used to isolate RNA, which was quantified using the Qubit Quantitation Platform (Invitrogen). RNA was reverse-transcribed for 50 min at 37 °C and 15 min at 65 °C (inactivation) using M-MLV Reverse Transcriptase.
For quantitative polymerase chain reaction (qPCR) experiments, total RNA was isolated and reverse transcribed, from independently prepared control, MGF in media, E-rods and MGF-rods. Using SYBR Green PCR Master Mix and a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). Amplification was achieved by the following protocol: 1 cycle of 50 °C for 2 min; 1 cycle of 95 °C for 10 min; 0 cycles of 95 °C for 15 s and 60 °C for 1 min. To ensure specificity of PCR, melt-curve analyses were performed at the end of all PCRs. The relative amount of target cDNA was determined from the appropriate standard curve and normalized to the amount of Histone H2B cDNA present in each sample. Each sample was analyzed in triplicate, and results were expressed relative to the control condition.
2.11 Statistical analysis
A statistically significant difference among groups was detected by analysis of variance (ANOVA). Sequential Holm t-tests were then performed to identify differences between specific pairs of conditions.
3 Results
Slow degradation of microrods
Continuous shaking in saline at 37 °C did not degrade microrods over 2 months. At day 1 and at 2 months, the width of microrods was approximately 15 μm. There was no significant difference in the mean width value of the microrods, implying no degradation under these conditions (data not shown).
MGF elution time course
Delivery of MGF was sustained over 2 weeks and no more MGF elution occurred in the third week [Fig. 3]. The majority of this release (~80 %) occurred over the first 7 days before MGF release began to taper. Background detection of signal from empty microrods was small, reaching less than 20 % of the total signal from the MGF-loaded rods over 14 days. The theoretical total MGF content in each aliquot of 450,000 microrods was approximately 4,600 ng with an average total of approximately 570 ng released over 14 days (theoretical payload delivery efficiency ~12.4 %). However, there are many factors that must be considered in order to calculate the delivery efficiency that require adjustment of the total MGF loaded in the microrods and is discussed further below, and additional data on MGF elution is given in Online Resource 1.
Fig. 3.
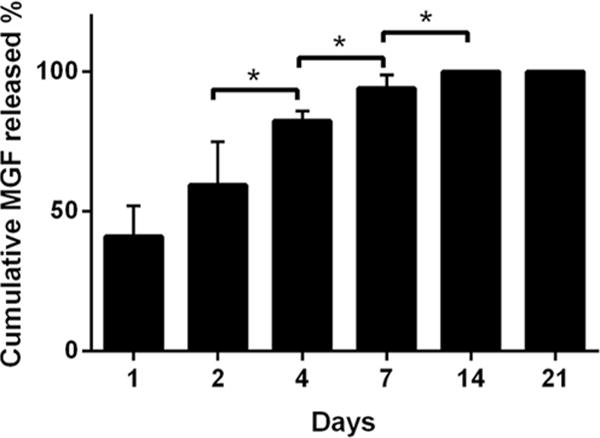
Time course of elution of MGF from the microrods detected by HPLC method. The cumulative MGF was measured at 0, 1, 2, 4, 7, 14 and 21 days. Each measurement was normalized to the 14 day MGF release. Mean ± SE, n=5, * p<0.05
Subcellular structure and proliferation
Microrods remodeled morphology and adhesion of the hMSCs after 48 h of culture [Fig. 4]. The hMSCs interacted with the microrods as determined by immunocytochemistry. In fact, the actin cytoskeleton of hMSCs elongated on the microrods, and focal adhesions (assessed by paxillin localization) distributed along the microrods. On the other hand, MGF in the media on flat and MGF-rod surfaces had no effect on the cell morphology [Fig. 4]. Microrods with or without MGF significantly blunted proliferation of hMSCs after 2 days of culture [Fig. 5] (p<0.05 between microrods and flat surface), however, MGF on flat surfaces had no effects on the proliferation of hMSCs (Flat vs. MGF in media, NS) [Fig. 5]. This result was expected since our lab previously showed no effect of MGF on hMSCs proliferation (Collins et al. 2010). Thus, proliferation is regulated by the microrods independently of MGF.
Fig. 4.
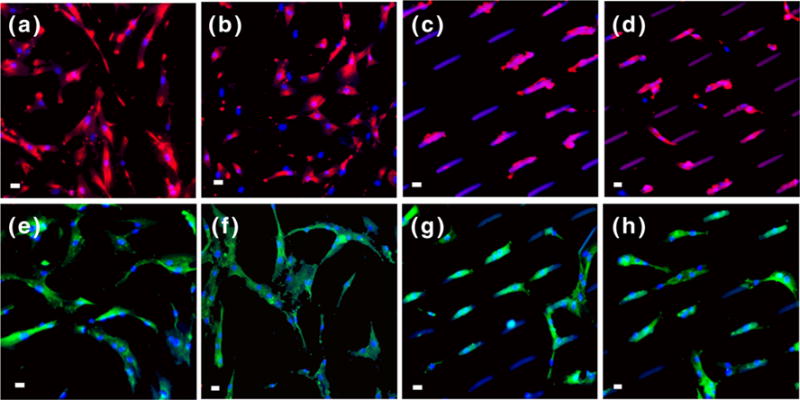
Microrods remodel hMSC morphology, the actin cytoskeleton and focal adhesions, but MGF does not. Stem cells grown on (a, e) flat, (b, f) MGF in media flat, (c, g) empty microrods, and (d, h) MGF microrods. The actin cytoskeleton (a, b, c, d) and the focal adhesions (e, f, g, h) are organized differently on cells near the microrods than on flat surfaces, but the cell morphology is not affected by the presence of MGF. Actin (red), paxillin (green), nuclei (blue) and microrods (blue/purple), Scale bar, 20 μm
Fig. 5.
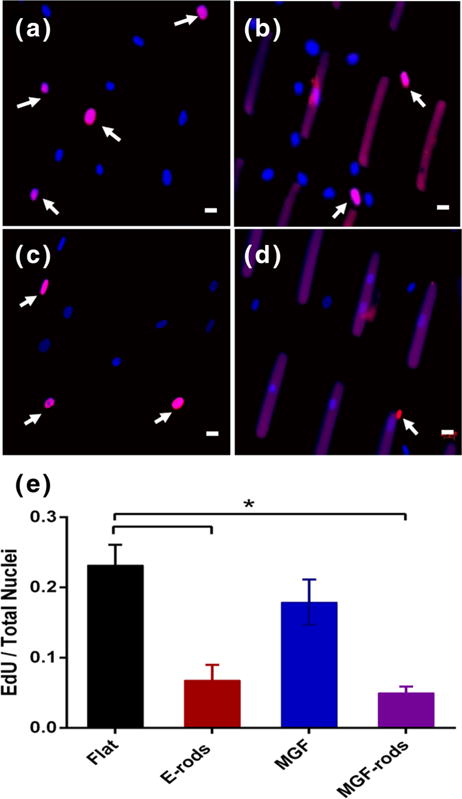
Microrods blunt proliferation of hMSCs. Newly dividing cells (pink) vs. non-dividing cells (blue) on (a) flat surfaces, (b) with empty microrods (E-rods), (c) on flat surface with MGF in the media, or (d) with MGF eluting microrods (MGF-rods), as seen by fluorescence microscopy. (e) EdU/total nuclei per condition show that microrods inhibit new synthesis of DNAwith or without MGF. Dividing nuclei (arrows) stained with EdU (pink), non-dividing with DAPI (blue), and microrods artificially stained with DAPI and EdU (Purple). Mean ± SE, n=4, * p<0.05. Scale bar, 20 μm
Migration
MGF in the media and MGF eluted (30–70 ng) overnight by the microrods was bioactive and induced stem cell migration compared to the microrods without MGF (1.72 ±0.23, p<0.05), and no migration of hMSCs was seen with E-rods [Fig. 6].
Fig. 6.
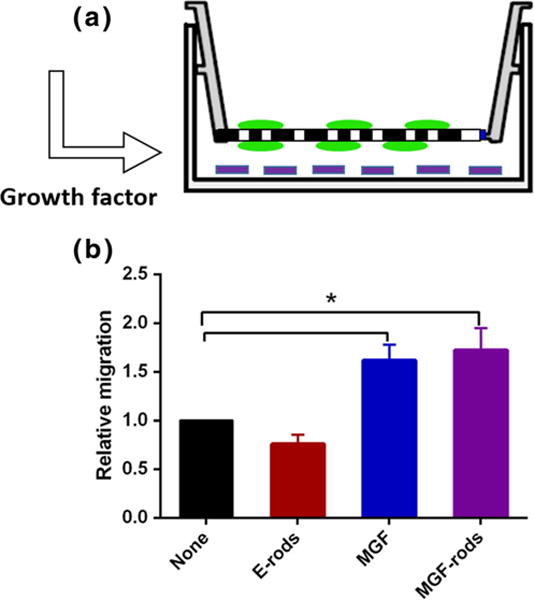
Eluted MGF is bioactive and regulates hMSCs migration. a Schematic diagram of Boyden chamber with cells (green) on top and the microrods (purple) on the bottom eluting MGF below. b Migration overnight is increased both by MGF in media and MGF eluted from microrods. Mean ± SE, n=3, # p<0.05
Hypoxic induction of apoptosis
After induction of hypoxic stress to NRVMs, the extent of cell death was measured by TUNEL positive nuclei (DNA fragmentation). Treatment of hypoxic heart cells with MGF showed an increase in viable cells (p<0.05 for experimental groups: MGF in media, MGF-rods vs. control groups: E-rods and Hypoxia) [Fig. 7a]. In addition, Bcl-2 gene expression (cell signaling) measured by qPCR also confirmed the anti-apoptotic role of MGF peptide [(Fig. 7b], demonstrating that both MGF and MGF-rods increased relative expression of Bcl-2. Furthermore, the eluted MGF peptide retained bioactivity after the fabrication process into the microrod.
Fig. 7.
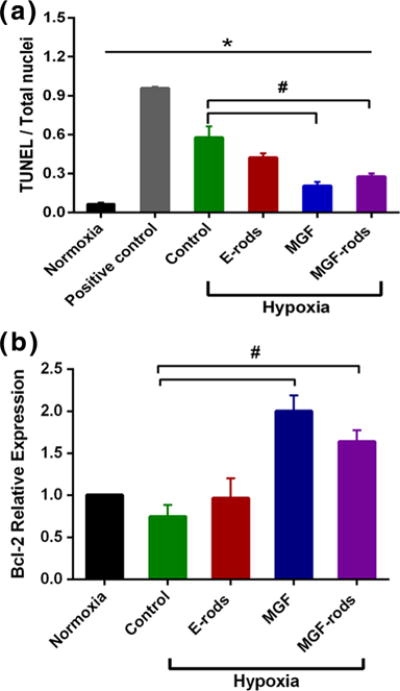
MGF protects NRVM from apoptosis induced by hypoxia. a NRVMs after 8 h of hypoxia (1 % O2) are apoptotic as assessed by TUNEL positive nuclei. Apoptosis is reduced by MGF added to the media (MGF) or eluted form the microrods (MGF-rods) but not by empty microrods (E-rods). Positive control is with DNAse I recombinant; b Increased relative gene expression of Bcl-2 of NRVMs after 8 h of hypoxia treated with MGF or MGF-rods confirms that MGF improves cell survival. Mean ± SE, n=4, * All vs. normoxia, # MGF and MGF-rods vs. control in hypoxia, p<0.05
4 Discussion
The major findings of this study were the successful encapsulation of native MGF peptide within microrods that sustained delivery of MGF up to 2 weeks. The native MGF eluted from the microrods retained bioactivity as assessed by induction of hMSC migration and, moreover, the inhibition of the apoptotic pathway in NRVMs subjected to hypoxia. Microrods alone without MGF regulated the cytoskeleton, adhesion, and proliferation of hMSCs. Therefore, the combination microdevice provides the mechanical cues and MGF bioactivity after fabrication, which may provide for potential therapeutic delivery and cardiac repair and regeneration in vivo.
4.1 PEGDMA microrods in drug elution applications
For effective application in the heart, a hydrogel material was chosen for the microrods that was biocompatible, able to be modulated in terms of stiffness for cell anchorage, compatible with high–throughput photolithographic processing, exhibits harmless long-term degradation byproducts and, finally, capable of drug elution over a biologically relevant time period of several days in the wake of acute cardiac injury. Many studies model the mechanical microenvironment using polyacrylamide substrates with tunable mechanical properties. However, due to cytotoxicity, polyacrylamide substrates are unsuitable for long-term in vitro studies or eventual in vivo applications. PEGDMA, on the other hand, is used extensively for tissue engineering and drug delivery applications and displays excellent biocompatibility (Bryant and Anseth 2003; Diramio et al. 2005). It is photopolymerizable, allowing us to precisely control the microrod geometry. In fact, this shape feature of the microrods is chosen to provide a high aspect ratio microstructure to affect higher mechanical influence on cells interacting with the microrod, and the size of the feature is chosen to mimic the scale of normal cell size found physiologically to provide biologically relevant mechanical cues to cells that interact with the microstructure. In addition, we have previously shown that the stiffness of PEGDMA can be tuned by changing the concentration and ultimately the cross-linking density, which are important factors in degradation rate (Ayala et al. 2010).
4.2 Microrods degradation
Due to the nature of the polymeric cross-linking in PEGDMA hydrogel constructs, microrods fabricated from this material will undergo hydrolysis over time, leaving behind small, inert PEG chains. This process appears to be slow, and previous work has shown minimal degradation over 3 weeks (Tucker et al. 2012). Although our experience with these types of constructs has further shown relative morphological stability of microrods in non-physiological conditions for months, a thorough analysis of the long-term degradation of microrods under physiologic conditions in vivo would be valuable. Degradation rate is sensitive to the size scale, polymeric composition, and particular polymerization process of the construct and understanding how each of these components affects PEGDMA microstructure stability will enable precision temporal tuning of incorporated drug release and microstructure deterioration for various applications. Conceivably, the incorporation of more rapidly hydrolysable cross-linking agents could also accelerate the drug release or microstructure degradation process as may be required in certain physiologic settings.
4.3 Physiological effect of the microrods alone
Microrods in a 3D system inhibited fibroblast proliferation and down-regulated expression of key extracellular matrix proteins involved in scar tissue formation (Norman et al. 2008; Ayala et al. 2010). hMSCs attached to 30 kPa microrods displayed elongated morphology as compared to cells not exposed to discrete micromechanical cues. The local stresses induced by microrods were recognized by the cells, which consequently altered the cytoskeletal architecture adjacent to the microrod (Norman et al. 2005; Collins et al. 2010; Ayala et al. 2010). This remodeling of cells by the external topography of microrods is due to the interactions that link the transmembrane integrin receptors to the actin cytoskeleton via adaptor proteins such as vinculin, paxillin, and α-actinin (Samarel 2005). This similar behavior by hMSCs was also observed in this study. In addition, a previously developed in vitro model system has shown that the inclusion of SU-8 microrods in 3D can alter long-term growth responses of neonatal ventricular myocytes (Curtis and Russell 2011; Curtis et al. 2013) the mechanism of which depends on aspects of RhoA/ROCK and PKC signaling. The use of geometric boundaries that force neonatal rat ventricular myocytes to spread into an elongated shape, similar to that of cardiomyocytes in vivo, leads to more myofibril alignment and clear axes of contraction (Motlagh et al. 2003; Bray et al. 2008). As expected, however, (Collins et al. 2010) MGF did not affect hMSC proliferation or the subcellular architecture.
4.4 Microrods retain bioactivity of MGF
Encapsulation of native MGF in the microrods is one way to protect it from rapid degradation in vivo. Early work on MGF in muscle tissue described this form of the IGF-1 gene as being unglycosylated and probably having a shorter half-life time in the serum or tissue due to proteolytic cleavage (Goldspink and Yang 2001). Therefore, native MGF was protected from degradation by chemically modifying the E-domain to stabilize the peptide by pegylatation and replacement of an L-arginine with a D-arginine to withstand the interstitial cleavage enzymes (Dluzniewska et al. 2005). Unfortunately, there are concerns regarding side effects with systemic delivery of the stable form due to stimulation of the IGF pathway systemically: an issue especially in women with certain types of breast cancer (Nahta et al. 2005). The incorporation and protection of MGF within microrod hydrogel constructs was achieved in this study, which allowed us to further study the potential for local delivery and bioactivity of MGF.
4.5 Sustained delivery of MGF for 2 weeks
Using E-peptide specific probes, the mRNA of MGF was found to be markedly increased during the acute stress (24 h) but then declined in skeletal muscle or cardiac cells following injury (Hill and Goldspink 2003; Mavrommatis et al. 2013; Stavropoulou et al. 2009). Two days of cyclic stretch at high strain (20 %) at 1 Hz caused increased MGF gene expression in NRVMs, again suggesting MGF is related to stress or injury and showing that myocytes produce MGF (Collins et al. 2010). Thus, the native MGF delivery window of 1 day to at least 2 weeks may be effective for cardiac repair in vivo.
We were able to attain this objective with a delivery profile of MGF from the PEGDMA microrods from 1 day up to at least 14 days. However, it is important to note that the actual delivery efficiency of the MGF containing microrod system is likely considerably higher than the calculated theoretical efficiency. The theoretical MGF content potentially overestimates the available drug to be delivered in the microrods in several ways. Firstly, it requires an assumption that the microrods are solid objects with a material volume equal to the spatial volume of the structure itself. However, the highly porous nature of the microstructures can significantly decrease the volume of material available to hold and deliver the peptide. In addition, a considerable portion of the MGF may be damaged or eluted prior to the start of the measured elution during UV exposure, cold storage, or water and alcohol rinses after fabrication of the microstructures. MGF might be further destroyed throughout the course of the experiment by general hydrolytic processes, obscuring our ability to detect the cumulative eluted peptide at later time points by HPLC. Lastly, inhomogeneities in the microrod population may lead to different loading capacities of each microrod, so the presumed total MGF content is taken as an average potential drug capacity. These factors were mitigated to an extent by isolating only microrods of the proper size and shape and minimizing time spent in water or ethanol solutions to reduce the amount of drug released prior to the start of the experiment. In addition, elution may vary considerably from the experimental conditions used in vitro with saline water at 37 °C. In the beating heart, body temperature is also 37 °C, but there is considerable agitation of the complex interstitial fluids, which may alter the elution rate.
Further experiments to understand the nature and stability with which MGF is incorporated into the matrix would be valuable as a tool to understand extended clinical applications of this technology.
4.6 Migration of hMSCs as a bioactivity assay
Elution of MGF from the microrods was able to establish a growth factor gradient, which induced migration of hMSCs. Thus, the slow release of MGF encapsulated in the microdevice has the potential for use in clinical applications to recruit stem cells if delivered to the site of injury. There is still controversy whether the migration mechanism of stem cells via these growth factors is dependent or independent of IGF-1 receptor (Yang and Goldspink 2002; Mills et al. 2007). However, previous work has suggested that the migration effect of MGF peptide on MSCs depends on IGF-1 receptor via Erk1/2 signal pathway (Cui et al. 2014; Wu et al. 2013). MGF promotes rat tenocyte migration by lessening cell stiffness and increasing pseudopodia formation via the FAK-ERK1/2 signaling pathway (Zhang et al. 2014), while the peptide intervention caused MSCs to stiffen (Wu et al. 2013). Our results suggest that the injectable MGF-microrods may foster stem cell homing in vivo, by generating a chemotactic gradient that is translated into a mechanotactic response through the IGF-1 receptor mediated pathways.
4.7 Prevention of NRVM apoptosis as a bioactivity assay
MGF is an anti-apoptotic peptide able to protect adult cardiomyocytes after infarction (Mavrommatis et al. 2013). The E-domain of MGF appears to have beneficiary effects in injured tissue either distinctly or synergistically to the mature peptide of IGF-1 (Carpenter et al. 2008; Dluzniewska et al. 2005). The bioactive MGF eluted from the microrods prevented apoptosis of hypoxic cardiac muscle cells in addition to the increased migration of hMSCs. Here, we altered the oxygenation conditions in vitro to establish changes in gene expression and cell survival of NRVMs under hypoxic conditions that mimic the ischemic heart. Results of the TUNEL assay and Bcl-2 expression confirm MGF protection for myocytes from apoptosis that should, confer longevity to progenitor and effector cells in tissues (Hockenberry et al. 1990; Korsmeyer 1992; Grünenfelder et al. 2001). By improving cell survival, the likelihood of regeneration will be improved and cell death of the pre-existing myocardium attenuated, which could translate into an improvement in function. Thus, the effective delivery of MGF from the microrods may permit myocytes to thrive in vivo even under hypoxic conditions.
5 Conclusion
Bioengineering approaches were able to achieve a therapeutically relevant 2-week time course of MGF delivery and to protect native MGF peptide in a bioactive state. Stem cell migration was preserved and myocyte apoptosis under hypoxic stress was reduced. The ability to incorporate and protect a peptide therapeutic in a monolithic hydrogel device to extend its half-life in vivo has great potential for countless translational applications. Thus, outcomes in a therapeutic setting of injection of this MGF-loaded microdevice into injured regions of the heart might be beneficial in reducing myocyte loss in ischemia and boosting repair by the chemo-attraction of stem cells to the damaged area.
By adapting a stable and well-understood polymer system to serve as both a mechanical stimulus as well as provide highly-localized, long-term delivery of bioactive peptides, we may be able to expand the therapeutic profile of many existing biomolecules.
Supplementary Material
Acknowledgments
The authors acknowledge funding support from the NIH (HL 062426, HL 090523) and T32 HL 07692. The work was made possible by a grant from the California Institute for Regenerative Medicine (Grant Number TG2-01153). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California.
Abbreviations
- hMSC
Human mesenchymal stem cells
- NRVM
Neonatal rat ventricular myocytes
- PEGDMA
poly(ethylene glycol) dimethacrylate
- EdU
5-ethynyl-2′-deoxyuridine
- DAPI
4′,6-Diamidino-2-phenylindole
- TUNEL
Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end-labeling
- qPCR
Quantitative polymerase chain reaction
- IGF-1
Insulin-like growth factor 1
- MGF
Mechano growth factor
- 3D
Three dimensions
- FBS
fetal bovine serum
- TFA
Trifluoroacetic acid
- CCM
Cell culture medium
- BSA
bovine serum albumin
- ECM
Extracellular matrix
- PBS
Phosphate buffered saline
- SEM
Scanning electron microscopy
- LGM
Low glucose media
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10544-014-9875-z) contains supplementary material, which is available to authorized users.
Contributor Information
Golnar Doroudian, Department of Bioengineering, University of Illinois at Chicago, Chicago, IL, USA.
James Pinney, Department of Physiology and Division of Bioengineering, University of California at San Francisco, San Francisco, CA, USA.
Perla Ayala, Department of Physiology and Division of Bioengineering, University of California at San Francisco, San Francisco, CA, USA.
Tamara Los, Department of Physiology and Biophysics, University of Illinois at Chicago, 835 S. Wolcott, Chicago, IL 60612, USA.
Tejal A. Desai, Department of Physiology and Division of Bioengineering, University of California at San Francisco, San Francisco, CA, USA
Brenda Russell, Email: russell@uic.edu, Department of Physiology and Biophysics, University of Illinois at Chicago, 835 S. Wolcott, Chicago, IL 60612, USA.
References
- Ates K, Yang SY, Orrell RW, Sinanan AC, Simons P, Solomon A, Beech S, Goldspink G, Lewis MP. FEBS Lett. 2007;581:2727–2732. doi: 10.1016/j.febslet.2007.05.030. [DOI] [PubMed] [Google Scholar]
- Ayala P, Lopez JI, Desai TA. Tissue Eng Part A. 2010;16(8):2519–2527. doi: 10.1089/ten.tea.2009.0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TI, Discher DE, Sweeney HL. Am J Physiol Heart Circ Physiol. 2006;290:H2196. doi: 10.1152/ajpheart.01017.2005. [DOI] [PubMed] [Google Scholar]
- Biehl JK, Yamanaka S, Desai TA. Dev Dyn. 2009;238(8):1964–1973. doi: 10.1002/dvdy.22030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng SY, Hartman TJ, Ahluwalia N, Vidula H, Desai TA, Russell B. Am J Physiol Cell Physiol. 2003;285:C171–C182. doi: 10.1152/ajpcell.00013.2003. [DOI] [PubMed] [Google Scholar]
- Bray MA, Sheehy SP, Parker KK. Cell Motil Cytoskeleton. 2008;65(8):641–651. doi: 10.1002/cm.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant SJ, Anseth KS. J Biomed Mater Res. 2003;64A(1):70–79. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- Carpenter V, Matthews K, Devlin G, Stuart S, Jensen J, Conaglen J, Jeanplong F, Goldspink P, Yang SY, Goldspink G, Bass J, McMahon C. Heart Lung Circ. 2008;17:33–39. doi: 10.1016/j.hlc.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Collins JM, Ayala P, Desai TA, Russell B. Small. 2010;6(3):355–360. doi: 10.1002/smll.200901757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JM, Goldspink PH, Russell B. J Mol Cell Cardiology. 2010;49(6):1042–1045. doi: 10.1016/j.yjmcc.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Yi Q, Feng J, Yang L, Tang L. J Mol Endocrinol. 2014;52(2):111–120. doi: 10.1530/JME-13-0157. [DOI] [PubMed] [Google Scholar]
- Curtis MW, Russell B. Eur J Physiol. 2011;462(1):105–117. doi: 10.1007/s00424-011-0931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MW, Budyn E, Desai TA, Samarel AM, Russell B. Biomech Model Mechanobiol. 2013;12(1):95–109. doi: 10.1007/s10237-012-0384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diramio JA, Kisaalita WS, Majetich GF, Shimkus JM. Biotechnol Prog. 2005;21(4):1281–1288. doi: 10.1021/bp0495670. [DOI] [PubMed] [Google Scholar]
- Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324(5935):1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dluzniewska J, Sarnowska A, Beresewicz M, Johnsomn I, Srai SK, Ramesh B, Goldspink G, Gorecki DC, Zablocka B. FASEB J. 2005;19:1896–1898. doi: 10.1096/fj.05-3786fje. [DOI] [PubMed] [Google Scholar]
- Donath MU, Zierhut W, Gosteli-Peter MA, Hauri C, Froesch ER. J Zapf, Eur J Endocrinol. 1998;139(1):109–117. doi: 10.1530/eje.0.1390109. [DOI] [PubMed] [Google Scholar]
- Doroudian G, Curtis MW, Gang A, Russell B. Biochem Biophys Res Commun. 2013;430(3):1040–1046. doi: 10.1016/j.bbrc.2012.11.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. J Clin Invest. 2006;116(7):1865–1877. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldspink G, Yang SY. Int J Sport Nutr Exerc Metab. 2001;11(Suppl):S21–7. doi: 10.1123/ijsnem.11.s1.s21. [DOI] [PubMed] [Google Scholar]
- Grünenfelder J, Miniati DN, Murata S, Falk V, Hoyt EG, Kown M, Koransky ML, Robbins RC. Circulation. 2001;104(12 Suppl 1):I202–I206. doi: 10.1161/hc37t1.094833. [DOI] [PubMed] [Google Scholar]
- Hattori K, Heissig B, Tashiro K, Honjo T, Tateno M, Shieh JH, Hackett NR, Quitoriano MS, Crystal RG, Rafii S, Moore MA. Blood. 2001;97(11):3354–3360. doi: 10.1182/blood.v97.11.3354. [DOI] [PubMed] [Google Scholar]
- Hill M, Goldspink G. J Physiol. 2003;549:409–418. doi: 10.1113/jphysiol.2002.035832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenberry D, Núñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- Kandalla PK, Goldspink G, Butler-Browne G, Mouly V. Mech Ageing Dev. 2011;132(4):154–162. doi: 10.1016/j.mad.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ. Immunol Today. 1992;13:285–288. doi: 10.1016/0167-5699(92)90037-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang Y, Wang P, Zhang B, Yan W, Sun J, Pan J. J Biomater Sci Polym Ed. 2013;24(7):849–864. doi: 10.1080/09205063.2012.723957. [DOI] [PubMed] [Google Scholar]
- Mavrommatis E, Shioura KM, Los T, Goldspink PH. Mol Cell Biochem. 2013;381(1–2):69–83. doi: 10.1007/s11010-013-1689-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills P, Lafreniere JF, Benabdallah BF, El Fahime M, El Tremblay JP. Exp Cell Res. 2007;313:527–537. doi: 10.1016/j.yexcr.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Motlagh D, Hartman TJ, Desai TA, Russell B. J Biomed Mater Res. 2003;67(1):148–157. doi: 10.1002/jbm.a.10083. [DOI] [PubMed] [Google Scholar]
- Musaro A, Giacinti C, Borsellino G, Dobrowolny G, Pelosi L, Cairns L, Ottolenghi S, Cossu G, Bernardi G, Battistini L, Molinaro M, Rosenthal N. Proc Natl Acad Sci U S A. 2004;101:1206–1210. doi: 10.1073/pnas.0303792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Cancer Res. 2005;65(23):11118–11128. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- Napier JR, Thomas MF, Sharma M, Hodgkinson SC, Bass JJ. J Endocrinol. 1999;163:63–68. doi: 10.1677/joe.0.1630063. [DOI] [PubMed] [Google Scholar]
- Norman JJ, Desai TA. Tissue Eng. 2005;11(3–4):378–386. doi: 10.1089/ten.2005.11.378. [DOI] [PubMed] [Google Scholar]
- Norman JJ, Collins JM, Sharma S, Russell B, Desai TA. Tissue Eng Part A. 2008;14:379–390. doi: 10.1089/tea.2007.0077. [DOI] [PubMed] [Google Scholar]
- Paul D, Samuel SM, Maulik N. Antioxid Redox Signal. 2009;11(8):1841–1855. doi: 10.1089/ars.2009.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarel AM. Am J Physiol Heart Circ Physiol. 2005;289(6):H2291–H2301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- Scadden DT. Nature. 2006;441(7097):1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- Schlegel W, Raimann A, Halbauer D, Scharmer D, Sagmeister S, Wessner B, Helmreich M, Haeusler G, Egerbacher M. PLoS ONE. 2013;8(10):e76133. doi: 10.1371/journal.pone.0076133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulou A, Halapas A, Sourla A, Philippou A, Papageorgiou E, Papalois A, Koutsilieris M. Mol Med. 2009;15:127–135. doi: 10.2119/molmed.2009.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RM, Parcher BW, Jones EF, Desai TA. AAPS PharmSciTech. 2012;13(2):605–610. doi: 10.1208/s12249-012-9780-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wu K, Lin F, Luo Q, Yang L, Shi Y, Song G, Sung KL. Biochem Biophys Res Commun. 2013;441(1):202–207. doi: 10.1016/j.bbrc.2013.10.031. [DOI] [PubMed] [Google Scholar]
- Yang SY, Goldspink G. FEBS Lett. 2002;522:156–160. doi: 10.1016/s0014-5793(02)02918-6. [DOI] [PubMed] [Google Scholar]
- Zhang B, Luo Q, Mao X, Xu B, Yang L, Ju Y, Song G. Exp Cell Res. 2014 doi: 10.1016/j.yexcr.2014.01.005. pii: S0014-4827(14)00013-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.