

Abstract
We previously demonstrated that heterozygous deletion of Gabra1, the mouse homolog of the human absence epilepsy gene that encodes the GABAA receptor (GABAAR) α1 subunit, causes absence seizures. We showed that cortex partially compensates for this deletion by increasing the cell surface expression of residual α1 subunit and by increasing α3 subunit expression. Absence seizures also involve two thalamic nuclei: the ventrobasal (VB) nucleus, which expresses only the α1 and α4 subtypes of GABAAR α subunits, and the reticular (nRT) nucleus, which expresses only the α3 subunit subtype. Here, we found that, unlike cortex, VB exhibited significantly reduced total and synaptic α1 subunit expression. In addition, heterozygous α1 subunit deletion substantially reduced miniature inhibitory postsynaptic current (mIPSC) peak amplitudes and frequency in VB. However, there was no change in expression of the extrasynaptic α4 or δ subunits in VB and, unlike other models of absence epilepsy, no change in tonic GABAAR currents. Although heterozygous α1 subunit knockout increased α3 subunit expression in medial thalamic nuclei, it did not alter α3 subunit expression in nRT. However, it did enlarge the presynaptic vesicular inhibitory amino acid transporter puncta and lengthen the time constant of mIPSC decay in nRT. We conclude that increased tonic GABAA currents are not necessary for absence seizures. In addition, heterozygous loss of α1 subunit disinhibits VB by substantially reducing phasic GABAergic currents and surprisingly, it also increases nRT inhibition by prolonging phasic currents. The increased inhibition in nRT likely represents a partial compensation that helps reduce absence seizures.
Keywords: biotinylation, brain, confocal microscopy, electrophysiology, epilepsy, GABA, immunohistochemistry, RNA editing, Western blot
Introduction
Epilepsy is a disorder in which the brain exhibits an enduring predisposition to generate seizures. It is a common disease that affects approximately 1% of the population and resists optimal medical therapy in approximately one third of cases. Typical absence seizures are a common seizure type that cause brief interruptions of consciousness and are associated with rhythmic 3 Hz bi-hemispheric spike-and-wave discharges on EEG. They occur in several different genetic generalized epilepsy syndromes (GGE) including childhood absence epilepsy (CAE), juvenile absence epilepsy, and juvenile myoclonic epilepsy. Although anticonvulsant medications are often effective in reducing absence seizures, approximately 50% of CAE patients fail optimal medical management (Glauser et al., 2013) and thus are at risk for injury as well as memory and behavioral deficits (Kernan et al., 2012;Lin et al., 2013).
Studies of rodent models of absence seizures suggested that typical absence seizures start in layer VI of the somatosensory cortex and quickly spread to the remainder of the cortex and two thalamic nuclei, the ventrobasal (VB) and reticular (nRT) nuclei (Meeren et al., 2002;Polack et al., 2007). Although the VB and nRT nuclei do not initiate the epileptic discharges in these models, unilateral VB/nRT lesions abolish absence seizures in rodents, a result that emphasizes the importance of these nuclei in seizure generation (Meeren et al., 2009). The cortex, VB, nRT, and interconnections among these regions comprise the thalamocortical network that is thought to be the core network involved in absence seizures.
Recent studies revealed that different pharmacological and genetic absence epilepsy models possess defects in different components of the thalamocortical network (Cope et al., 2009;Tan et al., 2007;Paz et al., 2011;Errington et al., 2011). Understanding the aberrant neurophysiology in different models of absence epilepsy will lead to the development of new pharmacological and, possibly, neurostimulation therapies for medically intractable absence seizures.
We have studied the pathophysiology of a human absence epilepsy gene, GABRA1, which encodes the α1 subunit of the GABAA receptor (GABAAR). The GABAAR, the major inhibitory ligandgated ion channel in the brain, is a pentamer composed of five subunits which arise from eight gene families; four of the gene families contain multiple isoforms (α1-6, β1-3, γ1-3, δ, ε, θ, π, p1-3). GABAARs composed of different subunit isoforms are expressed in different brain regions at different times in development and exhibit different physiological properties. Heterozygous loss-of-function mutations in the α1 subunit have been associated with three GGE syndromes that confer absence seizures (Cossette et al., 2002;Lachance-Touchette et al., 2011;Maljevic et al., 2006), and heterozygous Gabra1 knockout mice (Het‒KO) exhibit absence seizures (Arain et al., 2012). Previously, we found that neocortical neurons reduce endocytosis of GABAARs from the cell surface which compensates for the heterozygous α1 subunit deletion by: 1) increasing the surface expression of the residual α1 subunit protein driven by the wild type allele and 2) increasing both the total and surface expression of the α3 subunit (Zhou et al., 2013).
Here, we determined the effects of heterozygous α1 subunit deletion on GABAAR expression and GABAergic physiology in the thalamus with a particular focus on the VB and nRT nuclei. Unlike adult cortex which expresses α1-5, β1-3, γ, and δ GABAAR subunits, GABAAR subunit expression in adult VB and nRT is much more limited. Adult VB expresses only α1, α4, β1-2, γ and δ subunits (Hortnagl et al., 2013). Synaptic receptors in VB neurons predominantly consist of α1βγ receptors and mediate phasic GABAergic currents (Peden et al., 2008). Extrasynaptic receptors in VB neurons consist of α4βδ (Chandra et al., 2006;Porcello et al., 2003) and α1βγ (Peden et al., 2008) receptors, but it is predominantly the α4βδ receptors that mediate tonic GABAA currents (Chandra et al., 2006;Porcello et al., 2003;Cope et al., 2005). Adult nRT neurons express predominantly α3, β1-2, and γ subunits (Hortnagl et al., 2013) and conduct phasic, but not tonic GABAA currents (Cope et al., 2005).
We show that, unlike cortex, neither VB nor nRT compensate for the heterozygous loss of α1 subunit by increasing total or synaptic expression of residual α1 or α3 subunits. Tonic currents in VB are unaltered and mIPSC peak amplitudes are reduced. Het-KO nRT neurons increase the size of GABAergic vesicular inhibitory amino acid transporter (VIAAT) puncta and prolong the decay of phasic synaptic GABAA currents. These findings demonstrate that heterozygous α1 subunit deletion produces a unique alteration of intrathalamic GABAA neurotransmission with disinhibition in VB and increased GABAA inhibition in nRT.
Materials And Methods
Animals
All procedures were performed using protocols approved by the Vanderbilt University Institutional Animal Care and Use Committee. The mice were housed in a facility with a temperature and humidity controlled environment, a twelve hour light/dark schedule, and ad libitum food and water. We previously described the generation of mice with an unconditional deletion of the α1 subunit in a congenic C57BL/6J background (Arain et al., 2012). In addition, in some electrophysiology experiments, we used wild type and Het-KO mice that also expressed enhanced yellow fluorescent protein (EYFP) in parvalbumin-containing neurons (Jackson Laboratories, B6;SJL-Tg(Pvalb-COP4*H134R/EYFP)15Gfng/J, stock # 012355) in order to visualize the parvalbumin neurons in nRT in live brain slices. We mated Het-KO and wild type mice and used female pups at postnatal ages 33-37 because our previous EEG studies demonstrated frequent absence seizures in female Het-KO mice at this age (Arain et al., 2012). There is no difference in survival between wild type and Het-KO mice (Arain et al., 2012). On the day of the experiment, the mice were anesthetized with isoflurane and sacrificed. Brains were rapidly dissected and placed for sectioning in cutting solution kept at 0°C.
Antibodies
We used the primary antibodies listed in Table 1 and the secondary antibodies listed in Table 2. Previously, we verified the specificity of the anti α1 subunit and anti α3 subunit antibodies in immunohistochemistry studies using complete α1 subunit and α3 subunit deletion mice (Zhou et al., 2013). The specificity of the anti β2/3 and anti VIAAT antibodies in immunohistochemistry studies was demonstrated in other publications (Gutierrez et al., 1994;Zander et al., 2010).
Table 1. Primary antibodies.
| Target Protein | Species | Source. Clone/Catalog # | Application(s) | Dilution(s) |
|---|---|---|---|---|
| ATPase α subtmit (ATPase) | Mouse | The Developmental Studies Hvbridoma Databank. a6F | WB | 1:100 |
| GABAARαl | Mouse | UC Davis/NIH NeuroMab Facility. N95/35 | WB | 1:250 |
| GABAAR αl | Rabbit | Millipore. 06868 | IHC | 1:250 |
| GABAAR α3 | Rabbit | Alomone. AGA-003 | WB. IHC | 1:500, 1:500 |
| GABAAR α4 | Rabbit | Novus Biologicals, NB300-194 | WB | 1:500 |
| GABAAR β2/β3 | Mouse | Millipore. 62-3G1 | IHC | 1:100 |
| GABAAR δ | Rabbit | R&D Systems, PPS090 | WB | 1:300 |
| Glyceraldehyde-3 -phosphate dehydrogenase (GAPDH) | Rabbit | Abeam. AB9485 | WB | 1:2000 |
| VIAAT | Guinea pig | Synaptic Systems, 131004 | IHC | 1:250 |
WB = Western blot, EHC = Imrmmohistochemtstry
Table 2. Secondary antibodies.
| Target Protein, conjugation | Species | Source, Clone/catalog # | Applicatiou(s) | Dilution(s) |
|---|---|---|---|---|
| Guinea pig IgG, Alexa 488 | Donkey | Jackson Immtmoresearcli, 706-545-148 | IHC | 1:1000 |
| Mouse IgG, 800 | Goat | Ticor, 926-32210 | WB | 1:10,000 |
| Mouse IgG, Alexa 647 | Donkey | Jackson Immtmoresearcli, 715-605-150 | IHC | 1:500 |
| Rabbit IgG, 680 | Goat | Licor, 926-32221 | WB | 1:10.000 |
| Rabbit IgG, Cy3 | Donkey | Jackson Immtmoresearcli, 711-165-152 | IHC | 1:1000 |
WB = Western blot, IHC = Iimmmohistochemistry
Brain slice biotinylation assay and Western blots
We used a vibratome (Leica VT1200S) to make three to four coronal brain slices (300 μm) encompassing the thalamus. We then biotinylated the cell surface proteins using the procedures and solutions described previously (Zhou et al., 2013). After biotinylation, we dissected the thalami and, in some experiments, microdissected thalamic regions containing either the VB/nRT nuclei or the medial thalamic nuclei. The region we designated as “VB/nRT” was microdissected from the coronal sections as is shown in Figure 2A. We cut along the internal capsule from the hippocampus (dorsal point) to the hypothalamus (ventral point). We then located the midpoint, “M,” between the thalamic midline and the internal capsule and made two diagonal cuts from the dorsal point and the ventral point to “M” to isolate the VB/nRT region. The region we designated the “medial thalamic nuclei” was obtained by making vertical cuts from the hippocampus, through “M” to the hypothalamus bilaterally. We made protein lysates from the dissected brain tissue and purified the cell-surface and total protein as previously described (Zhou et al., 2013).
Figure 2. Heterozygous loss of α1 subunit reduces total and cell surface α1 subunit in VB/nRT and medial thalamic nuclei, and increases α3 subunit expression in the medial nuclei, but not VB/nRT.
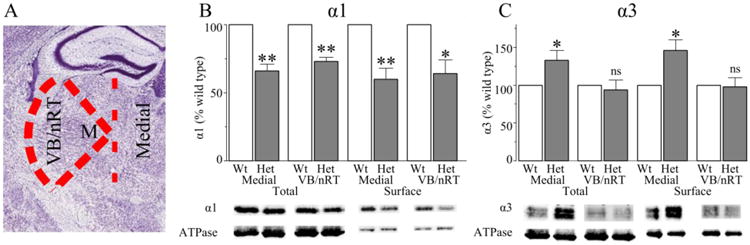
Panel A shows a coronal brain slice similar to the slices used in our study and illustrates the lines of dissection (dashed red lines = lines of dissection, image credit: Allen Mouse Brain Atlas). We first cut along the white matter in the internal capsule from the dorsal point (hippocampus) to the ventral point (hypothalamus). We then made diagonal cuts from the dorsal and ventral points to “M,” the midpoint between the white matter and thalamic midline to isolate “VB/nRT” and “medial thalamus.” Heterozygous loss of α1 subunit reduced total and surface α1 subunit expression in both the medial nuclei (N = 5, P < 0.008) and VB/nRT (N = 5, P < 0.020). In contrast, there was increased total and surface α3 subunit expression in the medial nuclei (N = 6, P < 0.020), but not VB/nRT (N = 6, P > 0.410) of Het-KO thalami. Wt = wild type, Het = Het-KO, * = P < 0.05, ns = not significant
The total and surface protein was analyzed on 10% SDS-PAGE gels followed by electrotransfer to nitrocellulose membranes. To ensure linearity of detection, 10 and 20 μg of total protein and 10, and 20 μl of surface protein were applied to the gel, and we verified that the signal from each protein increased in proportion to the amount loaded on the gel. The nitrocellulose membranes were blocked for one hour with 5% nonfat dry milk in Tris buffered saline containing 0.1% Tween pH 7.4. The membranes were then incubated with primary antibody at 4°C overnight and then with secondary antibody at room temperature for one hour. We imaged the blots using an infrared fluorescent imaging system (Licor).
Quantification of RNA editing of the α3 subunit
Because VB does not express the α3 subunit, we could quantify α3 RNA editing in VB/nRT to determine the fraction of edited α3 subunit RNA in nRT. We dissected VB/nRT regions from brain slices as described above and quantified Gabra3 RNA editing using a high-throughput multiplexed transcript analysis as described previously (Hood et al., 2014).
Immunohistochemistry and Confocal Microscopy
We used the immunohistochemistry protocol described by Schneider-Gasser et al. that allows for light fixation of cytoplasmic proteins but avoids over-fixation that prevents detection of clustered proteins in GABAergic synapses (Schneider Gasser et al., 2007). Briefly, we cut 2 mm coronal block slices (Zivic Instruments) in freshly-dissected brain tissue that encompassed the thalamus. We fixed the block slices in 4% paraformaldehyde dissolved in 100 mM sodium phosphate buffer for thirty minutes at 0°C and then cryoprotected them in 30% sucrose in phosphate buffered saline (PBS) at 4°C overnight. We generated 15 μm coronal sections using a cryostat (Leica) onto Colorfrost Plus glass slides (Thermo Scientific).
Nonspecific antibody binding was blocked with 10% donkey serum and 2% Triton X-100 in PBS for one hour at room temperature. The slides were then incubated overnight at 4°C with anti VIAAT, anti β2/β3 subunit and either anti α1 subunit or anti α3 subunit antibodies (Table 1) dissolved in blocking buffer overnight at 4°C. We then washed the slides and incubated them with the fluorescently conjugated secondary antibodies (Table 2) for one hour at room temperature. The slides were washed before covering and mounting with Vectashield mounting media (Vector Laboratories) that also contained 4′,6-diamidino-2-phenylindole (DAPI) that was used to identify cell nuclei.
We imaged the slides using an Olympus FV-1000 confocal microscope using a 100X, 1.40 NA SPlan-UApo oil immersion lens. The confocal aperture and zoom were set to image a slice of 1 μm thickness and with a resolution of 82 nm/pixel. We adjusted the laser intensity and gain to utilize the full dynamic range of the photomultipliers and the same scan settings were used for all the images acquired within an experiment. Images were made 1 μm below the surface of the tissue in the ventroposterior lateral (VPL) region of the VB nucleus, the genu of the caudal nRT midway between the dorsal and ventral points, and in the internal capsule adjacent to the nRT.
The confocal images were analyzed using ImageJ software (National Institutes of Health) using an adaptation of procedures described previously (Peden et al., 2008;Tyagarajan et al., 2011;Muller et al., 2004). First, the background level was defined as the mean pixel fluorescence intensity of the internal capsule. Next we measured the background-subtracted fluorescent intensity of the total image. Then, we measured the fluorescence intensity of diffuse staining in multiple regions of the image away from any VIAAT or GABAAR subunit puncta. Fourth, we automatically identified VIAAT and GABAAR subunit puncta by setting the image threshold to three times the value of the mean diffuse staining intensity and using ImageJ's particle counting algorithm to identify all puncta with an area between 0.1 and 2.0 μm2. The puncta found using these parameters had excellent correspondence with those identified by visual inspection of the images.
Using this procedure, we quantified the number, area, and background-subtracted fluorescence intensity of VIAAT, α1 subunit, α3 subunit, and β2/β3 subunit puncta. While the brief-fixation method employed here allows excellent visualization of clustered synaptic proteins (Schneider Gasser et al.,2007), it also results in staining variability among different slices. Therefore, the purpose of quantifying the confocal images is to compare the synaptic / extrasynaptic distribution of GABAAR subunit protein,as reflected in the value of the “synaptic clustering ratio,” and not to compare the relative fluorescence intensity among different slices. Previous confocal microscopy studies demonstrated that almost all VIAAT puncta overlap the puncta of the GABAAR clustering protein, gephyrin, but that only a fraction of gephyrin puncta are located near VIAAT puncta (Panzanelli et al., 2011;Studer et al., 2006). Therefore,we used VIAAT puncta to define “synapse-associated” GABAAR subunit protein. We made a binary mask of the VIAAT puncta and overlaid it on the images of the GABAAR subunits. We then used the particle counting algorithm to identify- and measure the fluorescence of- the synapse-associated GABAAR subunit protein. The “synaptic clustering ratio” in each brain slice was calculated as the average background-subtracted intensity of synapse-associated GABAAR subunit divided by the background-subtracted intensity of the entire image.
Electrophysiology
Brain slices for electrophysiology were made and treated as described previously (Zhou et al., 2013). Whole-cell patch clamp recordings were made at room temperature from VB or nRT neurons in the same regions in which the confocal images were obtained, namely, in VPL or in the genu of the caudal nRT. VB and nRT neurons were identified by anatomical location and morphology (Lubke, 1993) with an upright Nikon eclipse FN-1 IR-DIC microscope. In addition, for some experiments, the nRT neurons were also positively identified using the microscope's fluorescent imaging capabilities with slices made from mice that also expressed YFP under control of the parvalbumin promoter (Zhao et al., 2011). Recordings from YFP-expressing wild type and Het-KO nRT were indistinguishable from those that did not express YFP.
The electrophysiology pipettes and the internal recording solutions were identical to those previously described (Zhou et al., 2013). We also used the previously-described (Zhou et al., 2013) external recording solution for recording miniature inhibitory postsynaptic currents (mIPSCs). The external solution for recording tonic currents was identical to the mIPSC solution except that it lacked tetrodotoxin. We recorded and identified mIPSCs as we discussed previously (Zhou et al., 2013). The current decay of each mIPSC was fit to both a single exponential as well as to the sum of two exponentials and the decay constants (τ) were calculated. Although the decay phase of mIPSCs is often fit to one or two exponentials, we achieved our best fits with a single τ value.
To measure tonic GABAA currents, we recorded a stable baseline current for five minutes before adding 60 μM bicuculline (Sigma) into the external solution. We then recorded for another 20 minutes in the presence of bicuculline. To calculate the tonic current, we made all-points histograms of ten seconds of the recording under baseline and bicuculline conditions. The histograms demonstrated that, except for large synaptic events, the currents fit Gaussian distributions. From the histograms, we obtained the mean current in the presence and absence of bicuculline and calculated the tonic current amplitude as the difference between these two values (Glykys & Mody, 2007).
Data analysis and statistics
Results of parametric tests are presented as the mean ± standard error and results of nonparametric tests are presented as the median with an interquartile box plot. In addition, we depict the range of VIAAT puncta sizes, mIPSC amplitudes, and mIPSC decay constants on cumulative probability histograms to demonstrate the effects of α1 subunit deletion on the distributions of these values. We performed statistical analyses using the R 2.12.2 Statistical Package for Windows (R Foundation for Statistical Computing). Visual inspection of data histograms as well as the Shapiro-Wilk test were used to determine if the data were not normally distributed. We used the two-tailed single-sample t-test (vs. wild type at 100%) to determine the statistical significance of the effects of Het-KO on total and surface protein expression on Western blots. The independent samples two tailed t-test was used to compare the average immunohistochemistry puncta densities, and puncta size from each brain slice, as well as the fraction of α3 subunit RNA editing. We also used the independent samples two tailed t-test to compare the average mIPSC frequency, peak amplitude, rise time, and decay τ from each cell. We utilized the Wilcoxon rank sum test to compare the differences in synaptic clustering ratio. P values less than 0.05 were considered statistically significant.
Results
Effects of α1 subunit deletion on total and cell surface GABAAR subunit expression in the thalamus
We first determined the effects of partial loss of α1 subunit on total and surface expression of some of the most prominent GABAAR subunits in the entire thalamus. We biotinylated coronal brain slices and performed Western blots to quantify the relative amounts of total and surface α1, α3, α4, and δ subunit expression (Fig 1). We verified that the biotinylation reaction and neutravidin purification were selective for cell surface proteins by staining for the cytoplasmic protein, GAPDH. We also verified that there was no significant difference between wild type and Het-KO thalami in the expression of the loading control protein, ATPase. Next, we quantified the total and surface expression of the GABAAR subunits in wild type and Het-KO thalami. As expected, heterozygous α1 subunit deletion reduced the total expression of the α1 subunit (64 ± 3% vs wild type, P < 0.001, Fig 1A). However, in contrast to its effect in the cortex, Het-KO thalami did not partially compensate for the reduction of total thalamic α1 subunit expression by causing a significant increase in its relative surface expression (72 ± 4% vs wild type, P < 0.001, Fig 1A). However, similar to cortex, there was increased total and surface expression of the α3 subunit (total: 162 ± 7% vs wild type, surface: 187 ± 8% vs wild type, P < 0.001, Fig 1B) in Het-KO thalami. There was no significant change in the total or surface expression of the α4 or δ subunits (P > 0.056, Fig 1C-D).
Figure 1. Effects of heterozygous α1 subunit deletion on total and surface GABAAR subunit expression in the entire thalamus.
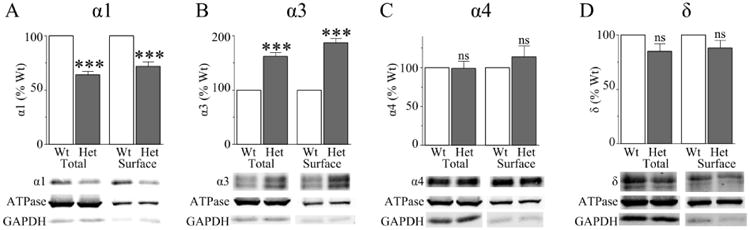
A) Heterozygous α1 subunit deletion reduced total and surface α1 subunit expression (N = 9, P < 0.001) and B) increased total and surface α3 subunit expression (N = 9, P < 0.001). There was no change the total (N = 9, p = 0.945) or surface (N = 5, P = 0.376) α4 subunit expression (C) or the total (N = 10, P = 0.056) or surface (N = 6, P = 0.176) δ subunit expression (D). *** = P < 0.001, ns = not significant
Two particular thalamic nuclei, nRT and VB, are intimately involved in the generation of the thalamocortical oscillations found in absence seizures (Beenhakker & Huguenard, 2009). Different GABAAR subunits are selectively expressed in different thalamic nuclei. The α1 subunit is expressed in VB, but not nRT, while α3 subunit is expressed in nRT, but not VB. Medial thalamic nuclei such as the paraventricular, mediodorsal, and centromedian nuclei express both α1 and α3 subunits (Hortnagl et al., 2013). Therefore, to determine the effects of α1 subunit deletion on total and surface α1 expression in VB and total and surface α3 subunit expression in nRT, we microdissected VB/nRT from the brain slices after the biotinylation reaction and, for comparison, we also microdissected the medial thalamic nuclei (Fig 2A). We purified the surface proteins from VB/nRT and medial thalamic regions and performed Western blots to quantify the effects of heterozygous loss of α1 subunit on the relative expression of α1 and α3 subunits in these specific thalamic regions.
Similar to the results obtained from the whole thalamic slice, heterozygous α1 subunit deletion significantly reduced total and surface α1 subunit expression in both VB/nRT (total: 73 ± 3% vs wild type, surface: 64 ± 10% vs wild type, P < 0.020, Fig 2B) as well as the medial thalamic nuclei (total: 66 ± 5% vs wild type, surface: 60 ± 3% vs wild type, P < 0.008, Fig 2B). There was no significant difference between the relative total and surface α1 subunit expression in either thalamic region.
In contrast to the results obtained from the whole thalamic slice, there was no change in total or surface α3 subunit expression in VB/nRT (P > 0.410, Fig 2C). However, heterozygous loss of α1 subunit did increase both total (133 ± 13% vs wild type, P = 0.015, Fig 2C) and surface (146 ± 14% vs wild type, P = 0.020, Fig 2C) α3 subunit expression in medial thalamic nuclei. Therefore, the increased α3 subunit expression found in the whole thalamus experiments (Fig 1) originated from medial and possibly other thalamic nuclei not including the nRT.
Extrasynaptic α1 subunit did not redistribute to the synapse in VB
The biotinylation and Western blot experiments demonstrated that neurons did not compensate for heterozygous α1 subunit deletion by increasing the relative amount of surface α1 subunit expression. However, biotinylation / Western blot experiments cannot determine if there is redistribution of residual surface extrasynaptic α1 subunit to the synapse. Therefore, we tested if heterozygous α1 subunit deletion altered the distribution of the GABAergic synaptic marker, VIAAT and/or redistributed residual surface α1 subunit and its assembly partner, β2/3 subunit, to GABAergic synapses.
We performed immunohistochemistry studies and stained for the α1 subunit, β2/3 subunit, and VIAAT in the VB region of the thalamus. We found that there was no difference (P = 0.921) between the density of VIAAT puncta between the VB of wild type (8.8 ± 0.4 puncta / 100 μm2, Fig 3A) and Het-KO (8.7 ± 0.6 puncta / 100 μm2, Fig 3B) mice. In addition, α1 subunit deletion did not significantly alter VIAAT puncta size (wild type 0.47 ± 0.02 μm2, Het-KO 0.51 ± 0.02 μm2, P = 0.156, Fig 3C).
Figure 3. Heterozygous α1 subunit deletion does not alter the fraction of α1 or β2/3 subunit associated with GABAergic synapses in VB.
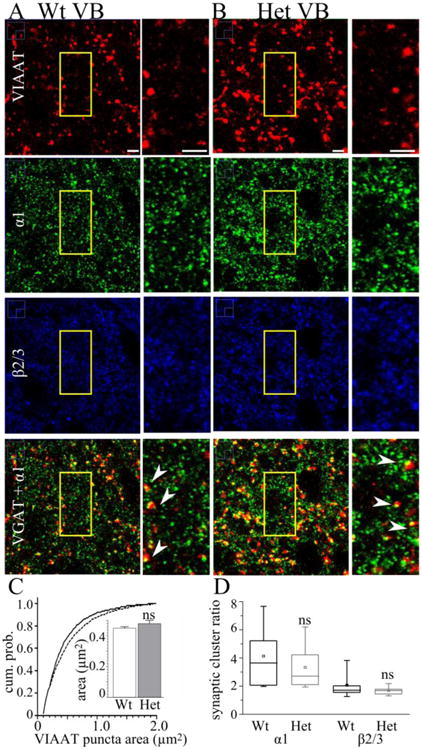
These are confocal microscopic images of wild type (A) and Het-KO (B) brain slices stained with antibodies directed to VIAAT (red), the α1 subunit (green), and β2/3 subunit (blue). In the fourth row, we depict the overlap of VIAAT and α1 staining (yellow). The field of view enclosed in the yellow boxes is shown on an expanded scale next to each image. The density of VIAAT puncta was not changed in Het-KO slices. The arrowheads in A and B show apposition between VIAAT puncta and the α1 subunit and demonstrate a similar extent of partial overlap between them. The cumulative probability plot (C, continuous line = wild type, dashed line = Het-KO) and the average of the mean VIAAT punctum sizes from each slice (inset in C) demonstrate that heterozygous α1 subunit deletion does not alter VIAAT punctum size (wild type = 0.45 ± 0.01 μm2, N = 20 slices from 6 mice, Het-KO = 0.48 ± 0.02 μm2, P = 0.199, N = 20 slices from 5 mice). D) Box plots depict the α1 and β2/3 synaptic clustering ratios. The box length extends from the 25th to 75th percentile and the whiskers extend from the 5th to 95th percentile. The square and horizontal line within the box marks the mean and median, respectively. Heterozygous α1 subunit deletion did not cause significant differences in either the α1 synaptic cluster ratio (wild type median = 3.6, N = 11 slices, Het-KO median = 2.7, P = 0.543, N = 13 slices) or β2/3 synaptic cluster ratio (wild type median = 1.7, N = 14 slices, Het-KO median = 1.7, P = 0.456, N = 12 slices). Scale bars = 3 μm, ns = not significant.
As expected, the majority of α1 and β2/3 subunit puncta did not localize to GABAergic synapses in VB (Fig 3A, B). However, a fraction of α1 and β2/3 puncta were present at GABAergic synapses and quantification of these puncta revealed that heterozygous α1 subunit deletion did alter the fraction of the total α1 or β2/3 subunit associated with the synapse (synaptic clustering ratio, wild type median α1 = 3.6; Het-KO median α1 = 2.7; P = 0.543; wild type median β2/3 = 1.7; Het-KO median β2/3 = 1.7; P = 0.456, Fig 3D). These results, combined with the biotinylation/Western blot data, demonstrated that heterozygous α1 subunit deletion reduces total α1 subunit expression and does not elicit partial compensation either by increasing the relative surface expression of the α1 subunit or by increasing the association of residual α1 or β2/3 subunits with GABAergic synapses.
Heterozygous α1 subunit deletion increases VIAAT puncta size in nRT and reduces the synaptic clustering ratio
In contrast to its effect on α1 subunit in VB, heterozygous loss of the α1 subunit does reduce the synaptic clustering ratio of both the α3 subunit (wild type median = 15.6, Het-KO median = 11.9, P = 0.026, Fig 4 A,B,D) and the β2/3 subunit (wild type median = 10.7, Het-KO median = 7.7, P = 0.037, Fig 4 A,B,D) in nRT. This result was not due to an increase in VIAAT puncta without an associated increase in α3 subunit puncta. While heterozygous α1 subunit deletion produces a non-statistically significant 38% increase in the VIAAT puncta density (wild type = 2.1 ± 0.2 puncta/100 μm2; Het-KO= 2.9 ± 0.3 puncta / 100 μm2, P = 0.063, Fig 4 A, B,), it also produces a corresponding (38%) increase in the density of α3 subunit puncta (wild type = 3.4 ± 0.4 puncta / 100 μm2; Het-KO= 4.7 ± 0.5 puncta / 100 μm2 P = 0.047, Fig 4 A, B).
Figure 4. The mean VIAAT punctum size is increased and α3 subunit synaptic cluster ratios are reduced in Het-KO nRT.
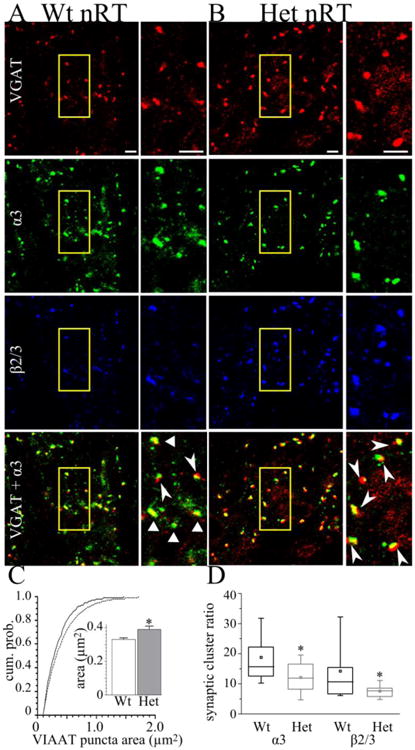
These are confocal microscopic images of wild type (A) and Het-KO (B) nRT stained with antibodies directed to VIAAT (red), the α3 subunit (green), and β2/3 subunit (blue). The bottom row depicts overlap between VIAAT and α3 subunit (yellow). The field of view enclosed in the yellow boxes is shown on an expanded scale next to each image. Het-KO did not change the density of VIAAT puncta. A cumulative plot (C, solid line = wild type, dashed line = Het-KO) shows that Het-KO changes the distribution of VIAAT puncta sizes and increases the size of medium and large nRT VIAAT puncta. The average of the mean VIAAT punctum size from all slices reveals that Het-KO increases VIAAT punctum size in nRT from 0.33 ± 0.01 μm2 (N = 13 slices from 6 mice) to 0.39 ± 0.02 μm2 (inset in C, P = 0.008, N = 12 slices from 5 mice). The triangles in wild type (A), but not Het-KO (B), show puncta with nearly complete overlap of VIAAT and α3 subunit puncta. The arrowheads in (A) and (B) show partial overlap between VIAAT and α3 subunit. D) Box plots depict the α3 and β2/3 synaptic cluster ratios. The box length extends from the 25th to 75th percentile and the whiskers extend from the 5th to 95th percentile. The square and horizontal line within the box marks the mean and median, respectively. Heterozygous α1 subunit deletion significantly reduces both the α3 synaptic cluster ratio (wild type median = 15.6, N = 13, Het-KO median = 11.9, P = 0.026, N = 11 slices) and β2/3 synaptic cluster ratio (wild type median = 10.7, N = 12 slices, Het-KO median = 7.7, P = 0.037, N = 11 slices). Four mice from each genotype were analyzed. Scale bars = 3 μm. * = P < 0.05.
One mechanism by which heterozygous loss of α1 subunit decreases the synaptic clustering ratio is by increasing VIAAT puncta area. Heterozygous α1 subunit deletion asymmetrically increases the VIAAT puncta size without increasing the size of α3 puncta. Visual examination of the apposition between the α3 subunit and VIAAT puncta reveals that in the nRT of Het-KO mice, a smaller fraction of each VIAAT punctum overlapped with each associated α3 subunit punctum (Fig 4 A, B). Quantification revealed that in the nRT of Het-KO mice, there was increased mean VIAAT punctum size (wild type = 0.33 ± 0.01 μm2; Het-KO = 0.39 ± 0.02 μm2, P = 0.008, Fig 4 C,), but no change in mean α3 or β2/3 punctum size (wild type α3 = 0.33 ± 0.02 μm2; Het-KO α3 = 0.33 ± 0.01 μm2, P = 0.897, wild type β2/3 = 0.28 ± 0.01 μm2; Het-KO β2/3 = 0.30 ± 0.02 μm2, P = 0.452, not shown). Therefore, this increase in mean VIAAT punctum size leaves a portion of each VIAAT punctum that does not overlap α3 subunit which thus reduces the value of the synaptic clustering ratio. It is important to note that this cause of reduced synaptic clustering ratio does not reflect a decrease in the ability of α3 or β2/3 subunits to target to GABAergic synapses. Rather, it demonstrates an asymmetric reorganization of the presynaptic component of these inhibitory synapses, a result that suggests that heterozygous α1 subunit deletion may alter GABAergic neurotransmission in nRT, even though this nucleus that does not express α1 subunit.
The extent of RNA editing of the α3 subunit is not altered in Het-KO nRT
RNA editing is a posttranscriptional mechanism by which enzymes called adenosine deaminases that act on RNA (ADAR), cause a site-specific conversion of adenosine to inosine which can result in an amino acid change or alternative splicing. Altered RNA editing of potassium channels and glutamate receptors has been found in animal models of pharmacologically-evoked status epilepticus and in surgical tissue resected from human temporal lobe epilepsy patients (Kortenbruck et al., 2001;Krestel et al., 2013;Russo et al., 2013). Altered RNA editing of GABAAR subunit mRNA has not yet been reported in human epilepsy patients or animal models of epilepsy.
Editing of the GABAAR α3 subunit mRNA produces the conversion of genomically encoded isoleucine to a methionine at the extracellular portion of the M3 transmembrane domain. Cells expressing GABAAR containing α3 subunits with a methionine rather than an isoleucine at this position exhibited GABA-evoked currents with slower activation times and faster deactivation times (Rula et al., 2008).
We microdissected the VB/nRT regions from four wild type and four Het-KO mice. RNA editing analysis of the α3 subunit revealed that there was no significant difference in the extent of α3 subunit RNA editing (wild type: 96.51% ± 0.47%; Het-KO: 95.45% ± 0.56%, P = 0.197, not shown).
Effects of heterozygous α1 subunit deletion on tonic and synaptic GABAA currents in VB neurons
GABAARs mediate both phasic and tonic currents. Phasic currents result from the transient activation of postsynaptic GABAARs to produce inhibitory postsynaptic currents while tonic currents result from the persistent activation of extrasynaptic or perisynaptic GABAAR by ambient GABA or by GABA that flows outside of the synapse (Belelli et al., 2009). Enhanced tonic GABAA currents in VB may be important in different types of absence epilepsy. Genetic rat (Cope et al., 2009) and mouse (Cope et al., 2009;Errington et al., 2011) models of absence epilepsy exhibited increased tonic GABAAR currents in VB neurons and selective activation of extrasynaptic thalamic GABAAR caused absence seizures in normal rats (Cope et al., 2009). Therefore, we determined if Het-KO mice possessed increased tonic GABAA currents in VB.
Tonic GABAA currents in VB are primarily mediated GABAAR containing α4 and δ subunits (Chandra et al., 2006;Porcello et al., 2003). Although we demonstrated that α4 or δ subunit expression was not altered in Het-KO thalami (Fig. 1 C, D), it was still possible that other mechanisms, such as the reduced GABA transporter activity found in a rat model of absence seizures (Cope et al., 2009), could enhance VB tonic currents in Het-KO mice. Therefore, we directly determined the effects of α1 subunit deletion on thalamic tonic currents in VB. We found that, in contrast to several other models of rodent absence epilepsy, there is no significant difference in the amplitude of the tonic currents between wild type (-41 ± 8 pA, Fig. 5A, C) and Het-KO (-49 ± 10 pA, P = 0.535, Fig. 5B, C) mice.
Figure 5. Tonic GABAA currents are not altered in Het-KO VB neurons.
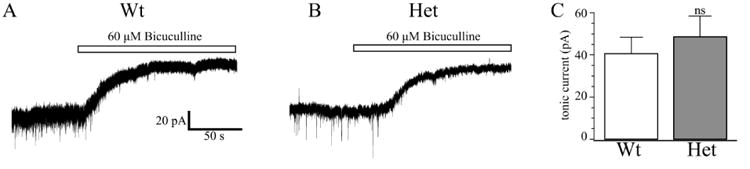
These are tonic currents from wild type (A) and Het-KO (B) VB neurons. The white bar indicates the application of 60 μM bicuculline. There was no significant difference (C) in tonic current amplitudes between wild type neurons (-41 ± 8 pA, N = 8 cells from six mice) and Het-KO neurons (-49 ± 10 pA, P = 0.705, N = 7 cells from six mice).
We next recorded phasic synaptic GABAA currents in thalamocortical VB neurons in wild type and Het-KO mice (Fig. 6). Heterozygous α1 subunit deletion reduces the magnitude of mIPSC peak current amplitudes from -32.7 ± 3.7 pA, to -18.1 ± 2.7 pA (P = 0.006, Fig 6C), a decrease that can be attributed to the reduction of α1 subunit expression in Het-KO thalami (Fig 1A, 2B). Heterozygous loss of α1 subunit also reduces the mIPSC frequency (wild type = 5.4 ± 1.2 Hz; Het-KO = 2.5 ± 0.5 Hz; P = 0.033, Fig 6 A, B), a result that could be consistent with a reduction in presynaptic activity and/or fewer events detected in Het-KO mice due to their lower amplitude. However, there was no change in the time course of VB mIPSC current kinetics; the 10-90% rise times for the wild type and Het-KO mIPSCs are 2.5 ± 0.1 ms and 2.4 ± 0.4 ms (P = 0.755), respectively, and the decay time constants, τ, are 21.4 ± 1.5 ms and 24.4 ± 7.6 ms (P = 0.716, Fig. 6D).
Figure 6. The frequency and amplitudes of mIPSCs are reduced in Het-KO VB neurons.
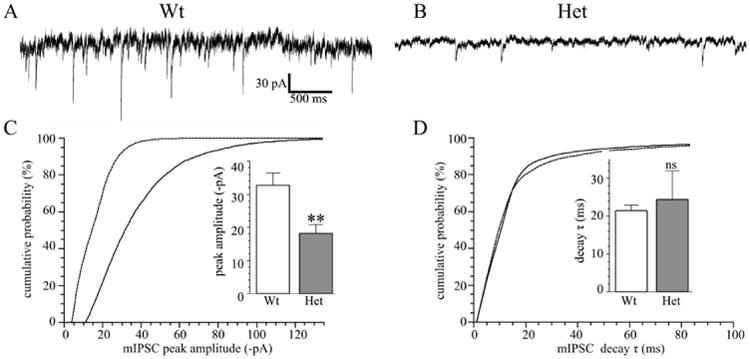
Panels A and B show sample mIPSC recordings from wild type (A, N = 7 cells from five mice) and Het-KO (B, N = 7 cells from five mice) VB neurons. The mIPSC frequency in Het-KO neurons was reduced from 5.4 ± 1.2 Hz to 2.5 ± 0.5 Hz (P = 0.033). In addition, the magnitude of mIPSC amplitudes was reduced on a cumulative plot (C, solid line wild type, dashed line Het-KO). The mean mIPSC peak currents averaged among different cells (C inset) was reduced from -32.7 ± 3.7 pA to -18.1 ± 2.7 pA (P = 0.006). There was no significantly difference in the decay time constants as seen on a cumulative probability plot (D) or upon comparing the averages of the mean decay constant (D inset, wild type = 21.4 ± 1.5 ms; Het-KO = 24.4 ± 7.6 ms, P = 0.716). ** = P < 0.01, ns = not significant.
Heterozygous α1 subunit deletion prolongs mIPSC decay in nRT
Next, we determined the effects of the partial loss of the α1 subunit on mIPSCs in nRT. We found that the heterozygous deletion does not change the amplitude (wild type = -13.8 ± 1.1 pA, Het-KO = -13.3 ± 0.8 pA, P = 0.706, Fig. 7 A-C) frequency (wild type = 1.5 ± 0.3 Hz, Het-KO = 1.2 ± 0.3 Hz, P = 0.519, not shown) or mean 10-90% rise times (wild type = 4.8 ± 0.5 ms, Het-KO = 6.4 ± 0.8 ms, P = 0.098, not shown). However, Het-KO prolongs the mean decay constant, τ, from 40.9 ± 4.3 ms to 55.9 ± 5.0 ms (P = 0.042, Fig. 5D), a result that demonstrates that heterozygous deletion of the α1 subunit alters GABAergic neurotransmission in the thalamus even in a nucleus (nRT) that does not express the α1 subunit.
Figure 7. Heterozygous α1 subunit deletion prolongs mIPSC decay in nRT neurons.
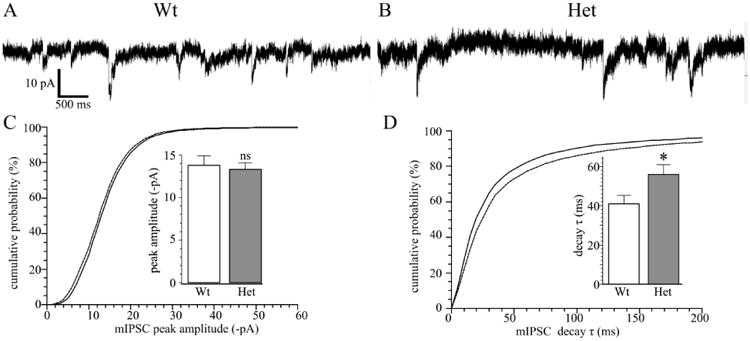
Panels A and B show mIPSC recordings from wild type (A, N = 8 cells from six mice) and Het-KO (B, N = 9 cells from seven mice) nRT neurons. There was no change in mIPSC frequency (wild type = 1.5 ± 0.3 Hz, Het-KO = 1.2 ± 0.3 Hz, P = 0.519) or in the mIPSC amplitudes as seen in the cumulative plot (C solid line wild type, dashed line Het-KO) or in the averages of the mean peak magnitudes (C inset: wild type = -13.8 ± 1.1 ms; Het-KO = -13.3 ± 0.8 ms, P = 0.706). However, the decay time constant, τ, was increased in Het-KO nRT neurons as seen cumulative probability plot (D) and in the mean of the average τ values from different cells (D inset, wild type = 40.9 ± 4.3 ms, Het-KO = 55.9 ± 5.0 ms, P = 0.042). * = P < 0.05, ns = not significant
Discussion
Here, we determined the effects of the heterozygous deletion of the human absence epilepsy gene, Gabra1, on GABAAR expression and function in the thalamus, a critical brain region for the maintenance of absence seizures. Our study produced three main findings. First, unlike other models of absence epilepsy (Cope et al., 2009;Errington et al., 2011), the Het-KO model does not increase tonic GABAA currents in VB. Second, unlike Het-KO cortex, VB does not increase the trafficking of residual α1 subunit to the cell surface and thus mIPSC peak amplitudes in VB are substantially reduced. Finally, also in contrast to Het-KO cortex, there is no increase in α3 subunit expression in nRT. However, similar to cortex, heterozygous α1 subunit deletion modifies GABAergic neurotransmission in nRT by prolonging the time course of mIPSC decay. These three effects on thalamic GABAAR expression and neurotransmission in conjunction with our previous findings regarding the effects of the deletion on cortical GABAergic transmission (Zhou et al., 2013) are summarized in Figure 8.
Figure 8. Altered thalamocortical GABAA neurotransmission in the Het-KO model of absence epilepsy.
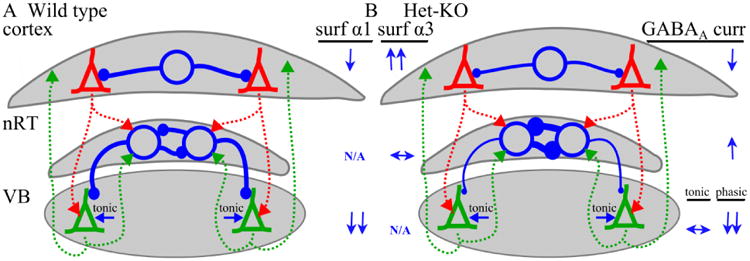
The effects of heterozygous α1 subunit deletion on GABAAR subunit expression and physiology are depicted on simplified models of wild type (A) and Het-KO (B) thalamocortical circuitry containing the cortex, thalamic reticular nucleus (nRT) and ventrobasal nucleus (VB). Excitatory neurons are pyramidal shaped and those in the cortex are colored red and those in VB are colored green. The axons from the excitatory neurons are dashed lines in the same color as the neurons. Inhibitory neurons are depicted as large open blue circles and their axons are the blue lines extending from the circles. Inhibitory synapses are small closed blue circles. The change in thickness of the inhibitory axons and the diameter of the synaptic circles represents the relative strength of GABAA inhibition in the Het-KO circuit. The relative increase (↑), decrease (↓), or no change (↔) in GABAAR α1 and α3 subunit expression and GABAAR current is listed next to the Het-KO circuit. In Het-KO cortex, there is a robust increase (↑↑) in surface α3 subunit expression and, because of increased surface trafficking of the residual α1 subunit, only a small decrease (↓) in surface α1 subunit expression resulting in an only modest decrease (↓) in phasic GABAA currents. Reticular nucleus does not express α1 subunit (N/A) and there is no change (↔) in surface α3 subunit expression. GABAA currents are modestly increased (↑) due to the prolonged time course of current decay. In VB, there is no α3 subunit (N/A) and surface α1 subunit expression is robustly decreased (↓↓) as are phasic GABAA currents. Tonic GABAA currents are unaffected (↔) in Het-KO VB.
Tonic GABAA currents are not increased in Het-KO VB
Prior investigations of homozygous α1 subunit knockout thalami demonstrated that homozygous deletion increased α4 subunit expression in adult (Kralic et al., 2006), but not P20 (Peden et al., 2008) VB. Therefore, it was possible that at age P33-37, the age at which we identified absence seizures in the Het-KO mice, heterozygous α1 subunit deletion would raise α4 subunit expression in the VB. Such a result could increase tonic GABAAR current amplitudes in the VB which would might contribute to the formation of absence seizures (Cope et al., 2009). However, we found that, at age P33-37, heterozygous α1 subunit deletion did not increase α4 or δ subunit expression in the thalamus or alter the amplitude of GABAAR tonic currents in VB. Possibly, the heterozygous loss of α1 subunit is not as sufficient a stimulus as homozygous α1 subunit deletion to increase α4 subunit expression in the thalamus or, perhaps, heterozygous α1 subunit deletion increases α4 subunit expression at ages beyond P33-37. Nonetheless, our data demonstrate that in contrast to other models of absence epilepsy (Cope et al., 2009;Errington et al., 2011), elevated tonic GABAA currents are not necessary to produce seizures in Het-KO mice of this age. This result suggests that novel therapeutic strategies that target elevated tonic GABAA currents in VB will not be universally effective in all types of absence epilepsy.
The role of VB disinhibition in Het-KO mice
We were initially surprised that, unlike cortical neurons (Zhou et al., 2013), Het-KO VB neurons do not increase the surface trafficking of the residual wild type α1 subunit as a method for raising phasic GABAAR activity to partially restore GABAAR homeostasis (Rannals & Kapur, 2011) and to normalize feed-forward inhibition from cortex through nRT to VB (Paz et al., 2011). One would expect that the disinhibition of VB neurons would increase their firing rate and propagate discharges to the cortex. In fact, in vivo extracellular recordings in genetic absence epilepsy rats from Strasbourg (GAERS) revealed higher VB neuron firing rates compared with nonepileptic controls (Carcak et al., 2014). However, the roles of GABAA inhibition in VB neurons and the relationship of VB inhibition to absence seizures are complex. Although GABAA neurotransmission can prevent VB neuronal firing, hyperpolarization is also needed by VB neurons to reactivate the T-type calcium channels and initiate the hyperpolarization-activated current – both of which are critical for burst firing seen in absence seizures (Coulter et al., 1989). The complex interaction between VB GABAA neurotransmission and absence seizures can be appreciated by pharmacological experiments. When VB nuclei of lethargic mice were microinfused with clonazepam, a benzodiazepine that enhances the synaptic α1βγ GABAAR that conduct phasic currents, but not the extrasynaptic α4βδ GABAAR that conduct tonic currents, absence seizures were unaffected (Hosford et al., 1997). Similarly, applying clonazepam to thalamic slices in which the α3, but not α1, subunits were mutated to be unresponsive to benzodiazepines, did not produce any significant change in the duration of evoked thalamic oscillations (Sohal et al., 2003). Finally, carbamazepine, an anticonvulsant drug that reduces focal seizures, but exacerbates absence seizures, potentiates synaptic-type (α1β3γ2) GABAAR currents and worsens epileptiform discharges in GAERS when microinfused in VB (Liu et al., 2006). Not only was the pro-absence effect of carbamazepine blocked by the GABAAR antagonist, bicuculline, but the disinhibition of VB by the infusion of bicuculline did not worsen the incidence of absence seizures (Liu et al., 2006). In total, these studies demonstrate that increased phasic GABAA currents in VB either worsens absence seizures or does not protect against them and suggests a reason why VB neurons, unlike cortical neurons (Zhou et al., 2013), do not actively increase the expression of residual α1 subunit to the cell surface.
Altered intrathalamic neurotransmission in Het-KO nRT and its relationship to seizures
We previously showed that in addition to increasing the surface expression of α1 subunit driven from the wild type allele, Het-KO cortical neurons also increase α3 subunit expression (Zhou et al., 2013). This finding is consistent with observations from homozygous α1 subunit deletion (Hom-KO) mice that found that Hom-KO neurons increase the protein expression of other α subunit isoforms that were normally expressed in that brain region and cell type (Kralic et al., 2006;Ogris et al., 2006;Peden et al., 2008;Schneider Gasser et al., 2007). However the effect of heterozygous or homozygous α1 subunit deletion had not been examined in nRT or the medial thalamic nuclei. Here, we were surprised to find that heterozygous α1 subunit deletion did not increase α3 subunit expression in nRT but did increase α3 subunit expression in the medial nuclei. One important difference between nRT and the medial thalamic nuclei and the brain regions examined by investigators studying Hom-KO mice (Kralic et al., 2006;Ogris et al., 2006;Peden et al., 2008;Schneider Gasser et al., 2007) is that the nRT is one of the few brain areas that does not express α1 subunit. Perhaps, reduced (Het-KO) or absent (Hom-KO) α1 subunit increases α3 subunit expression in the medial thalamic nuclei and other regions because the reduction/absence of α1 subunit expression reduces the competition of α3 subunit for assembly with partnering β and γ subunits into GABAAR pentamers. Possibly, GABAAR subunit substitution and compensation not only require that particular neurons already express the compensating subunit, but that those neurons also must normally express the subunit that is reduced.
Despite not increasing α3 subunit expression, we found that heterozygous α1 subunit deletion prolongs the decay phase of mIPSCs in nRT. The mechanistic basis underlying the prolonged mIPSC decay in nRT is unknown. While it is intriguing to speculate that the increased average VIAAT punctum size reflects altered presynaptic terminals that increase GABA concentration or differentially affect GABA release, diffusion and reuptake resulting in prolonged mIPSC decay, there are no studies that have measured the effects of altered VIAAT puncta on mIPSCs.
Regardless of the mechanism by which the time course of current decay is lengthened, this change is likely to reduce the incidence of absence seizures. When the GABAA agonist, muscimol, the barbiturate phenobarbital, or clonazepam are microinfused into the nRT in the lethargic (lh/lh) model of absence epilepsy, seizures are reduced (Hosford et al., 1997). Conversely, infusion of the GABAAR competitive antagonist, bicuculline, in the caudal nRT (approximately the region in which we made our recordings) increases the duration of epileptiform spike-wave-discharges (Aker et al., 2006). In addition, in mice in which either the α1 or α3 subunit is made insensitive to benzodiazepines, clonazepam only inhibits evoked thalamic oscillations when it is applied to thalamic slices in which α3 subunit-containing GABAAR in nRT are sensitive to benzodiazepines (Sohal et al., 2003). Finally, Schofield et al. demonstrated that genetically modified mice that exhibit increased GABAA neurotransmission within nRT also show a reduced sensitivity to pharmacologically-evoked seizures (Schofield et al., 2009). Therefore, it is likely that the prolonged mIPSC decay time in nRT of Het-KO mice represents a compensatory response that reduces absence seizures.
In conclusion, we demonstrated a novel pattern of altered GABAAR expression and neurotransmission in the thalamus in the Het-KO mouse model of absence epilepsy. We showed that unlike other models of absence epilepsy, Het-KO mice do not exhibit enhanced tonic GABAA inhibition in VB, a result that demonstrated that enhanced tonic VB inhibition is not necessary to produce absence seizures. We also found that phasic GABAA currents are reduced in Het-KO VB without compensation and that, surprisingly, phasic GABAA currents are prolonged in nRT. This latter result is likely a partial compensatory mechanism that reduces absence seizures.
Highlights.
Increased thalamic VB GABAA tonic inhibition is not required for absence seizures.
Thalamic VB is disinhibited with a reduction in mIPSC amplitude and frequency.
Inhibition in thalamic nRT is increased with prolonged mIPSC decay.
Increased nRT inhibition is likely a compensatory response to reduce seizures.
Acknowledgments
We gratefully acknowledge the support of United States Public Health Service Grant R01 NS064286 (M.J.G.) and the Vanderbilt Joel G. Hardman Chair in Pharmacology (R.B.E.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aker RG, Ozyurt HB, Yananli HR, Cakmak YO, Ozkaynakci AE, Sehirli U, et al. GABAA receptor mediated transmission in the thalamic reticular nucleus of rats with genetic absence epilepsy shows regional differences: functional implications. Brain Res. 2006;1111:213–221. doi: 10.1016/j.brainres.2006.06.118. [DOI] [PubMed] [Google Scholar]
- Arain FM, Boyd KL, Gallagher MJ. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor alpha1 subunit. Epilepsia. 2012;53:e161–e165. doi: 10.1111/j.1528-1167.2012.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW. Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci. 2009;29:12757–12763. doi: 10.1523/JNEUROSCI.3340-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcak N, Zheng T, Ali I, Abdullah A, French C, Powell KL, et al. The effect of amygdala kindling on neuronal firing patterns in the lateral thalamus in the GAERS model of absence epilepsy. Epilepsia. 2014;55:654–665. doi: 10.1111/epi.12592. [DOI] [PubMed] [Google Scholar]
- Chandra D, Jia F, Liang J, Peng Z, Suryanarayanan A, Werner DF, et al. GABAA receptor alpha 4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc Natl Acad Sci U S A. 2006;103:15230–15235. doi: 10.1073/pnas.0604304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, DiGiovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Calcium currents in rat thalamocortical relay neurones: kinetic properties of the transient, low-threshold current. J Physiol. 1989;414:587–604. doi: 10.1113/jphysiol.1989.sp017705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington AC, Gibson KM, Crunelli V, Cope DW. Aberrant GABAA receptor-mediated inhibition in cortico-thalamic networks of succinic semialdehyde dehydrogenase deficient mice. PLoS ONE. 2011;6:e19021. doi: 10.1371/journal.pone.0019021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: initial monotherapy outcomes at 12 months. Epilepsia. 2013;54:141–155. doi: 10.1111/epi.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mody I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol. 2007;582:1163–1178. doi: 10.1113/jphysiol.2007.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Khan ZU, De Blas AL. Immunocytochemical localization of gamma 2 short and gamma 2 long subunits of the GABAA receptor in the rat brain. J Neurosci. 1994;14:7168–7179. doi: 10.1523/JNEUROSCI.14-11-07168.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JL, Morabito MV, Martinez CR, III, Gilbert JA, Ferrick EA, Ayers GD, et al. Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol Cell Neurosci. 2014;61C:97–109. doi: 10.1016/j.mcn.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 2013;236:345–372. doi: 10.1016/j.neuroscience.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford DA, Wang Y, Cao Z. Differential effects mediated by GABAA receptors in thalamic nuclei in lh/lh model of absence seizures. Epilepsy Res. 1997;27:55–65. doi: 10.1016/s0920-1211(97)01023-1. [DOI] [PubMed] [Google Scholar]
- Kernan CL, Asarnow R, Siddarth P, Gurbani S, Lanphier EK, Sankar R, et al. Neurocognitive profiles in children with epilepsy. Epilepsia. 2012;53:2156–2163. doi: 10.1111/j.1528-1167.2012.03706.x. [DOI] [PubMed] [Google Scholar]
- Kortenbruck G, Berger E, Speckmann EJ, Musshoff U. RNA editing at the Q/R site for the glutamate receptor subunits GLUR2, GLUR5, and GLUR6 in hippocampus and temporal cortex from epileptic patients. Neurobiol Dis. 2001;8:459–468. doi: 10.1006/nbdi.2001.0394. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Sidler C, Parpan F, Homanics GE, Morrow AL, Fritschy JM. Compensatory alteration of inhibitory synaptic circuits in cerebellum and thalamus of gamma-aminobutyric acid type A receptor alpha1 subunit knockout mice. J Comp Neurol. 2006;495:408–421. doi: 10.1002/cne.20866. [DOI] [PubMed] [Google Scholar]
- Krestel H, Raffel S, von LM, Jagella C, Moskau-Hartmann S, Becker A, et al. Differences between RNA and DNA due to RNA editing in temporal lobe epilepsy. Neurobiol Dis. 2013;56:66–73. doi: 10.1016/j.nbd.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H+, et al. Novel α1 and γ2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011;34:237–249. doi: 10.1111/j.1460-9568.2011.07767.x. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Siddarth P, Riley JD, Gurbani SG, Ly R, Yee VW, et al. Neurobehavioral comorbidities of pediatric epilepsies are linked to thalamic structural abnormalities. Epilepsia. 2013;54:2116–2124. doi: 10.1111/epi.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zheng T, Morris MJ, Wallengren C, Clarke AL, Reid CA, et al. The mechanism of carbamazepine aggravation of absence seizures. J Pharmacol Exp Ther. 2006;319:790–798. doi: 10.1124/jpet.106.104968. [DOI] [PubMed] [Google Scholar]
- Lubke J. Morphology of neurons in the thalamic reticular nucleus (TRN) of mammals as revealed by intracellular injections into fixed brain slices. J Comp Neurol. 1993;329:458–471. doi: 10.1002/cne.903290404. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, et al. A mutation in the GABAA receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol. 2006;59:983–987. doi: 10.1002/ana.20874. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren HK, Veening JG, Moderscheim TA, Coenen AM, van LG. Thalamic lesions in a genetic rat model of absence epilepsy: dissociation between spike-wave discharges and sleep spindles. Exp Neurol. 2009;217:25–37. doi: 10.1016/j.expneurol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Muller E, Triller A, Legendre P. Glycine receptors and GABA receptor alpha 1 and gamma 2 subunits during the development of mouse hypoglossal nucleus. Eur J Neurosci. 2004;20:3286–3300. doi: 10.1111/j.1460-9568.2004.03785.x. [DOI] [PubMed] [Google Scholar]
- Ogris W, Lehner R, Fuchs K, Furtmuller B, Hoger H, Homanics GE, et al. Investigation of the abundance and subunit composition of GABAA receptor subtypes in the cerebellum of alpha1-subunit-deficient mice. J Neurochem. 2006;96:136–147. doi: 10.1111/j.1471-4159.2005.03509.x. [DOI] [PubMed] [Google Scholar]
- Panzanelli P, Gunn BG, Schlatter MC, Benke D, Tyagarajan SK, Scheiffele P, et al. Distinct mechanisms regulate GABAA receptor and gephyrin clustering at perisomatic and axo-axonic synapses on CA1 pyramidal cells. J Physiol. 2011;589:4959–4980. doi: 10.1113/jphysiol.2011.216028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, et al. A new mode of corticothalamic transmission revealed in the Gria4(-/-) model of absence epilepsy. Nat Neurosci. 2011;14:1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden DR, Petitjean CM, Herd MB, Durakoglugil MS, Rosahl TW, Wafford K, et al. Developmental maturation of synaptic and extrasynaptic GABAA receptors in mouse thalamic ventrobasal neurones. J Physiol. 2008;586:965–987. doi: 10.1113/jphysiol.2007.145375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcello DM, Huntsman MM, Mihalek RM, Homanics GE, Huguenard JR. Intact synaptic GABAergic inhibition and altered neurosteroid modulation of thalamic relay neurons in mice lacking delta subunit. J Neurophysiol. 2003;89:1378–1386. doi: 10.1152/jn.00899.2002. [DOI] [PubMed] [Google Scholar]
- Rannals MD, Kapur J. Homeostatic Strengthening of Inhibitory Synapses Is Mediated by the Accumulation of GABAA Receptors. J Neurosci. 2011;31:17701–17712. doi: 10.1523/JNEUROSCI.4476-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rula EY, Lagrange AH, Jacobs MM, Hu N, Macdonald RL, Emeson RB. Developmental modulation of GABA(A) receptor function by RNA editing. J Neurosci. 2008;28:6196–6201. doi: 10.1523/JNEUROSCI.0443-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I, Bonini D, Via LL, Barlati S, Barbon A. AMPA receptor properties are modulated in the early stages following pilocarpine-induced status epilepticus. Neuromolecular Med. 2013;15:324–338. doi: 10.1007/s12017-013-8221-6. [DOI] [PubMed] [Google Scholar]
- Schneider Gasser EM, Duveau V, Prenosil GA, Fritschy JM. Reorganization of GABAergic circuits maintains GABA(A) receptor-mediated transmission onto CA1 interneurons in alpha1-subunit-null mice. Eur J Neurosci. 2007;25:3287–3304. doi: 10.1111/j.1460-9568.2007.05558.x. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Kleiman-Weiner M, Rudolph U, Huguenard JR. A gain in GABAA receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures. Proc Natl Acad Sci U S A. 2009;106:7630–7635. doi: 10.1073/pnas.0811326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Keist R, Rudolph U, Huguenard JR. Dynamic GABAA receptor subtype-specific modulation of the synchrony and duration of thalamic oscillations. J Neurosci. 2003;23:3649–3657. doi: 10.1523/JNEUROSCI.23-09-03649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer R, von BL, Haenggi T, Schweizer C, Benke D, Rudolph U, et al. Alteration of GABAergic synapses and gephyrin clusters in the thalamic reticular nucleus of GABAA receptor alpha3 subunit-null mice. Eur J Neurosci. 2006;24:1307–1315. doi: 10.1111/j.1460-9568.2006.05006.x. [DOI] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, et al. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A. 2007;104:17536–17541. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yevenes GE, Nikonenko I, Ebeling C, Schwerdel C, et al. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc Natl Acad Sci U S A. 2011;108:379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander JF, Munster-Wandowski A, Brunk I, Pahner I, Gomez-Lira G, Heinemann U, et al. Synaptic and vesicular coexistence of VGLUT and VGAT in selected excitatory and inhibitory synapses. J Neurosci. 2010;30:7634–7645. doi: 10.1523/JNEUROSCI.0141-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Z, Ding L, Deel ME, Arain FM, Murray CR, et al. Altered cortical GABAA receptor composition, physiology, and endocytosis in a mouse model of a human genetic absence epilepsy syndrome. J Biol Chem. 2013;288:21458–21472. doi: 10.1074/jbc.M112.444372. [DOI] [PMC free article] [PubMed] [Google Scholar]