

Abstract
Complement cascade is involved in several renal diseases and in renal transplantation. The different components of the complement cascade might represent an optimal target for innovative therapies. In the first section of the paper the authors review the physiopathology of complement involvement in renal diseases and transplantation. In some cases this led to a reclassification of renal diseases moving from a histopathological to a physiopathological classification. The principal issues afforded are: renal diseases with complement over activation, renal diseases with complement dysregulation, progression of renal diseases and renal transplantation. In the second section the authors discuss the several complement components that could represent a therapeutic target. Even if only the anti C5 monoclonal antibody is on the market, many targets as C1, C3, C5a and C5aR are the object of national or international trials. In addition, many molecules proved to be effective in vitro or in preclinical trials and are waiting to move to human trials in the future.
Keywords: Complement cascade, Complement and glomerulopathies, Eculizumab, Targeting complement, Complement and renal transplantation
Core tip: Our therapeutical armamentarium is to date limited in many kidney diseases and in several aspects of renal transplantation. The findings that complement cascade is involved in many kidney diseases and in renal transplantation offer the availability of new therapeutical targets basing on the pathogenesis. The anti C5 monoclonal antibody, eculizumab, is now used to treat the atypical hemolytic uremic syndrome (aHUS), but 24 trials are ongoing in different renal diseases and in renal transplantation. Other targets as C1, C3, C5a, and C5aR are innovative treatments for diseases as aHUS, membranoproliferative glomerulonephritis, ischemia-reperfusion injury, and objects of ongoing trials.
INTRODUCTION
The complement system serves as first line defense against invading pathogens and is a component of the innate immune system[1,2]. The complement system represents a link between the innate and adaptive immunity. In addition, several studies examined the cross-talk between complement and toll-like receptors, another component of the innate immune system and the complement system[3]. The complement system is composed of three distinct activation pathways: classic pathway (CP), alternative pathway (AP), and mannose-binding lectin pathway (LP). Any pathway activates the complement cascade generating C3-convertase which cleaves C3 into C3a and C3b[4]. In normal conditions a small amount of C3-convertase is activated by the AP and is necessary to have regulators to prevent complement attack on healthy self cells[5] (Figure 1). This regulation is provided by a combination of plasma and cell surface inhibitory proteins. Fluid phase regulators include C1-inhibitor (C1-INH) that prevents the auto activation of the initial complex of the CP, the decay-accelerating factor that binds to C4b, and acts as co-factor for factor I (CFI) cleavage of C4b opsonin. Another regulator for AP is factor H (CFH), which also acts as a co-factor for CFI in the inactivation of C3b. Other regulators are Clusterin and Vitronectin that inhibit the insertion of terminal complexes into the cell membranes. Finally, carboxypeptidase N acts as anaphylatoxin inhibitor.
Figure 1.
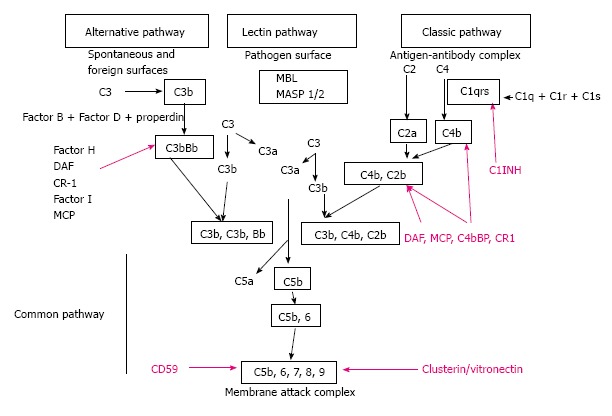
Representation of the classical, lectin and alternative pathways of complement activation, including regulatory molecules (purple). MBL: Mannose binding lectin; MASP 1/2: Mannan-binding lectin-associated serine protease-1; C1INH: C1 inhibitor; DAF: Decay accelerating factor; CR-1: Complement receptor 1; MCP: Membrane co-factor protein.
Finally, cell surface regulatory proteins, including regulators complement receptor 1 (CR1) (C3b receptor), membrane co-factor protein (MCP, CD46), and decay-accelerating factor (DAF, CD55) act inhibiting the C3 and C5 convertases activity. Complement-mediated injury is the result of the prevalence of the activating factor over the complement regulators[6,7].
Independently from complement involvement, serum complements levels may are low or normal. The pathogenesis of hypocomplementemia is related to the high consumption rate due to immune deposits, but other factors are the presence of hereditary complement deficiency and the presence of circulating factors that promote complement activation and consumption. When the CP is activated both C3 and C4 may be low. C3 levels low alone may be expression of the activation of the AP.
In kidney diseases and in kidney transplantation, the complement cascade is frequently involved and might represent a first line therapeutic strategy.
RESEARCH
We have analyzed the available data on complement and renal diseases and renal transplantation by careful revision of the currently available data. Literature research was performed using PubMed (NCBI/NIH) under employment of the search terms “complement cascade”, “complement and glomerulopathies”, “dense deposit disease”, “membranoproliferative glomerulonephritis”, “C3 glomerulonephritis”, “complement and renal transplantation”, “targeting complement”, “eculizumab”. Studies currently under way were sought for in “clinicaltrials.gov” and the European EUDRACT register. The papers published in the last three years on international journals on transplantation and kidney disease were carefully examined. Almost 160 papers were selected for this review.
PHYSIOPATHOLOGY OF COMPLEMENT INVOLVEMENT IN KIDNEY DISEASES
There are 2 broad categories of kidney diseases in which the complement system has a pathogenic role. The first is associated with complement over activation, and the second with complement dysregulation. Moreover the complement system is frequently involved in the kidney injuries after kidney transplantation[8] and in the progression of kidney diseases. The principal complement abnormalities leading to renal diseases are summarized in Table 1[9].
Table 1.
Representative abnormalities in complement leading to renal disease
| Components/related molecules | Diseases |
| Complement C3 | C3 glomerulopathy (DDD), aHUS |
| Factor H | C3 glomerulopathy (DDD/C3GN), aHUS |
| Factor I | C3 glomerulopathy (C3GN), aHUS |
| MCP | aHUS |
| Factor B | aHUS |
| CFHR5 | Familial C3 glomerulopathy (CFHR5 nepropathy) |
| CFHR3-1 | Familial C3 glomerulopathy |
| CFHR1/3 | IgA nephropathy, aHUS |
| Factor B autoantibody | C3 glomerulopathy (DDD) |
| Factor H autoantibody | C3 glomerulopathy (DDD/C3GN) |
| Bb (activated factor B) | HUS, ANCA-associated vasculitis |
| C3Nef | C3 glomerulopathy (DDD, C3GN) |
| Soluble C5b-9 | HUS, TTP, ANCA-associated vasculitis |
| C3a | ANCA-associated vasculitis, TTP |
| C5a | ANCA-associated vasculitis |
| C1q/C1qR | C1q nephropathy |
| Properdin | TI injury due to massive proteinuria |
| C5 | ANCA-associated vasculitis |
| Factor B | ANCA-associated vasculitis |
| CRaR | TI inflammation, IRI |
| C5aR | IRI |
| Factor H | IRI |
| C5b-9 | IRI |
| CD59 | IRI |
DDD: Dense deposit disease; aHUS: Atypical hemolytic uremic syndrome; C3GN: C3 glomerulonephritis; MCP: Membrane co-factor protein; CFHR5: Complement factor H-related protein; ANCA: Anti neutrophil cytoplasmic antibody; C3Nef: C3 nephritic factor; TTP: Thrombotic thrombocytopenic purpura; TI: Tubulo-interstitial; IRI: Ischemia-reperfusion-injury.
OVERACTIVATION OF COMPLEMENT
Lupus nephritis
Deficiencies in the early components of the CP including C1q, C2 and C4 are associated with the development of systemic lupus erythematosus (SLE)[10]. Familial C1q deficiency has been found to be a relevant genetic risk factor for the development of SLE, indeed C1q deficiency results in impaired phagocytosis[11]. The apoptotic cells are not immunologically benign and the reduced phagocytic clearance of these cells increases the likelihood that auto antigens are presented to lymphocytes and induce the development of the autoimmunity. Thirty percent of patients with lupus nephritis have C1q antibodies[12]. The products of C5 metabolism may also contribute directly to glomerular injury and in studies of murine models of lupus nephritis, a monoclonal antibody that blocked C5 cleavage significantly ameliorated the glomerulonephritis and prolonged survival[13].
Anti glomerular basement membrane glomerulonephritis
The complement system is involved in anti glomerular basement membrane glomerulonephritis either through CP and enhancing the inflammatory response through C5a activation[14] and/or cell lysis effect of C5b-9[15].
Antineutrophil cytoplasmic antibody associated vasculitis
Several authors[16,17] documented the involvement of complement in antineutrophil cytoplasmic antibody (ANCA) glomerulonephritis. Chen et al[16] documented C3 deposits in the glomeruli of patients with high levels of proteinuria and poor renal function. C5-9, C3d and complement factor B (CFB) were also reported in biopsies from patients with myeloperoxidase (MPO)-ANCA-associated pauci-immune glomerulonephritis. Xing et al[17], from the same group, observed that C4d was negative in biopsies of patients with MPO-ANCA glomerulonephritis. These studies suggest that this model of glomerulonephritis requires the activation of the AP, not the CP or the LP. Further studies in patients with active ANCA associated vasculitis documented high levels of C3a, C5a, soluble C5b-9 and Bb[18]. Recently other authors documented[19] that the C5a specific receptor (C5aR) expressed on neutrophils is involved in the pathogenesis of ANCA-induced glomerulonephritis (GN). Therefore targeting the C5a-C5aR receptor interaction in such patients might represent a therapeutical strategy[20]. A clinical trial to evaluate the safety and efficacy of an inhibitor of the C5a receptor (CCX168) is ongoing and an interim analysis reported promising results[21].
Membranous nephropathy
Several studies have identified that autologous antigens are the target of antibody response in idiopathic membranous nephropathy (MN). According to the latest studies[22] M-type phospholipase A2 receptor (PLA2R) located on podocytes has been identified as the target antigen in idiopathic MN. The predominant anti PLA2R IgG subclass activates the alternative or the mannose binding lectin (MBL) pathway[23]. This is confirmed by some studies documenting glomerular MBL and C4b deposition in MN[24,25].
In human secondary MN, C1q, C3, C4, CFB, MBL, and C5b-9 typically are present and co-deposited with IgG, suggesting that the LP and the AP could play the relevant role[26,27].
C1Q nephropathy
C1q nephropathy is characterized by the presence of conspicuous C1q immune deposits in glomeruli with no evidence of SLE MPGN type I. C1q nephropathy is characterized by the C1q binding to poly-anionic substances (DNA, RNA, viral proteins) or to C1q receptors, according some authors C1q nephropathy has been thought to be a subgroup of primary focal segmental glomerular sclerosis[28].
IgA nephropathy
Two distinct mechanisms of complement activation are involved in IgA nephropathy. The AP is the key pathway in 75% of cases[29,30]. In 25% of biopsy specimens, the presence of glomerular IgA1 and C3 is associated with MBL and MBL-associated serine protease 1 (MASP-1) deposition. MBL binds to the abnormally galactosylated region of the IgA1 through its carbohydrate binding domain resulting in complement catabolism through the lectin binding pathway. The presence of MBL and MASP-1 is associated with disease severity and poor histological prognostic features[31].
Immune complexes-associated membranoproliferative glomerulonephritis
In the past on immunopathological basis the MPGN was classified, on the basis of immunopathological findings, into three subtypes: MPGN type I, II [also known as dense deposit disease (DDD), and III]. Recently, the classification of MPGN has been completely reviewed by Bomback et al[32] on pathogenetic basis[33]. This led to a new understanding of the pathogenesis and to a reclassification of MPGN (Figure 2). The former MPGN type I and III belong to the chapter of complement over activation, while C3GN and the DDD will be described in the chapter of complement dysregulation.
Figure 2.
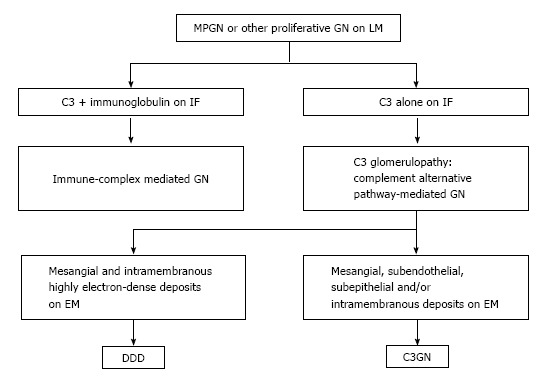
Reclassification of membrano-proliferative glomerulonephritis. C3GN: C3 glomerulonephritis; DDD: Dense deposit disease; EM: Electron microscopy; GN: Glomerulonephritis; IF: Immunofluorescence; LM: Light microscopy; MPGN: Membrano-proliferative glomerulonephritis.
In immune complex associated MPGN, the CP is activated by antibodies. Monoclonal antibodies or immune complexes precipitate catabolism, resulting in the chemo attraction of leukocytes and the direct cell injury by the MAC. The leukocyte generation of cytokines and proteases stimulate the mesangial cell proliferation and the matrix expansion[34].
DYSREGULATION OF COMPLEMENT
Atypical hemolytic uremic syndrome
The dysregulation of the AP cascade due to acquired or genetic factors leads to defective complement control that may cause a range of complement associated glomerulopathies (Figure 3). The better known among them is the atypical HUS (aHUS). aHUS classically is a triad of microangiopathic hemolytic anemia, acute kidney injury and thrombocytopenia caused by failure to regulate the alternative complement pathway. In over 60% of cases mutations have been identified in genes encoding complement regulatory proteins: CFH, CFI, MCP, thrombomodulin (THBD), and in genes encoding complement activators: CFB and C3. Complement cascade dysregulation causes a damage of endothelium leading to thrombosis and microangiopathic hemolytic anemia[35,36]. CFH mutations are observed in 25%-30% of patients with an aHUS[37]. Up to now, more than 80 mutations have been identified. Patients with aHUS and anti factor H antibodies have also been reported. These antibodies bind to short consensus repeats, thus reducing the CFH activity[38]. Reduction in MCP expression is reported in over 80% of cases with mutation in this gene[36,39]. Genetic disorders are rarely related to CFI[40]. THBD mutations with hyperactivity have been found in only 3%-5% of patients[41].
Figure 3.
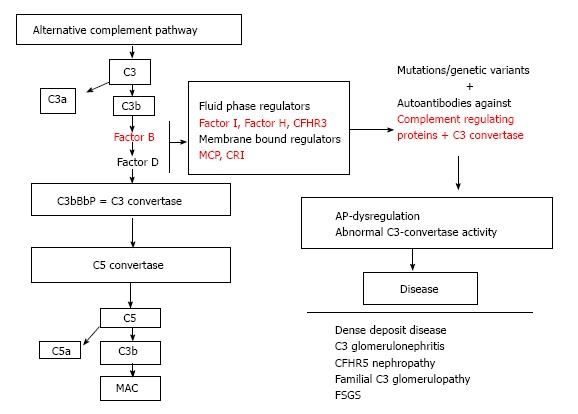
Dysregulation of the alternative complement cascade due to acquired or genetic factors leads to defective complement control causing a range of complement-associated glomerulopathies. CFHR3: Complement factor H-related protein 3; MCP: Membrane co-factor protein; CR1: Complement receptor 1; AP: Alternative pathway; CFHR5: Complement factor H-related protein 5; FSGS: Focal segments glomerular sclerosis; MAC: Membrane attack complex.
Recently, Noris et al[42] have documented that the classical HUS caused by Shiga toxin producing escherichia coli (STEC-HUS) and thrombotic thrombocytopenic purpura (TTP) are caused by inappropriate complement activation. Even if STEC-HUS, aHUS and TTP are all diseases of complement activation and recognize a common pathogenesis, we should remember that aHUS is linked to the complement dysregulation, while STEC-HUS and TTP are linked to the complement over activation and, on a pathogenetic basis, belong to the previous chapter.
In the HUS-SYNSORB Pk trial, children with STEC-HUS had increased plasma levels of Bb and C5-9 at the beginning of the study, which normalized after one month[43]. This suggests that patients with acute onset STEC-HUS have an activation of the AP in the acute phase of the disease, which normalizes within 1 mo. In the initial phases of STEC-HUS, the toxin triggers the endothelial complement deposition and interferes with the activity of the complement regulatory molecules[44]. Moreover, lack of the lectin-like domain of THBD, worsen STEC-HUS in mice[45].
Recent studies that further document the involvement of complement in STEC-HUS are those reporting the beneficial effect of Eculizumab (an anti C5 monoclonal antibody) in the outbreak of STEC-HUS induced by E. Coli 0104: H4 in Germany[46] and in the outbreak of STEC-HUS induced by the same strain in France[47].
Réti et al[48] recently reported increased levels of C3a and C5b-9 associated with decreased complement C3 levels during the acute phase of TTP. This fact indicates a complement consumption, which occurs in some TTP patients. To confirm complement involvement in TTP, in some patients refractory to treatment, eculizumab has been used with good results. These patients had severe TTP and a deficiency of disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) due to high titers of anti ADAMTS13 antibodies[49].
C3 glomerulopathies
Isolated C3 deposition within the glomerulus is the defining histological criterion for C3 glomerulopathy. C3 glomerulopathy is a recently introduced pathological entity defined by a glomerular pathology characterized by C3 accumulation with absent or scanty immunoglobulin deposition. In August 2012, an invited group of experts met to discuss the C3 glomerulopathy in the first C3 glomerulopathy meeting[50]. According the conclusions of the meeting and the recent paper from Barbour et al[51] on the basis of histological and clinical features, C3 glomerulopathies may be distinguished into: (1) DDD; and (2) C3 glomerulonephritis (C3GN). A familial form of C3GN has been recently described: the CFHR5 nephropathy.
From the patho-physiological point of view, three different mechanisms may be operating in the different conditions:
Auto antibodies: C3 nephritic factor (C3NeF) is an autoantibody that binds to a neoepitope on the AP C3 convertase. C3NeF stabilizes convertase against the CFH-mediated decay resulting in an uncontrolled C3 activation[52]. C3NeF is common in DDD, less in C3GN and absent in CFHR5 nephropathy.
Other auto-antibodies have also been described. Two patients with DDD and auto antibodies targeting both CFB and C3b have been recently identified[53]. Auto antibodies anti CFH also occur in DDD and in C3GN[54,55].
Genetic sequence variations: On genetic basis, heterozygous mutations in the CFH, CFI and MCP genes have been documented in C3GN[56] and also in MPGN type I and DDD[57]. Some of these heterozygous mutations have been also observed in patients with aHUS[58] showing similarities between the two diseases.
Genetic structure variation: As aforementioned, CFHR5 nephropathy has been described as a familial form of C3GN[59]. A mutation in complement factor H-related protein 5 in patients with glomerulonephritis has been identified via a genome-wide linkage study. The mutant CFHR5 protein present in patient serum had reduced affinity for surface-bound complement. Genetic abnormalities in the CFHR3 and CFHR1 loci were also recently reported. Such patients develop an autosomal dominant complement-mediated GN similar to CFHR5 nephropathy[60].
As aforementioned, the complement cascade is involved also in other renal conditions as the progression of renal disease and renal transplantation.
COMPLEMENT AND PROGRESSION OF RENAL DISEASE
The progression of renal disease may be mediated by tubule interstitial inflammation. Several studies have confirmed this datum and the involvement of complement activation[61]. Complement activation by tubular cells is mediated by the properdin binding. This fact is principally relevant in the case of proteinuric renal disorders. Studies in vitro have documented that the complement system is activated by the AP[62,63].
Complement activation may occur also in non proteinuric renal diseases as documented by Bao et al[64]. In this condition the C3a receptor is involved in causing renal inflammation and fibrosis.
Another important factor is the “in situ” produced chemokines. Genomic studies performed by Bao et al[65] documented that in some murine models several pro inflammatory and pro fibrotic chemokine genes are up-regulated. This activation occurs upon complement activation.
According to the aforementioned studies, targeting complement might be a useful therapeutical approach for chronic kidney disease in the future. Further studies are necessary for a better understanding of the role of complement in mediating tubule interstitial damage and consequent fibrosis.
COMPLEMENT IN RENAL TRANSPLANTATION
Transplanted kidneys principally suffer from injuries such as ischemia reperfusion (I/R) injury and rejection. Complement may mediate all these conditions.
Ischemia-reperfusion injury
The short-term consequences injuries as I/R injury and hyper acute rejection are principally related to innate immunity, while later injuries such as the antibody mediated rejection (ABMR) and the cell mediated rejection (CMR) are related either to the innate and the adaptive immune system.
I/R causes in the transplant both a vascular and parenchyma cell injury. In I/R complement is principally activated through the AP as a consequence of in situ generation of C3[66]. Other studies suggest the activation of MBL[67]. The majority of transplanted kidneys are retrieved from cadaveric donors. In such kidneys C3 may be present in the organ before retrieval because of donor suffering. Damman et al[68] found higher gene expression of C3 and increased deposition of C3d in kidney biopsies obtained from deceased grafts. Now a large scale study in the United Kingdom is analyzing in renal from deceased donors, the soluble form of C3-1 as a protecting agent for IRI and to improve graft outcomes[69]. Going in molecular details, Simone et al[70] documented that in renal I/R injury complement activates nicotinamide adenine dinucleotide phosphate-oxidase (NAPDH oxidase) enzymes. During renal IRI an endothelial-to-mesenchymal transition (EndMT) may occur, mediated by complement activation. EndMT may have a critical role in generating renal fibrosis[71]. Curci et al[72] documented that, during I/R injury, an activation of the CP and LP of the complement system occurs primarily at the endothelial cell level. In a recent study, the same authors[73] analyzed in large mammals the role of complement in the induction of EndMT by using recombinant C1 inhibitor in vivo.
Their data documented that the activation of the serine/threonine-specific protein kinase (Akt) pathway was essential to induced EndMT in vitro. In accordance, inhibition of complement in vivo abrogated the Akt signaling, with inhibition of EndMT and of tissue fibrosis.
Pratt et al[74] documented that C3 produced by a graft and by recruited immune cells is a two phases trigger that in the early period produces a post-perfusion injury, later may contribute to late rejection associated-allograft injury. Indeed a recent study[75] documented that I/R injury can affect the systemic immune response to antigens requiring a functional alternative pathway of complement. C3 split products, C3b and C3d, deposited on antigen presenting cells (APCs), can increase allo-antigens uptake and their presentation to the T cells. So doing, C3 positive APCs potentiate the T cell response in vitro[74]. The role of C3 in activating T cells is confirmed by studies documenting that the macrophages deficient for the C3 have impaired capability to stimulate the T cells[76,77]. The enhancement of the effector T cell expansion by complement should be ascribed to the limited antigen induced apoptosis[78]. In addition, other studies have documented the role of complement on the iTreg. Indeed, the iTreg-mediated tolerance to alloantigen in humans[79] might be mediated by the signaling through C5a receptor and C3a receptor.
As already mentioned, complement is involved both in the CMR and in the ABMR.
Cell-mediated rejection
The complement activation through any pathway generates C3a and C5a. These anaphylatoxins bind to both APCs and T cells to stimulate and activate T cells[80]. Li et al[81] demonstrated that the deficiency of the C5a receptor limited the adaptive response of recipient T cells to alloantigen. C1q appears to have a regulatory role in the threshold for the T cell activation by dendritic cells[82]. Moreover, in human kidney transplants with acute rejection, C5aR expression was increased in the renal tissue and in the cells infiltrating the tubular interstitium[83]. The same authors documented that in mice treated with a C5aR antagonist the infiltration of monocyte-macrophage was significantly attenuated, perhaps as a result of reduced levels of monocyte chemo attractant protein 1 and the intercellular adhesion molecule 1. However a murine model of kidney transplantation with C4 deficiency demonstrated that a CMR can occur in the absence of the CP or of the LP activation[84]. This suggests that the AP may play the key role in CMR.
Antibody-mediated rejection
The antibody-mediated renal allograft rejection often involves either donor specific antibodies (DSAs) and the CP of complement system activation. After binding to DSAs, complement is activated. C4d is a degradation product of C4 and, because it binds and remains covalently attached at the site of complement activation, represents a useful diagnostic tool[85]. Haidar et al[86] discovered that the deposition of C4d on erythrocytes was even more related to histological rejection signs, thus representing a possible not invasive diagnostic tool.
C3a and C5a likely act as potent chemo tactic factors promoting the infiltration of proinflammatory cells. In addition, the MAC might directly damage the allograft[85]. Expression of the membrane-bound regulator, CD55, is inversely related to C4d staining in biopsy specimens. Indeed CD 55 (also known as DAF), which accelerates the decay of C3 and C5 convertases, might modify the severity of the rejection. An increased CD55 expression is associated with an improved long-term kidney transplant outcomes in recipients without antibody-mediated rejection, suggesting a possible role for CD55 in the kidney protection[87].
In addition the kidney, after transplantation, may be involved in clinical conditions as recurrence of some renal diseases. Recently complement involvement has been documented also in chronic antibody mediated rejection.
Atypical hemolytic uremic syndrome is associated with a high rate of recurrence and poor outcomes after kidney transplantation. Acquired or inherited dysregulation of the alternative complement pathway, thought to be the driving force of the disease, is identified in most aHUS patients[88]. Recurrent thrombotic microangiopathy is very rare in patients who had developed end stage renal failure following HUS caused by Shiga-toxin producing STEC, whereas disease recurrence is common in patients with aHUS[89]. The recurrence rate[90] of C3 glomerulopathy on renal transplantation could be approximately estimated on about 60% as derived from two small case series of Servais et al[57] and Little et al[91] and confirmed in the recent paper of Zand et al[92]. In such conditions anticomplement therapy could be useful.
Moreover recent data document the complement involvement also in antibody mediated chronic rejection where the “bad” activity of antibodies can also be involved in previously considered “chronic” lesions (i.e., transplant glomerulopathy[93,94].
TARGETING COMPLEMENT: THERAPEUTIC STRATEGIES
Complement is clearly involved in many kidney diseases as well as in kidney transplantation. Hence targeting complement cascade at different levels may represent a new therapeutic strategy directed against the pathogenetic mechanisms.
Since the end of 2000’s several review papers reported the efficacy in vitro or in preclinical studies of anti complement molecules[95-97]. Unfortunately in the setting of renal diseases only an anti C5 monoclonal antibody is on the market. All others molecules are either on the market for diseases not involving the kidney or are still in preclinical phases (Table 2) or failed their efficacy.
Table 2.
Some of the molecules aimed to target complement, phase of the trial and renal diseases related
| Compound name | Complement target | Compound class | Phase/indication |
| C1 inhibitor (Berinert) | C1r, C1s, MASP1, MASP2 | Regulator | P 1/2 transplant |
| Cp40, AMY 101 | PC transplant , aHUS, DDD | ||
| sCR1, CDX-1135 | C3 conv, C4b, C3b | Regulator | P 1 DDD |
| Mirococept, APT070 | C3 conv, C4b, C3b | Regulator | P 1/2 transplant |
| Eculizumab | C5 | Ab | P 4 aHUS, P 2/3 STEC-HUS, P 2 ANCA vasculitis, P 1 transplant |
| Mubodina | C5 | Ab | PC aHUS, DDD |
| Ergidina | C5 | Ab | PC transplant |
| CCX168 | C5aR | Small molecule | P 2 ANCA vasculitis |
| ADC-1004 | C5aR | Protein | PC transplant |
MASP1: Mannan-binding lectin-associated serine protease; P: Phase; PC: Preclinical; aHUS: Atypical hemolytic uremic syndrome; DDD: Dense deposit disease; sCR1: Soluble complement receptor 1; Ab: Antibodies; STEC-HUS: Shiga toxin producing Escherichia Coli-hemolytic uremic syndrome.
TARGETING C5
The first available anti-complement therapy is eculizumab, a fully humanized monoclonal antibody that binds with high affinity to C5 and prevents the generation of MAC[98]. Eculizumab was first approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH)[99] and more recently, for the treatment of aHUS and other kidney diseases.
To date[100] 11 trials are ongoing for aHUS, 2 trials for STEC-HUS, 2 trials for MPGN and 1 trial for ANCA related diseases. Trials with eculizumab are also ongoing in the field of kidney transplantation, in particular 8 trials for the prevention and/or the treatment of acute or chronic ABMR, 3 trials for the prevention of delayed graft function (DGF), 1 trial for the prevention of I/R injury and 1 trial for the prevention of glomerular disease recurrence after transplantation.
After initial reports of the possible beneficial effects of eculizumab in treating patients with aHUS[101,102], more recently the beneficial effect of treating aHUS by eculizumab has been documented by two studies. One study[103] reported the data from 27 patients treated in off label studies. The other study[104] reported the data of 37 patients enrolled in 2 phase II trials. The second of these phase II trials was notable because 80% of subjects achieved thrombotic microangiopathy event-free status. These studies were the object of debate for the issues concerning the duration and the optimal dosing of therapy[105-108]. Indeed, although eculizumab revolutioned the treatment of aHUS, several unresolved issues remain, among which whether eculizumab should be always the first line therapy for aHUS and whether the drug should be considered as a life-long therapy also taking in account the treatment high cost. In addition, in PNH, but not by now in aHUS, patients have been described with unexplained eculizumab resistance. A recent study[109,110] documented that such resistance was due to C5 variants with mutations at Arg885.
As aforementioned also STEC-HUS and TTP are caused by inappropriate complement activation[42]. The eculizumab treatment has proved effective in these conditions. After the report of 3 cases[111], the eculizumab effectiveness has been documented in two STEC-HUS outbreaks occurring in Germany and in France[46,47]. The efficacy of eculizumab in treating the TTP has also been reported[49,112], even if others advocate rituximab as the best option in treating TTP[113].
The pathogenetic similarities between aHUS and some C3 glomerulopathies might imply that eculizumab treatment could fit well in treating all these diseases[103]. The new classification of C3 glomerulopathies (previously MPGN) have already been described[51]. Only DDD and C3GN have some similarities with aHUS and eculizumab could be beneficial for such patients. To date, in literature the eculizumab use for C3 glomerulopathies is limited to 6 case reports[114-119] and the results from a 1-year, open-label study of eculizumab therapy in 6 subjects[120,121]. The treatment results differ and indicate that eculizumab may be a non adequate treatment for a subgroup of patients with DDD and C3GN. Although some investigators suggested that aHUS should be considered part of a spectrum that includes DDD and C3GN, the underlying defect is not always the same. In aHUS the endothelial damage is often due to complement dysregulation at the level of cell membrane in the solid phase[36]. The solid phase dysregulation in aHUS translates to C5 convertase dysregulation being at least equal and often greater than C3 convertase dysregulation. Hence the blockade of C5 in such conditions is expected to yield improvement. In contrast, in some cases of C3 glomerulopathies a dysregulation of the fluid phase occurs and, as a consequence, C3 convertase dysregulation is greater than C5 dysregulation. These cases, because of a feed-back effect on the C3 convertase activity, could potentially be aggravated by C5 blockade[122]. Therefore, one of the major challenges in treating patients with C3 glomerulopathy with the anti complement therapy is how to distinguish the patients with primarily C3 convertase dysregulation from the patients with primarily C5 convertase dysregulation. Blockade at the level of C3 may be an alternative to eculizumab therapy, primarily in patients with C3 glomerulopathies associated with a C3 convertase dysregulation greater than the C5 convertase dysregulation.
As mice deficient in C5 have demonstrated resistance against anti-MPO-mediated glomelulonephritis[123], an open label phase II trial (NCT01275287), in which the patients with ANCA-associated glomerulonephritis were randomized to standard of care treatment vs standard of care plus eculizumab, was started. Unfortunately the trial was withdrawn because of lack of enrollment.
In the case of the recurrence after transplantation of a kidney disease susceptible to anti C5 therapy, eculizumab treatment is effective.
Zuber et al[124] successfully treated 22 renal transplant recipients with recurrence of aHUS.
McCaughan et al[117] reported a patient with DDD recurrence after kidney transplantation successfully treated by eculizumab. More recently Lonze et al[125] reported the cases of antiphospholipid antibody syndrome, two of them with the catastrophic variant, which were successfully treated at the time of transplantation with continuous systemic anticoagulation together with eculizumab prior to and following the live donor renal transplantation.
As aforementioned renal damage due to complement activation occurs in two phases after transplantation: during reperfusion after that the donor kidney has undergone a significant period of ischemia and during the acute rejection once the innate and adaptive immune system has recognized the donor antigens. In both conditions the complement may play a relevant role. Four clinical trials are now active aiming to control the ischemia-reperfusion injury and the consequent DGF. All these trials hypothesized that C5 cleavage is a key step in the pathogenesis of I/R injury following transplantation and its block could be an effective prophylactic tool to prevent acute kidney injury (NCT01919346, NCT01403389, NCT02145182, NCT01756508).
Eculizumab has also been successfully used in reducing antibodies in highly sensitized patients with positive cross-matches prior to transplantation[126-128]. In a larger case-control study the patients with DSAs were treated with eculizumab after transplantation and compared to the historical controls[129]. Eculizumab treatment was able in significantly lowering ABMR and in decreasing the 1-year transplant glomerulopathy incidence rate.
Table 3 summarizes all the trials ongoing with eculizumab in treating either glomerular disease and renal transplantation.
Table 3.
Trials ongoing with eculizumab in renal diseases and in renal transplantation
| Rank | Identifier | Status | Study name |
| 1 | NCT01221181 | Active | Eculizumab therapy for dense deposit disease and C3 nephropathy |
| 2 | NCT02093533 | Recruiting | Eculizumab in primary MPGN |
| 3 | NCT01275287 | Active | Targeting complement activation in ANCA-vasculitis |
| 4 | NCT00844545 | Completed | Open label controlled trial of eculizumab in adult patients with plasma therapy-resistant aHUS |
| 5 | NCT00844844 | Completed | Open label controlled trial of eculizumab in adolescent patients with plasma therapy-resistant aHUS |
| 6 | NCT00844428 | Unknown | Open label controlled trial of eculizumab in adolescent patients with plasma therapy-sensitive aHUS |
| 7 | NCT00838513 | Unknown | Open label controlled trial of eculizumab in adult patients with plasma therapy-sensitive aHUS |
| 8 | NCT01194973 | Unknown | An open-label, multi center clinical trial of eculizumab in adult patients with aHUS |
| 9 | NCT01193348 | Unknown | An open label, Multi center clinical trial of eculizumab in pediatric patients with aHUS |
| 10 | NCT01755429 | Unknown | The safety and efficacy of eculizumab in Japanese patients with aHUS |
| 11 | NCT01522170 | Enrolling | aHUS observational long term follow up |
| 12 | NCT01522183 | Recruiting | aHUS registry |
| 13 | NCT01770951 | Completed | A retrospective, observational, non-interventional trial to assess eculizumab treatment effect in patients with aHUS |
| 14 | NCT02205541 | Not yet recruiting | Eculizumab in shiga-toxin related hemolytic and uremic syndrome pediatric patients |
| 15 | NCT01410916 | Completed | Safety and efficacy study of eculizumab in shiga-toxin producing Escherichia coli (STEC-HUS) |
| 16 | NCT01406288 | Completed | Completed outbreak of HUS linked to Escherichia coli of serotype 0104:H4 |
| 17 | NCT01756508 | Recruiting | Eculizumab for prevention and treatment of kidney graft reperfusion injury |
| 18 | NCT01919346 | Recruiting | Eculizumab for prevention of DGF in kidney transplantation |
| 19 | NCT01403389 | Active | A study of the activity of eculizumab for prevention of DGF in deceased donor transplant |
| 20 | NCT02142182 | Recruiting | A trial for prevention of DGF after kidney transplantation |
| 21 | NCT01567085 | Active | Safety and efficacy of eculizumab in the prevention of AMR in sensitized recipients of a kidney transplant from a deceased donor |
| 22 | NCT02113891 | Not yet recruiting | Eculizumab therapy for subclinical antibody-mediated rejection in kidney transplantation |
| 23 | NCT01095887 | Active | Eculizumab added to conventional treatment in the prevention of antibody-mediated rejection in blood group incompatible living donor kidney transplantation |
| 24 | NCT01106027 | Active | Dosing regimen of eculizumab added to conventional treatment in positive crossmatch deceased kidney yransplant |
| 25 | NCT01895127 | Recruiting | Efficacy and safety of eculizumab for treatment of antibody-mediated rejection following renal transplantation |
| 26 | NCT00670774 | Active | Dosing regimen of eculizumab added to conventional treatment in positive crossmatch living kidney transplant |
| 27 | NCT01399593 | Active | Safety and efficacy of eculizumab to prevent AMR in living donor kidney transplant recipients receiving desensitization |
| 28 | NCT01327573 | Active | Eculizumab therapy for chronic complement-mediated injury in kidney transplantation |
| 29 | NCT01029587 | Recruiting | Eculizumab to enable renal transplantation in patients with history of catastrophic antiphospholipid antibody syndrome |
MPGN: Membrano-proliferative glomerulonephritis; ANCA: Anti neutrophil cytoplasmic antibody; aHUS: Atypical hemolytic uremic syndrome; STEC-HUS: Shiga-toxin producing escherichia; DEGF: Delayed graft function; AMR: Antibody mediated rejection.
TARGETING C5a AND C5aR
C5a is a powerful anaphylatoxin that stimulates the cytokine production, enhances the T-cell activation and augments the leukocyte adhesion and the vascular permeability (Figure 4). There is an increased expression of the C5aR in transplanted kidneys with IRI or acute rejection[83,130]. Recently Cravedi et al[131] documented that pharmacological C5aR blockade in mice reduces the graft versus host disease, prolongs the survival and inhibits the T-cell responses. This provides the basis for future studies aimed to target C5aR. Several studies have documented that the activation of the C5-C5a receptor axis is involved in several human diseases[132]. In addition to eculizumab that to date is the only specific complement inhibitor approved for clinical use, several therapeutics targeting the C5a-C5aR axis are in different stages of clinical development ranging from preclinical studies to phase II studies. These agents may target the axis at different levels, ranging from conversion of C5 to C5a and C5b, to inactivation of C5a, or to the inhibition of the two C5a receptors C5aR (CD88) and C5L2[132,133]. As aforementioned, after the findings that C5aR blockade protects against MPO-ANCA GN in mice[21] a clinical trial (NCT01363388) was started with a planned enrollment of 60 subjects affected by ANCA associated glomerulonephritis.
Figure 4.
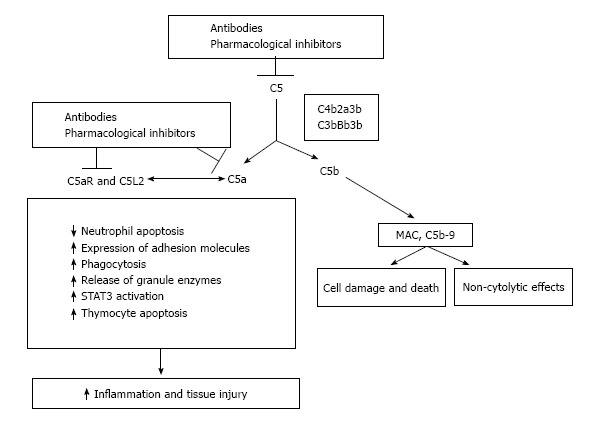
Significance of inhibiting the C5-C5a receptor axis. C5 convertases formed during activation of complement cascade cleave C5 into its active products. Inhibition is feasible pharmacologically or with neutralizing antibodies. MAC: Membrane attack complex; STAT3: Signal transducers and activators of transcription 3.
An important consideration, and a possible drawback, in blocking the C5a-C5aR axis is that the block itself might adversely affect the host defense and might counteract some of the useful recently identified functions of complement. Indeed, C5a may protect against neuron apoptosis[134], might act as an inhibitor of angiogenesis[135], and is essential for liver regeneration[136].
C1 INHIBITION
The beneficial effect of C1 inhibition on IRI has been widely studied by Castellano et al[71] and Curci et al[72]. These studies have been recently commented by Carney[73]. Purified or recombinant C1-INH is a host serine protease inhibitor that is able to block complement cascade acting either at level of classical and lectin pathway[137]. The first clinical indication of C1-INH has been hereditary angioedema. To date C1-INH has shown effects in several disease as myocardial ischemia and reperfusion injury[138], renal transplantation[139] and sepsis[140].
To date three clinical trials are ongoing in the field of kidney transplantation. The 2 first clinical trials (NCT01147302 and NCT01134510) have been made for the prevention of acute ABMR adding C1-INH to post-transplant treatment. The investigators observed that no patients developed ABMR during treatment with C1-INH and, in addition, noted a reduction in DGF due to I/R injury. As a consequence, recently a third trial with C1-INH was started (NCT02134314) to prevent DGF in patients receiving deceased donor kidney transplant.
TARGETING C3
In theory, the blockade at the level of C3 may be more effective than the anti C5 therapy, in particular for the C3 glomerulopathies when the C3 convertase activation is prevalent over the C5 convertase. Soluble CR1 (sCR1) is among the proteins that regulate the C3 convertase. CR1 is a cell-surface glycoprotein expressed on several cells among which monocytes, APCs, T and B cells and podocytes. As a consequence sCR1 may modulate the complement cascade on all cells expressing on their surface CR1, follicular dendritic cells and a small T cells population[141-143]. In addition, CR1 is the only co-factor of factor I able to promote cleavage of inactive C3b and inactive C4b into their inactive protein fragments[144]. In normal condition only small quantities of sCR1 are in circulation. Lazar et al[145] and Li et al[146] administered high sCR1 in patients undergoing cardiac surgeries or cardiopulmonary by-pass to inhibit complement activity. These studies documented that sCR1 is effective and safe. sCR1 has been recently used in renal diseases and in renal transplantation.
Recently, Zhang et al[147] from Iowa University reported the results using sCR1 in mice deficient in factor H. sCR1 increases C3 serum levels and decreases C3 deposition. In the same report Zhang et al[147] reported the beneficial effect of treating by sCR1 a young patient affected by ESRD due to DDD. This group is currently enrolling patients for a small phase I trial of sCR1 (also called CDX-1135) in patients with DDD (NCT01791686).
C3 and C5 convertases decay is influenced by CR1. Treatment with sCR1 improved kidney transplant survival after a period of cold storage and when kidneys were transplanted across a complete major histocompatibility complex mismatch[148,149].
The effects of Mirococept (APT070) (sCR1) has been widely described by Sacks et al[69] and is currently the subject of a large scale study in kidney transplantation to test the superiority of Mirococept in the prevention of IRI in cadaveric renal allografts[150].
CONCLUSION
Emerging evidence has recently documented that the complement cascade as a common pathogenetic mechanism in many kidney diseases, in the chronic progression of the kidney diseases and in the kidney transplantation.
This finding led us to an improved understanding of the molecular mechanisms that are at the basis of the kidney diseases and, as a consequence allowed us to formulate a new classification of some renal diseases as the different kinds of hemolytic uremic syndromes and the membranoproliferative GN.
Among the new drugs aimed to target the complement in the renal diseases only the C5 monoclonal antibody, eculizumab, is to date on the market. Others are on clinical trials for the C3 glomerulopathies and the ANCA-mediated GN.
The use of eculizumab in treating the patients affected by aHUS has some limitations and is an example for the need of other drugs targeting the complement cascade. Indeed if eculizumab should be considered for all the patients with aHUS, because the dysregulation at C5 level is largely prevalent in this disease, the C3 glomerulopathies have variable degrees of the C5 convertase dysregulation.
In the cases were the C3 glomerulopathies are associated with a greater C3 convertase dysregulation, the blockade at the level of C3 should be the alternative to the eculizumab treatment.
The complement system is now recognized a pervasive, multifaceted mediator of transplant injury in animal models and in human transplant recipients[151]. The development of pharmacologic agents that block human complement components and receptors in the field of renal transplantation[152,153] now represents the basis of the concept that targeting complement in kidney transplant recipients will improve graft and patient survival rate.
Footnotes
P- Reviewer: Gheith O, Ohashi NA S- Editor: Gong XM L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: The authors declare to have no conflict of interest in relation to the present manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 30, 2014
First decision: December 2, 2014
Article in press: January 19, 2015
References
- 1.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 3.Holst B, Raby AC, Hall JE, Labéta MO. Complement takes its Toll: an inflammatory crosstalk between Toll-like receptors and the receptors for the complement anaphylatoxin C5a. Anaesthesia. 2012;67:60–64. doi: 10.1111/j.1365-2044.2011.07011.x. [DOI] [PubMed] [Google Scholar]
- 4.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 5.Heeringa SF, Cohen CD. Kidney diseases caused by complement dysregulation: acquired, inherited, and still more to come. Clin Dev Immunol. 2012;2012:695131. doi: 10.1155/2012/695131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kościelska-Kasprzak K, Bartoszek D, Myszka M, Zabińska M, Klinger M. The complement cascade and renal disease. Arch Immunol Ther Exp (Warsz) 2014;62:47–57. doi: 10.1007/s00005-013-0254-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCaughan JA, O’Rourke DM, Courtney AE. The complement cascade in kidney disease: from sideline to center stage. Am J Kidney Dis. 2013;62:604–614. doi: 10.1053/j.ajkd.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 9.Wada T, Nangaku M. Novel roles of complement in renal diseases and their therapeutic consequences. Kidney Int. 2013;84:441–450. doi: 10.1038/ki.2013.134. [DOI] [PubMed] [Google Scholar]
- 10.Pickering MC, Walport MJ. Links between complement abnormalities and systemic lupus erythematosus. Rheumatology (Oxford) 2000;39:133–141. doi: 10.1093/rheumatology/39.2.133. [DOI] [PubMed] [Google Scholar]
- 11.Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, Gingras AR, Mantovani A, Hack EC, Roos A. Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol. 2002;32:1726–1736. doi: 10.1002/1521-4141(200206)32:6<1726::AID-IMMU1726>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 12.Seelen MA, Trouw LA, Daha MR. Diagnostic and prognostic significance of anti-C1q antibodies in systemic lupus erythematosus. Curr Opin Nephrol Hypertens. 2003;12:619–624. doi: 10.1097/00041552-200311000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Hu Q, Madri JA, Rollins SA, Chodera A, Matis LA. Amelioration of lupus-like autoimmune disease in NZB/WF1 mice after treatment with a blocking monoclonal antibody specific for complement component C5. Proc Natl Acad Sci USA. 1996;93:8563–8568. doi: 10.1073/pnas.93.16.8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turnberg D, Cook HT. Complement and glomerulonephritis: new insights. Curr Opin Nephrol Hypertens. 2005;14:223–228. doi: 10.1097/01.mnh.0000165887.75501.24. [DOI] [PubMed] [Google Scholar]
- 15.Ma R, Cui Z, Liao YH, Zhao MH. Complement activation contributes to the injury and outcome of kidney in human anti-glomerular basement membrane disease. J Clin Immunol. 2013;33:172–178. doi: 10.1007/s10875-012-9772-2. [DOI] [PubMed] [Google Scholar]
- 16.Chen M, Xing GQ, Yu F, Liu G, Zhao MH. Complement deposition in renal histopathology of patients with ANCA-associated pauci-immune glomerulonephritis. Nephrol Dial Transplant. 2009;24:1247–1252. doi: 10.1093/ndt/gfn586. [DOI] [PubMed] [Google Scholar]
- 17.Xing GQ, Chen M, Liu G, Heeringa P, Zhang JJ, Zheng X, E J, Kallenberg CG, Zhao MH. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–291. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 18.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83:129–137. doi: 10.1038/ki.2012.313. [DOI] [PubMed] [Google Scholar]
- 19.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kallenberg CG, Heeringa P. Complement is crucial in the pathogenesis of ANCA-associated vasculitis. Kidney Int. 2013;83:16–18. doi: 10.1038/ki.2012.371. [DOI] [PubMed] [Google Scholar]
- 21.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, Seitz LC, Penfold ME, Gan L, Hu P, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol. 2014;25:225–231. doi: 10.1681/ASN.2013020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beck LH, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck LH, Salant DJ. Membranous nephropathy: recent travels and new roads ahead. Kidney Int. 2010;77:765–770. doi: 10.1038/ki.2010.34. [DOI] [PubMed] [Google Scholar]
- 24.Lhotta K, Würzner R, König P. Glomerular deposition of mannose-binding lectin in human glomerulonephritis. Nephrol Dial Transplant. 1999;14:881–886. doi: 10.1093/ndt/14.4.881. [DOI] [PubMed] [Google Scholar]
- 25.Val-Bernal JF, Garijo MF, Val D, Rodrigo E, Arias M. C4d immunohistochemical staining is a sensitive method to confirm immunoreactant deposition in formalin-fixed paraffin-embedded tissue in membranous glomerulonephritis. Histol Histopathol. 2011;26:1391–1397. doi: 10.14670/HH-26.1391. [DOI] [PubMed] [Google Scholar]
- 26.Ma H, Sandor DG, Beck LH. The role of complement in membranous nephropathy. Semin Nephrol. 2013;33:531–542. doi: 10.1016/j.semnephrol.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takano T, Elimam H, Cybulsky AV. Complement-mediated cellular injury. Semin Nephrol. 2013;33:586–601. doi: 10.1016/j.semnephrol.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Mii A, Shimizu A, Masuda Y, Fujita E, Aki K, Ishizaki M, Sato S, Griesemer A, Fukuda Y. Current status and issues of C1q nephropathy. Clin Exp Nephrol. 2009;13:263–274. doi: 10.1007/s10157-009-0159-5. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22:1795–1803. doi: 10.1681/ASN.2011050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai KN. Pathogenesis of IgA nephropathy. Nat Rev Nephrol. 2012;8:275–283. doi: 10.1038/nrneph.2012.58. [DOI] [PubMed] [Google Scholar]
- 31.Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001;167:2861–2868. doi: 10.4049/jimmunol.167.5.2861. [DOI] [PubMed] [Google Scholar]
- 32.Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. 2012;8:634–642. doi: 10.1038/nrneph.2012.213. [DOI] [PubMed] [Google Scholar]
- 33.Sethi S, Nester CM, Smith RJ. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int. 2012;81:434–441. doi: 10.1038/ki.2011.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol. 2010;25:1409–1418. doi: 10.1007/s00467-009-1322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. doi: 10.1186/1750-1172-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roumenina LT, Loirat C, Dragon-Durey MA, Halbwachs-Mecarelli L, Sautes-Fridman C, Fremeaux-Bacchi V. Alternative complement pathway assessment in patients with atypical HUS. J Immunol Methods. 2011;365:8–26. doi: 10.1016/j.jim.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Kavanagh D, Goodship TH. Atypical hemolytic uremic syndrome, genetic basis, and clinical manifestations. Hematology Am Soc Hematol Educ Program. 2011;2011:15–20. doi: 10.1182/asheducation-2011.1.15. [DOI] [PubMed] [Google Scholar]
- 38.Skerka C, Józsi M, Zipfel PF, Dragon-Durey MA, Fremeaux-Bacchi V. Autoantibodies in haemolytic uraemic syndrome (HUS) Thromb Haemost. 2009;101:227–232. [PubMed] [Google Scholar]
- 39.Provaznikova D, Rittich S, Malina M, Seeman T, Marinov I, Riedl M, Hrachovinova I. Manifestation of atypical hemolytic uremic syndrome caused by novel mutations in MCP. Pediatr Nephrol. 2012;27:73–81. doi: 10.1007/s00467-011-1943-5. [DOI] [PubMed] [Google Scholar]
- 40.Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, Jablonski M, Renault N, Rameix-Welti MA, Loirat C, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int. 2010;77:339–349. doi: 10.1038/ki.2009.472. [DOI] [PubMed] [Google Scholar]
- 41.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–357. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–633. doi: 10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 43.Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, Holers VM, Lesser M, Kline M, Hoffman C, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4:1920–1924. doi: 10.2215/CJN.02730409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol. 2011;187:172–180. doi: 10.4049/jimmunol.1100491. [DOI] [PubMed] [Google Scholar]
- 45.Zoja C, Locatelli M, Pagani C, Corna D, Zanchi C, Isermann B, Remuzzi G, Conway EM, Noris M. Lack of the lectin-like domain of thrombomodulin worsens Shiga toxin-associated hemolytic uremic syndrome in mice. J Immunol. 2012;189:3661–3668. doi: 10.4049/jimmunol.1102118. [DOI] [PubMed] [Google Scholar]
- 46.Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, Kuhlmann U, Bramstedt J, Panzer U, Vischedyk M, et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104: H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant. 2012;27:3807–3815. doi: 10.1093/ndt/gfs394. [DOI] [PubMed] [Google Scholar]
- 47.Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Hélou S, Bordes C, Reffet A, Llanas B, et al. Outbreak of Escherichia coli O104: H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant. 2014;29:565–572. doi: 10.1093/ndt/gft470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Réti M, Farkas P, Csuka D, Rázsó K, Schlammadinger Á, Udvardy ML, Madách K, Domján G, Bereczki C, Reusz GS, et al. Complement activation in thrombotic thrombocytopenic purpura. J Thromb Haemost. 2012;10:791–798. doi: 10.1111/j.1538-7836.2012.04674.x. [DOI] [PubMed] [Google Scholar]
- 49.Chapin J, Weksler B, Magro C, Laurence J. Eculizumab in the treatment of refractory idiopathic thrombotic thrombocytopenic purpura. Br J Haematol. 2012;157:772–774. doi: 10.1111/j.1365-2141.2012.09084.x. [DOI] [PubMed] [Google Scholar]
- 50.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barbour TD, Pickering MC, Cook HT. Recent insights into C3 glomerulopathy. Nephrol Dial Transplant. 2013;28:1685–1693. doi: 10.1093/ndt/gfs430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daha MR, Fearon DT, Austen KF. C3 nephritic factor (C3NeF): stabilization of fluid phase and cell-bound alternative pathway convertase. J Immunol. 1976;116:1–7. [PubMed] [Google Scholar]
- 53.Chen Q, Müller D, Rudolph B, Hartmann A, Kuwertz-Bröking E, Wu K, Kirschfink M, Skerka C, Zipfel PF. Combined C3b and factor B autoantibodies and MPGN type II. N Engl J Med. 2011;365:2340–2342. doi: 10.1056/NEJMc1107484. [DOI] [PubMed] [Google Scholar]
- 54.Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S. Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol. 1999;163:4590–4596. [PubMed] [Google Scholar]
- 55.Sethi S, Fervenza FC, Zhang Y, Nasr SH, Leung N, Vrana J, Cramer C, Nester CM, Smith RJ. Proliferative glomerulonephritis secondary to dysfunction of the alternative pathway of complement. Clin J Am Soc Nephrol. 2011;6:1009–1017. doi: 10.2215/CJN.07110810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, Grünfeld JP, Lesavre P, Noël LH, Fakhouri F. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44:193–199. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 58.Barbour T, Johnson S, Cohney S, Hughes P. Thrombotic microangiopathy and associated renal disorders. Nephrol Dial Transplant. 2012;27:2673–2685. doi: 10.1093/ndt/gfs279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malik TH, Lavin PJ, Goicoechea de Jorge E, Vernon KA, Rose KL, Patel MP, de Leeuw M, Neary JJ, Conlon PJ, Winn MP, et al. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol. 2012;23:1155–1160. doi: 10.1681/ASN.2012020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodríguez-Iturbe B, García García G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin Pract. 2010;116:c81–c88. doi: 10.1159/000314656. [DOI] [PubMed] [Google Scholar]
- 62.Gaarkeuken H, Siezenga MA, Zuidwijk K, van Kooten C, Rabelink TJ, Daha MR, Berger SP. Complement activation by tubular cells is mediated by properdin binding. Am J Physiol Renal Physiol. 2008;295:F1397–F1403. doi: 10.1152/ajprenal.90313.2008. [DOI] [PubMed] [Google Scholar]
- 63.Siezenga MA, van der Geest RN, Mallat MJ, Rabelink TJ, Daha MR, Berger SP. Urinary properdin excretion is associated with intrarenal complement activation and poor renal function. Nephrol Dial Transplant. 2010;25:1157–1161. doi: 10.1093/ndt/gfp630. [DOI] [PubMed] [Google Scholar]
- 64.Bao L, Wang Y, Haas M, Quigg RJ. Distinct roles for C3a and C5a in complement-induced tubulointerstitial injury. Kidney Int. 2011;80:524–534. doi: 10.1038/ki.2011.158. [DOI] [PubMed] [Google Scholar]
- 65.Bao L, Wang Y, Chang A, Minto AW, Zhou J, Kang H, Haas M, Quigg RJ. Unrestricted C3 activation occurs in Crry-deficient kidneys and rapidly leads to chronic renal failure. J Am Soc Nephrol. 2007;18:811–822. doi: 10.1681/ASN.2006101176. [DOI] [PubMed] [Google Scholar]
- 66.Pratt JR, Abe K, Miyazaki M, Zhou W, Sacks SH. In situ localization of C3 synthesis in experimental acute renal allograft rejection. Am J Pathol. 2000;157:825–831. doi: 10.1016/S0002-9440(10)64596-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Møller-Kristensen M, Wang W, Ruseva M, Thiel S, Nielsen S, Takahashi K, Shi L, Ezekowitz A, Jensenius JC, Gadjeva M. Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol. 2005;61:426–434. doi: 10.1111/j.1365-3083.2005.01591.x. [DOI] [PubMed] [Google Scholar]
- 68.Damman J, Daha MR, van Son WJ, Leuvenink HG, Ploeg RJ, Seelen MA. Crosstalk between complement and Toll-like receptor activation in relation to donor brain death and renal ischemia-reperfusion injury. Am J Transplant. 2011;11:660–669. doi: 10.1111/j.1600-6143.2011.03475.x. [DOI] [PubMed] [Google Scholar]
- 69.Sacks S, Karegli J, Farrar CA, Asgari E, Schwaeble W, Zhou W, Smith RA. Targeting complement at the time of transplantation. Adv Exp Med Biol. 2013;735:247–255. doi: 10.1007/978-1-4614-4118-2_17. [DOI] [PubMed] [Google Scholar]
- 70.Simone S, Rascio F, Castellano G, Divella C, Chieti A, Ditonno P, Battaglia M, Crovace A, Staffieri F, Oortwijn B, et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic Biol Med. 2014;74:263–273. doi: 10.1016/j.freeradbiomed.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 71.Castellano G, Melchiorre R, Loverre A, Ditonno P, Montinaro V, Rossini M, Divella C, Battaglia M, Lucarelli G, Annunziata G, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176:1648–1659. doi: 10.2353/ajpath.2010.090276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Curci C, Castellano G, Stasi A, Divella C, Loverre A, Gigante M, Simone S, Cariello M, Montinaro V, Lucarelli G, et al. Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol Dial Transplant. 2014;29:799–808. doi: 10.1093/ndt/gft516. [DOI] [PubMed] [Google Scholar]
- 73.Carney EF. Acute kidney injury: critical role of complement in EndMT. Nat Rev Nephrol. 2014;10:183. doi: 10.1038/nrneph.2014.19. [DOI] [PubMed] [Google Scholar]
- 74.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8:582–587. doi: 10.1038/nm0602-582. [DOI] [PubMed] [Google Scholar]
- 75.Fuquay R, Renner B, Kulik L, McCullough JW, Amura C, Strassheim D, Pelanda R, Torres R, Thurman JM. Renal ischemia-reperfusion injury amplifies the humoral immune response. J Am Soc Nephrol. 2013;24:1063–1072. doi: 10.1681/ASN.2012060560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li K, Patel H, Farrar CA, Hargreaves RE, Sacks SH, Zhou W. Complement activation regulates the capacity of proximal tubular epithelial cell to stimulate alloreactive T cell response. J Am Soc Nephrol. 2004;15:2414–2422. doi: 10.1097/01.ASN.0000135974.06478.7B. [DOI] [PubMed] [Google Scholar]
- 77.Peng Q, Li K, Patel H, Sacks SH, Zhou W. Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J Immunol. 2006;176:3330–3341. doi: 10.4049/jimmunol.176.6.3330. [DOI] [PubMed] [Google Scholar]
- 78.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting edge: Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol. 2013;190:5921–5925. doi: 10.4049/jimmunol.1300847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Q, Peng Q, Xing G, Li K, Wang N, Farrar CA, Meader L, Sacks SH, Zhou W. Deficiency of C5aR prolongs renal allograft survival. J Am Soc Nephrol. 2010;21:1344–1353. doi: 10.1681/ASN.2009090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Castellano G, Woltman AM, Schlagwein N, Xu W, Schena FP, Daha MR, van Kooten C. Immune modulation of human dendritic cells by complement. Eur J Immunol. 2007;37:2803–2811. doi: 10.1002/eji.200636845. [DOI] [PubMed] [Google Scholar]
- 83.Gueler F, Rong S, Gwinner W, Mengel M, Bröcker V, Schön S, Greten TF, Hawlisch H, Polakowski T, Schnatbaum K, et al. Complement 5a receptor inhibition improves renal allograft survival. J Am Soc Nephrol. 2008;19:2302–2312. doi: 10.1681/ASN.2007111267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin T, Zhou W, Farrar CA, Hargreaves RE, Sheerin NS, Sacks SH. Deficiency of C4 from donor or recipient mouse fails to prevent renal allograft rejection. Am J Pathol. 2006;168:1241–1248. doi: 10.2353/ajpath.2006.050360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol. 2012;8:670–678. doi: 10.1038/nrneph.2012.212. [DOI] [PubMed] [Google Scholar]
- 86.Haidar F, Kisserli A, Tabary T, McGregor B, Noel LH, Réveil B, Toupance O, Rieu P, Thervet E, Legendre C, et al. Comparison of C4d detection on erythrocytes and PTC-C4d to histological signs of antibody-mediated rejection in kidney transplantation. Am J Transplant. 2012;12:1564–1575. doi: 10.1111/j.1600-6143.2012.04003.x. [DOI] [PubMed] [Google Scholar]
- 87.Brodsky SV, Nadasdy GM, Pelletier R, Satoskar A, Birmingham DJ, Hadley GA, Obeidat K, Nadasdy T. Expression of the decay-accelerating factor (CD55) in renal transplants--a possible prediction marker of allograft survival. Transplantation. 2009;88:457–464. doi: 10.1097/TP.0b013e3181b0517d. [DOI] [PubMed] [Google Scholar]
- 88.Zuber J, Le Quintrec M, Morris H, Frémeaux-Bacchi V, Loirat C, Legendre C. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev (Orlando) 2013;27:117–125. doi: 10.1016/j.trre.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 89.Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. 2010;10:1517–1523. doi: 10.1111/j.1600-6143.2010.03156.x. [DOI] [PubMed] [Google Scholar]
- 90.Mella A, Messina M, Lavacca A, Biancone L. Complement cascade and kidney transplantation: The rediscovery of an ancient enemy. World J Transplant. 2014;4:168–175. doi: 10.5500/wjt.v4.i3.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Little MA, Dupont P, Campbell E, Dorman A, Walshe JJ. Severity of primary MPGN, rather than MPGN type, determines renal survival and post-transplantation recurrence risk. Kidney Int. 2006;69:504–511. doi: 10.1038/sj.ki.5000084. [DOI] [PubMed] [Google Scholar]
- 92.Zand L, Lorenz EC, Cosio FG, Fervenza FC, Nasr SH, Gandhi MJ, Smith RJ, Sethi S. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol. 2014;25:1110–1117. doi: 10.1681/ASN.2013070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG, Kaplan B, Halloran PF. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. 2009;9:2520–2531. doi: 10.1111/j.1600-6143.2009.02799.x. [DOI] [PubMed] [Google Scholar]
- 94.Sis B, Mengel M, Haas M, Colvin RB, Halloran PF, Racusen LC, Solez K, Baldwin WM, Bracamonte ER, Broecker V, et al. Banff ‘09 meeting report: antibody mediated graft deterioration and implementation of Banff working groups. Am J Transplant. 2010;10:464–471. doi: 10.1111/j.1600-6143.2009.02987.x. [DOI] [PubMed] [Google Scholar]
- 95.Kulkarni PA, Afshar-Kharghan V. Anticomplement therapy. Biologics. 2008;2:671–685. doi: 10.2147/btt.s2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Makrides SC. Therapeutic inhibition of the complement system. Pharmacol Rev. 1998;50:59–87. [PubMed] [Google Scholar]
- 97.Mollnes TE, Kirschfink M. Strategies of therapeutic complement inhibition. Mol Immunol. 2006;43:107–121. doi: 10.1016/j.molimm.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 98.Bomback AS. Anti-complement therapy for glomerular diseases. Adv Chronic Kidney Dis. 2014;21:152–158. doi: 10.1053/j.ackd.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 99.Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, Röth A, Szer J, Elebute MO, Nakamura R, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355:1233–1243. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- 100.Hillmen P ClinicalTrials. gov. Available from: https: //www.clinicaltrials.gov.
- 101.Westra D, Wetzels JF, Volokhina EB, van den Heuvel LP, van de Kar NC. A new era in the diagnosis and treatment of atypical haemolytic uraemic syndrome. Neth J Med. 2012;70:121–129. [PubMed] [Google Scholar]
- 102.Ariceta G, Arrizabalaga B, Aguirre M, Morteruel E, Lopez-Trascasa M. Eculizumab in the treatment of atypical hemolytic uremic syndrome in infants. Am J Kidney Dis. 2012;59:707–710. doi: 10.1053/j.ajkd.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 103.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 104.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 105.Ring T. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369:1377–1378. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 106.Amadio A, Tejani AM. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369:1378. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 107.Kistler AD. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369:1378. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 108.Tanimoto T, Oshima Y, Kami M. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369:1378–1379. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 109.Razzak M. Anaemia: mutations in C5 explain eculizumab resistance. Nat Rev Nephrol. 2014;10:182. doi: 10.1038/nrneph.2014.30. [DOI] [PubMed] [Google Scholar]
- 110.Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370:632–639. doi: 10.1056/NEJMoa1311084. [DOI] [PubMed] [Google Scholar]
- 111.Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364:2561–2563. doi: 10.1056/NEJMc1100859. [DOI] [PubMed] [Google Scholar]
- 112.Tsai E, Chapin J, Laurence JC, Tsai HM. Use of eculizumab in the treatment of a case of refractory, ADAMTS13-deficient thrombotic thrombocytopenic purpura: additional data and clinical follow-up. Br J Haematol. 2013;162:558–559. doi: 10.1111/bjh.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scully M, Goodship T. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol. 2014;164:759–766. doi: 10.1111/bjh.12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Radhakrishnan S, Lunn A, Kirschfink M, Thorner P, Hebert D, Langlois V, Pluthero F, Licht C. Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med. 2012;366:1165–1166. doi: 10.1056/NEJMc1106619. [DOI] [PubMed] [Google Scholar]
- 115.Vivarelli M, Pasini A, Emma F. Eculizumab for the treatment of dense-deposit disease. N Engl J Med. 2012;366:1163–1165. doi: 10.1056/NEJMc1111953. [DOI] [PubMed] [Google Scholar]
- 116.Daina E, Noris M, Remuzzi G. Eculizumab in a patient with dense-deposit disease. N Engl J Med. 2012;366:1161–1163. doi: 10.1056/NEJMc1112273. [DOI] [PubMed] [Google Scholar]
- 117.McCaughan JA, O’Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant. 2012;12:1046–1051. doi: 10.1111/j.1600-6143.2011.03923.x. [DOI] [PubMed] [Google Scholar]
- 118.Kerns E, Rozansky D, Troxell ML. Evolution of immunoglobulin deposition in C3-dominant membranoproliferative glomerulopathy. Pediatr Nephrol. 2013;28:2227–2231. doi: 10.1007/s00467-013-2565-x. [DOI] [PubMed] [Google Scholar]
- 119.Gurkan S, Fyfe B, Weiss L, Xiao X, Zhang Y, Smith RJ. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol. 2013;28:1975–1981. doi: 10.1007/s00467-013-2503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D‘Agati VD, Canetta PA, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7:748–756. doi: 10.2215/CJN.12901211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, Appel GB, D‘Agati VD. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol. 2012;23:1229–1237. doi: 10.1681/ASN.2011121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nester CM, Smith RJ. Treatment options for C3 glomerulopathy. Curr Opin Nephrol Hypertens. 2013;22:231–237. doi: 10.1097/MNH.0b013e32835da24c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, Heyne N, Ardissino G, Chatelet V, Noël LH, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12:3337–3354. doi: 10.1111/j.1600-6143.2012.04252.x. [DOI] [PubMed] [Google Scholar]
- 125.Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, Singer AL, Carter-Monroe N, Nazarian SM, Segev DL, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459–465. doi: 10.1111/ajt.12540. [DOI] [PubMed] [Google Scholar]
- 126.Lonze BE, Dagher NN, Simpkins CE, Locke JE, Singer AL, Segev DL, Zachary AA, Montgomery RA. Eculizumab, bortezomib and kidney paired donation facilitate transplantation of a highly sensitized patient without vascular access. Am J Transplant. 2010;10:2154–2160. doi: 10.1111/j.1600-6143.2010.03191.x. [DOI] [PubMed] [Google Scholar]
- 127.Cohney SJ, Hughes P, Rosemary M, Walker RG, Cantwell L, Vanhardeveld E, Blecker K, Finlay M, Landgren A, Murugasu A. C5 inhibition with eculizumab to prevent antibody mediated rejection (AbMR) in patients with donor specific anti-HLA antibody (DSAb) and a positive cross match. Am J Transplant. 2011;11:483. [Google Scholar]
- 128.Hardinger KL, Brennan DC. Novel immunosuppressive agents in kidney transplantation. World J Transplant. 2013;3:68–77. doi: 10.5500/wjt.v3.i4.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, Cosio FG, Gandhi MJ, Kremers W, Gloor JM. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]
- 130.Lewis AG, Köhl G, Ma Q, Devarajan P, Köhl J. Pharmacological targeting of C5a receptors during organ preservation improves kidney graft survival. Clin Exp Immunol. 2008;153:117–126. doi: 10.1111/j.1365-2249.2008.03678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant. 2013;13:2530–2539. doi: 10.1111/ajt.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–1642. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 133.Li R, Coulthard LG, Wu MC, Taylor SM, Woodruff TM. C5L2: a controversial receptor of complement anaphylatoxin, C5a. FASEB J. 2013;27:855–864. doi: 10.1096/fj.12-220509. [DOI] [PubMed] [Google Scholar]
- 134.Mukherjee P, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem. 2001;77:43–49. doi: 10.1046/j.1471-4159.2001.00167.x. [DOI] [PubMed] [Google Scholar]
- 135.Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, Alatsatianos M, DeAngelis RA, Roche PA, Magotti P, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010;116:4395–4403. doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–923. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol. 2013;190:3839–3847. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lu F, Fernandes SM, Davis AE. The effect of C1 inhibitor on myocardial ischemia and reperfusion injury. Cardiovasc Pathol. 2013;22:75–80. doi: 10.1016/j.carpath.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tillou X, Poirier N, Le Bas-Bernardet S, Hervouet J, Minault D, Renaudin K, Vistoli F, Karam G, Daha M, Soulillou JP, et al. Recombinant human C1-inhibitor prevents acute antibody-mediated rejection in alloimmunized baboons. Kidney Int. 2010;78:152–159. doi: 10.1038/ki.2010.75. [DOI] [PubMed] [Google Scholar]
- 140.Igonin AA, Protsenko DN, Galstyan GM, Vlasenko AV, Khachatryan NN, Nekhaev IV, Shlyapnikov SA, Lazareva NB, Herscu P. C1-esterase inhibitor infusion increases survival rates for patients with sepsis. Crit Care Med. 2012;40:770–777. doi: 10.1097/CCM.0b013e318236edb8. [DOI] [PubMed] [Google Scholar]
- 141.Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol. 1998;160:5273–5279. [PubMed] [Google Scholar]
- 142.Rødgaard A, Christensen LD, Thomsen BS, Wiik A, Bendixen G. Complement receptor type 1 (CR1, CD35) expression on peripheral T lymphocytes: both CD4- and CD8-positive cells express CR1. Complement Inflamm. 1991;8:303–309. doi: 10.1159/000463200. [DOI] [PubMed] [Google Scholar]
- 143.Weiss L, Fischer E, Haeffner-Cavaillon N, Jouvin MH, Appay MD, Bariety J, Kazatchkine M. The human C3b receptor (CR1) Adv Nephrol Necker Hosp. 1989;18:249–269. [PubMed] [Google Scholar]
- 144.Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, Kavanagh D, Mertens HD, Svergun DI, Johansson CM, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. 2011;18:463–470. doi: 10.1038/nsmb.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lazar HL, Keilani T, Fitzgerald CA, Shapira OM, Hunter CT, Shemin RJ, Marsh HC, Ryan US. Beneficial effects of complement inhibition with soluble complement receptor 1 (TP10) during cardiac surgery: is there a gender difference? Circulation. 2007;116:I83–I88. doi: 10.1161/CIRCULATIONAHA.106.677914. [DOI] [PubMed] [Google Scholar]
- 146.Li JS, Sanders SP, Perry AE, Stinnett SS, Jaggers J, Bokesch P, Reynolds L, Nassar R, Anderson PA. Pharmacokinetics and safety of TP10, soluble complement receptor 1, in infants undergoing cardiopulmonary bypass. Am Heart J. 2004;147:173–180. doi: 10.1016/j.ahj.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 147.Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, Meyer NC, Hunsicker LG, Sethi S, Smith RJ. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–1829. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Patel H, Smith RA, Sacks SH, Zhou W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol. 2006;17:1102–1111. doi: 10.1681/ASN.2005101116. [DOI] [PubMed] [Google Scholar]
- 149.Pratt JR, Hibbs MJ, Laver AJ, Smith RA, Sacks SH. Allograft immune response with sCR1 intervention. Transpl Immunol. 1996;4:72–75. doi: 10.1016/s0966-3274(96)80041-4. [DOI] [PubMed] [Google Scholar]
- 150. Available from: http: //www.controlled-trials.com/ISRCTN49958194 [DOI 10.1186/ISRCTN49958194]
- 151.Cravedi P, Heeger PS. Complement as a multifaceted modulator of kidney transplant injury. J Clin Invest. 2014;124:2348–2354. doi: 10.1172/JCI72273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Banz Y, Rieben R. Role of complement and perspectives for intervention in ischemia-reperfusion damage. Ann Med. 2012;44:205–217. doi: 10.3109/07853890.2010.535556. [DOI] [PubMed] [Google Scholar]
- 153.Chen G, Chen S, Chen X. Role of complement and perspectives for intervention in transplantation. Immunobiology. 2013;218:817–827. doi: 10.1016/j.imbio.2012.09.002. [DOI] [PubMed] [Google Scholar]