

Abstract
Haematuria was known as a benign hallmark of some glomerular diseases, but over the last decade, new evidences pointed its negative implications on kidney disease progression. Cytotoxic effects of oxidative stress induced by hemoglobin, heme, or iron released from red blood cells may account for the tubular injury observed in human biopsy specimens. However, the precise mechanisms responsible for haematuria remain unclear. The presence of red blood cells (RBCs) with irregular contours and shape in the urine indicates RBCs egression from the glomerular capillary into the urinary space. Therefore glomerular haematuria may be a marker of glomerular filtration barrier dysfunction or damage. In this review we describe some key issues regarding epidemiology and pathogenesis of haematuric diseases as well as their renal morphological findings.
Keywords: Haematuria, Pathogenesis, Glomerular filtration barrier, Dysmorphic red blood cells, Chronic kidney disease, Microscopic haematuria
Core tip: Recent advances suggest that glomerular haematuria may be a negative prognostic factor for renal function outcome. A more fragile and easily ruptured glomerular filtration barrier (GFB) may be responsible for glomerular bleeding. Several factors have been associated to this pathogenic process, including: (1) genetic alteration of GFB components, leading to a more fragile and easily ruptured GFB structure; (2) aberrant deposition of toxic molecules in the GFB; and (3) enhanced inflammatory response, as reported in autoimmune diseases, infections, or primary glomerulonephritis. In this review we fully describe these pathological mechanisms, with special interest in haematuric diseases and their renal morphological findings.
INTRODUCTION
Haematuria is a common presenting feature of renal and urological diseases. It is described as the presence of more than 2 red blood cells (RBCs) per high-power field in the urine sediment. When the presence of RBCs in the urine is massive, the urine colour is red and is called macroscopic haematuria. Microscopic haematuria (MH) is detected by microscopical examination or dipstick, so its real incidence is unknown[1,2]. According with its origin haematuria can be glomerular or non-glomerular, however in this review we will focus exclusively on glomerular haematuria due to its implications in renal prognosis. The precise pathogenic mechanisms responsible of glomerular haematuria remain unclear. However, the identification of the specific molecular defect responsible of different genetic disorders commonly associated with haematuria has highlighted possible mechanisms. These genetic diseases originate glomerular filtration barrier (GFB) damage, leading to a more fragile and easily ruptured structure. Sometimes by directly alteration of the glomerular basement membrane (GBM) [as reported in Alport syndrome (AS), thin basement membrane nephropathy (TBMN) or hereditary angiopathy, nephropathy, aneurysms, and muscle cramps (HANAC) syndrome] or podocyte structure [Myosin heavy chain 9 (MYH9)-associated kidney disease], and others by aberrant deposition of toxic compounds like in storage disorders (Fibronectin glomerulopathy, Immunotactoid and Fibrillary glomerulonephritis). Inherit genetic mutations can also lead in an abnormal regulation of the complement alternative pathway and therefore C3 glomerular deposition [C3 glomerulopathies as complement factor H-related protein 5 (CFHR5) nephropathy or dense deposit disease], inducing a potent inflammatory response that results in phagocyte chemotaxis, with opsonization and lysis of cells which can easily explain haematuria. Haematuria can also be produced by inflammatory status as reported in autoimmune diseases [anti-neutrophil cytoplasmic antibodies (ANCA), Vasculitis, GBM disease, systemic lupus erithematosus or Cryoglobulinemia], infections (Endocapillary glomerulonephritis), or primary glomerulonephritis [IgA Nephropathy (IgAN), Membranoproliferative, Crescentic] (Table 1). In this review we will describe the molecular mechanisms responsible for histopathological findings in these diseases in order to explain the pathogenesis of haematuria and their relation with renal outcome.
Table 1.
Classification of haematuric diseases by histopathological findings
| Glomerular endothelial cell layer | GBM disorders | Mesangial deposits | Podocytary slit diaphragm disorders | Subendothelial/subepithelial deposit | Others |
| ANCA | Primary | IgAN | MYH9 disease | Primary GN | WRN |
| Endocapillary | Alport | HSP | Fabry disease | MBP | SCD |
| TBMD | Endocapillary | ||||
| HANAC | Crescentic | ||||
| ? LPHS | |||||
| Secondary GN | |||||
| Secondary | SLE | ||||
| Anti-GBM disease | Cryoglobulinemia | ||||
| C3 glomerulopathy | |||||
| CFHR5 nephropathy | Fibrillar deposit | ||||
| Fibronectin | |||||
| Fibrillary | |||||
| Immunotactoid |
GBM: Glomerular basement membrane; ANCA: Antineutrophil cytoplasmic antibodies; CFHR5: Complement factor H–related 5 nephropathy; GN: Glomerulonephritis; HANAC: Hereditary angiopathy, nephropathy, aneurysms, and muscle cramps syndrome; IgAN: IgA nephropathy; LPHS: Loin back pain syndrome; MBP: Membranoproliferative; SCD: Sickle cell disease; HSP: Henoch-Schönlein purpura; SLE: Systemic lupus erythematous; TBMD: Thin basement membrane disease; WRN: Warfarin related nephropathy.
PROGNOSTIC VALUE OF HAEMATURIA
Haematuria has traditionally been considered as a hallmark of some glomerular diseases, without repercussion on short and long-term kidney function[3]. However, over the last decade new evidences reported negative prognostic implications for both microscopic[4] and macroscopic haematuria[5] on the progression of renal disease. Thus, Vivante et al[4] reported that persistent asymptomatic isolated microscopic haematuria in 1 million young Israeli adults was significantly associated with increased risk of end stage renal disease (ESRD) after 22 years of follow up. Moreover, persistent glomerular hematuria in kidney donors has been associated with an increased risk of proteinuria and kidney disease progression at 2.3 years after donation[6].
Acute kidney injury (AKI) is a common complication of severe macroscopic haematuria, with an incidence of around 30% in IgAN patients with gross macrohaematuria bouts[7,8] and around 20% in warfarin-related nephropathy (WRN)[9]. Gutiérrez et al[5] reported that around 25% of IgAN patients did not recover baseline serum creatinine after cessation of macroscopic haematuria-associated AKI. In this study, duration of macroscopic bout was the more important prognostic factor determining incomplete recovery of renal function. Similarly in CFHR5-nephropathy almost all male patients who reached ESRD had episodes of macroscopic haematuria episodes after upper respiratory tract infections in childhood and adolescence[10].
The most important renal guidelines, Kidney Disease Outcomes Quality Initiative and Kidney Disease: Improving Global Outcome give a contradictory advice about haematuria management. These guidelines recommend to assess every chronic kidney disease (CKD) patient with dipstick[11,12], but haematuria is not recognized as a risk factor of CKD progression, and not recommend further monitoring or treatment in glomerulonephritis patients with isolated microscopic haematuria[13]. However they recognize that IgAN with haematuria and minimal proteinuria is a progressive disease[14], indicating that although clinical outcome for many haematuric patients is good, the lifetime risk for CKD patients may be elevated depending on the specific underlying disease.
CONSEQUENCES OF GLOMERULAR HAEMATURIA
Clinical data and basic research evidences suggest that haematuria induces renal damage. Acute tubular necrosis and intraluminal obstructive RBC casts are the most characteristic histologic findings in AKI during macroscopic hematuria. The principal mechanism of damage is the direct tubular toxicity of hemoglobin (Hb), heme, iron, or other molecules released from RBCs. It has been proposed that RBC passage throughout the GFB induces distortion of erythrocyte cytoskeleton, which is unable to maintain the cellular integrity, leading to RBC rupture. As consequence, the toxic molecules normally lock inside RBC’s cytoplasm, such as Hb, heme, or iron, are released into the urinary space.
Hb is internalized into the epithelial tubular cell by the megalin/cubilin complex. Hb under the epithelial cell oxidant conditions dissociates into heme and globin. Heme oxigenase-1 (HO-1) catalyzes the conversion of heme to biliverdin, iron and carbon monoxide[15]. At that time, the bilirubin reductase converts biliverdin in bilirubin and the iron is stored as Ferritin (Figure 1). HO-1 is now recognized as a protective molecule with anti-oxidant and anti-inflammatory properties against diverse insults in different tissues[16].
Figure 1.
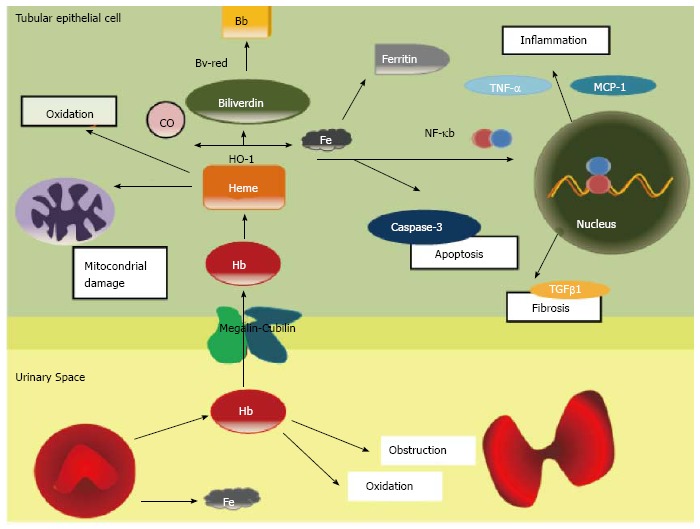
Haematuria-induced kidney injury in tubular cells. Hb: Haemoglobin; Bb: Bilirubin; Bv-red: Biliverdin reductase; CO: Carbon monoxide; Fe: Iron; HO-1: Heme oxygenase 1; MCP: Monocyte chemoattractant protein; NF-κb: Nuclear factor kappa b; TGF-β: Transforming growth factor beta; TNF-α: Tumour necrosis factor alpha.
Free heme is also extremely toxic. In plasma and intracellular membranes, heme can oxidize lipids, denature proteins and perturb the cellular integrity[17]. In large amounts, heme may be a source of iron that drives oxidant injury after hypoxic and nephrotoxic insults[18]. Indirectly heme can also induce renal injury by its proinflammatory effects, as inducing the chemokines such as monocyte chemoattractant protein-1 throughout the redox-sensitive transcription factor NF-KB[15]. The heme group of haemoglobin may also decrease nitric oxide availability, promoting intrarenal vasoconstriction and ischemia[19]. Finally, another possible mechanism involved in haematuria damage may be associated to a delayed dysmorphic RBC’s elimination, which may explain the prolonged recovery period in patient with macrohaematuria induced AKI.
PATHOGENESIS OF HAEMATURIA
The presence of dysmorphic RBCs with irregular contours and shape in the urine is almost pathognomonic of glomerular haematuria[20] and indicates RBCs egression from the glomerular capillary into the urinary space. Therefore glomerular haematuria is a marker of the GFB dysfunction or damage[21].
GFB is an extremely complex and specialized structure[22,23], with different constituents and cell types, which allows a free permeability to water, small and midsized plasma solutes, but keeps a highly specialized selectivity for proteins and larger molecules according with size and molecular weight[24]. GFB has five major components: (1) from the vascular side, the endothelial surface layer, a complex glycosaminoglycan net which cover the endothelial layer as well as the fenestrations; (2) the endothelial cell; (3) the GBM; (4) podocytes with its interdigitating foot processes and specialized intercellular junctions, the “slit diaphragms”; and (5) finally on the urinary side, the subpodocyte space, an area delimited between the podocyte cell body and the foot processes (Figure 2). Furthermore, mesangial cell also indirectly contributes to GFB structure regulating and supporting the blood flow and the glomerular capillary structure, as well as controlling the mesangial matrix turnover (Figure 2). The GFB integrity is maintained by a complex interplay of signaling interactions between the three constituent cell types[24-26].
Figure 2.
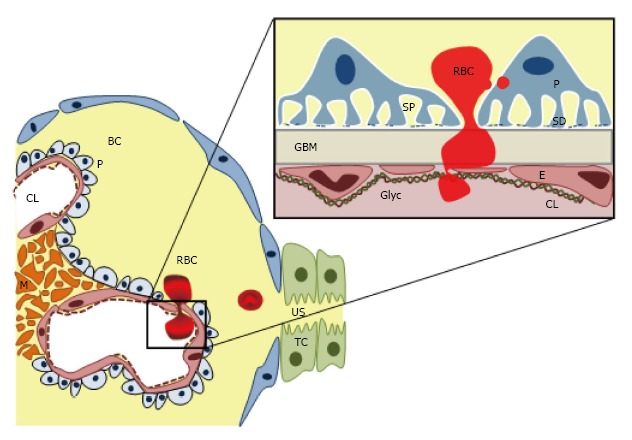
Glomerular filtration barrier structure and red blood cell egression leading to haematuria. CL: Capillary lumen; BC: Bowman’s capsule; E: Endothelial cell; GBM: Glomerular basement membrane; Gly: Glycosaminoglicans; M: Mesangium; P: Podocyte; RBC: Red blood cell; SD: Slit diaphragm; SP: Subpodocyte space; TC: Tubular cell; US: Urinary space.
It has been thought that under physiological conditions, the endothelium with its fenestrations (50-100 nm) acts as molecular size sieve, self-sufficient to maintain the RBCs (6.2-8.2 μm) away from the GBM. However, haematuria in some diseases such as TBMN, Fibrillar deposit diseases or MYH9-associated kidney disease with typically intact endothelium, highlighted the key integrative role of GFB complex as a RBCs sieve. Therefore, how the RBCs, 100-fold bigger than the glomerular endothelium’s pore, cross the GFB remains unclear. It is possible that a damaged GFB layer may release inflammatory or chemotactic signals promoting RBC passage throughout this layer, however the specific mechanisms have not yet found out.
According to its primary and histopathologic localization the haematuric disorders may be classified into: (1) Glomerular endothelial cell and surface layer injuries; (2) primary and secondary GBM disorders; (3) Diseases with mesangial deposition; (4) Diseases with subendothelial and subepithelial deposition; (5) Podocyte-associated disorders; and (6) Miscellaneous (Table 2).
Table 2.
Possible pathogenic mechanisms of haematuria
| Disease | Molecular defect | Prevalence | Main glomerular defect |
Clinical expression |
||
| Haematuria | Proteinuria | CKD progression | ||||
| Genetic disorder | ||||||
| GFB structural damage | ||||||
| Structural GBM damage | ||||||
| ALPORT | X-linked: COL4A5 AR: COL4A3/COL4A4 | 1/50000 | GBM | MH | Variable | 100% approximately 20-30 yr |
| TBMD | COL4A3/COL4A4 | 1% | GBM | MH | Usually absent | 20% CKD |
| HANAC | COL4A1 | 3 families | GBM | MH or gross | Not described | Variable |
| Structural podocyte damage | ||||||
| MYH9 | Non muscle myosin IIA heavy chain | 1:100000 | None | MH | Variable | ESRD by young adulthood |
| Storage disorders | ||||||
| Fibronectine GN | Fibronectin | 44 cases | Mesangial/subendoth | 60% MH | 93% variable degree | ESRD at 20-60 yr |
| Fibrillary | 10-30 nm fibrils | Rare | Mesangial /GBM | MH 47%-73% Gross 5% | Present 41%-55% nephrotic | 50% ESRD in few years |
| Immunotactoid | > 30 nm fibrils | 10-fold rarer than FGN | Mesangial/subepith/subendoth | MH 80% | 100% | 17% ESRD in 3 yr |
| Fabry’s disease | Lysosomal storage | 1:3100- 1:1600 | All the cells | MH | Usually nephrotic | ESRD after age 50 yr |
| Complement mediated | ||||||
| C3 glomerulopathy | Alternative pathway | 1-2 × 106 | Mesangial/GBM | MH 87% | 38% | Variable |
| Inflammatory disorders | ||||||
| Autoimmune | ||||||
| ANCA | Ab vs endothelium | 10-20 × 106 | Endothelium | MH | Variable | Variable |
| Anti GBM | Ab vs COL4 | 0.5-1 × 106/yr | GBM | MH | Variable | Variable |
| Infections (endocapillary) | ||||||
| Primary GN (IgAN, membranoproliferative, crescentic) | ||||||
| IgAN | Galactose-deficient IgA1 | 10%-16% | Mesangial | MH always 75% gross | Rare nephrotic Usual proteinuria | 20% ESRD 20 yr after diagnosis |
| Miscellaneous | ||||||
| WRN | Unknown | 16.5% non-CKD 33% CKD | None | Usually MH | None | Accelerated CKD progression |
| LPHS | Unknown | Unknown | GBM (?) | MH or gross | Absent or minimal | GFR > 60 |
ANCA: Antineutrophil cytoplasmic antibodies; AR: Autosomal recesive; CKD: Chronic kidney disease; COL4A1: Alpha 1 chain of type IV collagen; COL4A3: Alpha 3 chain of type IV collagen; COL4A4: Alpha 4 chain of type IV collagen; COL4A1: Alpha 1 chain of type IV collagen; ESRD: End stage renal disease; GFB: Glomerular filtration barrier; GFR: Glomerular filtration rate; HANAC: Hereditary angiopathy, nephropathy, aneurysms, and muscle cramps syndrome; IgAN: IgA nephropathy; LPHS: Loin back pain haematuria syndrome; MH: Microscopic haematuria; TBMD: Thin basement membrane disease; WRN: Warfarin related nephropathy.
Glomerular endothelial cell and surface layer injuries
In spite of the relatively big size of glomerular endothelial fenestrations, they play an important role in GFB perm selectivity due to its coating glycocalyx layer, composed principally by proteoglycans[27]. Glomerular endothelial cell glycocalyx and its associated surface layer retain more than 95% of the circulating proteins.
The glomerular endothelial layer is the main target of ANCAs, which attack small-vessel causing vasculitis, leading to necrotizing and crescentic glomerulonephritis. ANCA-vasculitis present an overall annual incidence of approximately 10-20 cases/million people, with an onset age peak of 65-74 years[28]. ANCA can induce the production and release of reactive oxygen species and lytic enzymes by infiltrated neutrophils[29], complement system via the alternative pathway[30] as well as endothelial cell as an amplification disease loop, resulting in endothelium lysis[31]. On early stages of ANCA-vasculitis the endothelial lesion could explain the onset of haematuria, although in advanced stages it could be explained by a severe GFB impairment usually involving all its layers. Although haematuria has been classically considered a marker of glomerular injury activity in ANCA, a recent report showed not repercussion of persistent haematuria (determined by dipstick) in GFR at 1 year[32]. However, in this study, persistent haematuria was associated to low baseline GFR and ANCA status.
Endothelial cell damage has been also reported in endocapillary glomerulonephritis (GN) and infection-associated GN. With a decreasing incidence over the last decades in developed countries[33], endocapillary GN is now more frequent in fragile patients, such as elder, alcoholics and intravenous drug users[34]. The typical presentation is nephritic syndrome or acute renal failure 15 d after an infection[35], in which haematuria is almost always present. Although the prognosis is excellent for children, in the 20%-74% of adults renal impairment persist[34-37]. The immune complexes produced in situ or deposited from circulation induce a severe inflammatory response resulting in neutrophils chemotaxis and endocapillary hypercellularity, leading to haematuria. Endocapillary GN has been recently proposed as a C3 glomerulopathy, because the nephrogenic antigen triggers the activation of the complement alternative pathway.
GBM disorders
As previously reported, GBM has a key role on the glomerular filtration barrier permeability. GBM is composed of a dense gel-like meshwork of type IV collagen (COL4) and laminin, along with sulfated proteoglycans. The causes of GBM injury can be categorized on primary GBM disorders, as the collagen nephropathies, and secondary GBM diseases, including diseases with GBM as a target.
Primary GBM disorders: Collagen nephropathies are the main primary GBM disorders. Type IV collagen is the main component of GBM, so its mutations produce abnormal tights winding of the collagen triple helix. Type IV collagen-associated diseases are the most common hereditary disorders presenting with isolated microscopic haematuria resulting from mutations in genes for type IV collagen[38], especially on its alpha 3 (COL4A3) and 4 chains (COL4A4)[39].
AS was the first characterized GBM collagen-disorder. AS has a prevalence of 1 case/50000 live births[10]. The 85% of AS cases are due to X-linked mutations in α5 collagen chain (COL4A5), whereas the remaining 15% are due to autosomal recessive mutations in COL4A3 or COL4A4, although a minority of cases have been described as autosomal dominant sporadic mutations. X-linked AS is characterized by sensorineural hearing loss, ocular abnormalities and progressing nephropathy. These alterations are more severe in males. Autosomal reccesive Alport has the same clinical features than X-linked AS, with more aggressive and early CKD impairment (mean age at ESRD is 21 years[40]) without gender preference, and with typically asymptomatic parents genetically related. The electron microscopy show GBM thickening and thinning plus splitting and lamellation of lamina densa[41]. These alterations render in a persistent expression of foetal COL4A1 and COL4A2, being fragile and sensitive to proteases, allowing RBCs egression in the urinary space, and therefore persistent microhematuria. Persistent microhematuria is more frequent in children, often with macroscopic haematuria bouts, which suggest an exacerbating trigger factor over this chronically damaged GBM, although this promoter agent has not been yet identified. Importantly, renal function decreases progressively to ESRD before the fourth decade in 90% of patients[42,43].
Heterozygous mutations in the COL4A3, COL4A4 or COL4A5 genes produce TBMN. TBMN has an incidence of 1% and is characterized by a GBM < 150 nm. The slightly more compact GMB, due to a lack of non-collagenous molecules, is more fragile, which could explain the persistent isolated haematuria. TBMN’s typical presentation include microhematuria and minimal or no proteinuria, with normal glomerular filtration rate and blood pressure. However, recent evidences showed a worse prognosis than it has been though[44], where microhematuria progress to proteinuria, to renal cysts[45] and to CKD in 26.6% of all patients, and in 48% of all patients > 50 years old[10,46].
HANAC syndrome is an extraordinary infrequent systemic basement-membrane disease, due to heterozygous mutation in COL4A1[47]. HANAC syndrome could present either with micro- or macroscopic bouts, related or not with impaired glomerular filtration rate and/or renal cysts. ESRD has not been yet described probably due to the low number of patients reported to date. Electron microscopy showed thickening and splitting on the basement membranes (including tubules, capillaries and GBM)[48]. The micro- and macroscopic haematuria bouts may be the result of the abnormal remodeling of the extracellular matrix and altered composition of all basement membranes[47].
Although loin pain haematuria syndrome (LPHS) is not a collagen nephropathy, we include it here due to its similarities with TBMN and AS histolopathologic features. More frequent in females (70%), LPHS presents with recurrent haematuria by the third decade of life. The electron microscopy showed abnormally thin or thick GBM[49]. It has been proposed that abnormalities on GBM allow RBCs leak into the urinary space causing intratubular obstruction and clots. The intratubular obstruction could induce interstitial edema and intraglomerular hypertension which originates further glomerular haemorrhage.
Secondary GBM disorders: Some disorders attack GBM, such as Anti-GBM disease and C3 glomerulopathy. Anti-GBM disease is characterized by autoantibodies against the alpha3 chain non-collagen 1 domain of type IV collagen. Anti-GBM disease shows an incidence of 0.5-1 case/million people per year[50]. It has been proposed that anti-GBM disease could be triggered over genetically predisposed patients (HLA-DRB1*1501 allele and genes of the FCGR and KLK families[51]) by environmental or cellular/humoral immunity factors. These auto-antibodies attack GBM disturbing its intrinsic structure, explaining the almost always present haematuria, with nephritic syndrome and crescentic glomerulonephritis.
On the other hand, the recently introduced C3 glomerulopathy, as a glomerular pathology with C3 accumulation with none significant immunoglobulin deposition[52]. C3 glomerulopathy clinically has been associated to haematuria, proteinuria and different degrees of renal dysfunction[53]. C3 glomerulopahty is secondary to an aberrant regulation of complement alternative pathway, either genetic or acquired. C3 glomerulopathies include dense deposit disease (DDD), C3 glomerulonephritis and complement factor H-related (CFHR) genes mutations[54], such as hybrid CFHR3-1 gene and an internal duplication within the CFHR5 gene[55]. C3 glomerulopathy’s incidence has been estimated in 1-2 cases/million people, independently of gender, although it has been reported an increased severity in males. DDD is characterized by linear, hyperosmiophilic, intramembranous dense deposit in lamina densa, restricted to both tubular and Bowman’s capsular basement membranes. Haematuria is observed in the 87% of the cases, mainly microscopic hematuria (68%)[53] and that persists during the follow up. Haematuria can be explained by the GBM impairment, although mesangial, subendothelial and subepithelial deposits has been also described[53]. Two evidences suggested the role of an infection triggering C3 glomerulopathies, firstly the concurrence of macroscopic bouts of haematuria with upper respiratory tract infections in CFHR5 nephropathy[54], and secondly the elevated antistreptolysin-O (a substance produced by group A Streptococcus bacteria) titters in C3 glomerulopathy[53]. Although proteinuria has pointed as the most important prognostic factor, in the Athanasiou cohort[56] all patients that reached ESRD presented macroscopic haematuria bouts associated with fever upper respiratory tract infections in the childhood and adolescence.
Mesangial deposit disorders
IgAN is the most common cause of glomerular haematuria. IgAN has an uncertain prevalence (10%-16%)[57] and is characterized by the presence of persistent isolated microscopic haematuria, with occasional macroscopical bouts associated to upper respiratory or gastrointestinal infections. Haematuria may be accompanied by proteinuria, sometimes in the nephrotic range. Although it has been consider benign, nearly 20% of patients develop ESRD within 20 years of diagnosis[58-61]. Mesangial hypercellulary is the usual histological finding, being the degree of interstitial fibrosis and tubular atrophy the strongest predictors of renal outcome[62]. However haematuria’s role over IgAN outcome has not been properly addressed. Macroscopic haematuria bouts has negative implications on long-term prognosis[5] and although the prognosis of IgAN patients with isolated MH have been reported as good, almost 50% of the largest cohort presented spontaneous remission of MH during the follow up[63].
Even though mechanism of haematuria is unknown, during the episodes of macroscopic haematuria it has been detected an increase in circulating immune complexes composed of galactose-deficient IgA1 complexed with antiglycan antibodies[64]. These circulating immune complexes are deposited in the mesangium, inducing cell proliferation and secretion of several inflammatory mediators (including cytokines, growth factors and aldosterone/angiotensin) which can be released to the urinary space and induce both, podocytes and proximal tubular epithelial cells damage[65]. Therefore, these mediators could compromise GBM filtration-barrier function, allowing RBCs egression. The same pathological mechanism has been observed in Henoch-Schönlein purpura (HSP), a systemic disorder characterized by the coincidence of IgAN and leukocytoclastic vasculitis. In HSP patients, the plasma concentration of galactose-deficient IgA1 complexes are also increased and the subendothelial deposits, crescents as well as glomerular-tuft necrosis are even more frequent than in IgAN[65].
Subendothelial and subepithelial deposits diseases
Many nephropathies are characterized by the presence of deposits in subendothelial and subepithelial spaces, inducing a significant impairment in the GFB integrity and therefore haematuria.
Primary glomerulonephritis: Membranoproliferative, Endocapillary and Crescentic GN are the main primary GN associated with subendothelial and subepithelial deposits. It has been proposed that leucocytes and immune-complex can produce a severe inflammatory response activating glomerular cells and interfering with GBM structure, leading haematuria.
Fibril deposit disease: Fibronectin glomerulopathy (GFND) is a rare autosomal-dominant nephropathy due to a mutation in Fibronectin 1 (FN1) gene expressed[66]. FN1 is a dimeric glycoprotein constituent of the extracellular matrix. Its mutations altered the protein-dimers assembly into fibrils in the extracellular matrix and produce a disbalance between soluble and insoluble fibronectin, leading to its pathognomonic deposition in mesangium and subendothelial area[66,67]. In addition to fibronectine deposition, it has been reported IgA, C1q and fibrinogen deposits[68]. GFND may present at different ages, although mostly in adolescence or early adulthood. GFND is characterized by microhaematuria, proteinuria and hypertension. GFND patients progress to ESRD from second to sixth decade of life[66]. In these patients ESRD can recur after renal transplantation[69].
Fibrillary and Immunotactoid GN presented fibrils or microtubules deposition in mesangium, GBM or both. Immunotactoid GN can be differentiated from Fibrillary due to its typically wider fibrils with focal parallel alignment. The pathogenesis is unclear, however its response to immunosuppression pointed an underlying autoimmune condition[70]. Fibrillary GN presents deposits infiltrating both mesangium and lamina densa[71], which implied a severe impairment in the GFB allowing RBCs egression, explaining the pathogenesis of haematuria in these diseases.
Podocyte associated disorders
The podocytes are highly specialized epithelial cells, with interdigitating foot processes and specialized intercellular junctions term the “slit diaphragms”, playing a key role in GFB integrity. Mutations in proteins involved in the slit diaphragm and foot processes have been mainly associated with familial nephrotic syndrome. A previously kind of familial benign haematuria, MYH9-associated kidney disease, has been recently described as a genetic variation on MYH9 gene. MYH9 encode non-muscle myosin IIA heavy chain, a major protein of the actin-myosin’s podocyte contractile apparatus, necessary to keep the capillary wall integrity[72]. Other autosomal-dominant syndromes as May-Hegglin anomaly and the Flechtner and Epstein syndromes also include abnormalities in MYH9 gene, with a total incidence < 1:100000[38]. MYH9 gene mutations presented variable degrees of sensorineural deafness and glomerulopathy[73], usually in African people. Haematuria and/or proteinuria are typically present since childhood, with a progression to ESRD by young adulthood[74]. Electron microscopy showed occasional focal thickening and splitting of the GBM[74,75]. MYH9 mutations produce a more fragile podocyte and capillary wall which allow RBCs egression, explaining the presence of haematuria[75].
Fabry’s disease also present haematuria. Fabry’s disease is a lysosomal storage X-linked disorder, much more common than it has been though (1:3100[76]-1:1600[77]) and more frequent and aggressive in males. This lysosomal impairment leads to intracellular accumulations of globotriaosylceramide in almost all the human cells[78]. The globotriaosylceramide accumulation induce autophagy in podocytes and endothelial cells damage, resulting in focal and segmental sclerosis, as well as a significant impairment in the GFB, therefore leading proteinuria and haematuria[79].
Miscellaneous
There are several diseases associated to haematuria without any obvious histopathological finding to justify it. Warfarin coagulopathy (international normalized ratio > 3.0) may induce AKI, the so called WRN[80]. AKI could be caused by intratubular obstruction of RBC casts during glomerular haemorrhage, although atheroembolism[81], interstitial nephritis[82], and direct effects of warfarin on the glomerulus[83] have been also pointed. The real WRN incidence could be 16% in non-CKD and 37% in CKD patients[84]. There has been described several risk factors for WRN, including: (1) aspirin therapy; (2) drugs that increase glomerular hydrostatic pressure, such as dihydropyridine calcium channel blockers; (3) low serum albumin levels; and (4) concurrent congestive heart failure[85]. The correction of the warfarin coagulopathy with vitamin K prevents WRN, and could promote the recovery on animal models[86]. In WRN, 66% of the patients with macroscopic haematuria bouts show impairment of renal function. WRN is associated with an accelerated CKD progression and mortality rate, although this was related with the patient¡’s comorbidities[9]. It has been speculated that this warfarin iatrogenic coagulopathy may be observed in patients with permeable and previous “fragile” GFB (like subclinical GN or TBMD), allowing RBC egression.
Sickle cell disease (SCD) is a multisystemic disorder with homozygous or heterozygous inheritance of β-globin mutated gene, leading in the production of hemoglobin S (HbS), with a global incidence of 30/million people. HbS produce abnormally dense and rigid RBCs with tendency to sickle. The 3%-4% of SCD patients presented haematuria, although it is more frequent on heterozygotes with the sickle cell trait. The tortuous sickle RBCs can easily extravasate the glomerulus capillary wall, raising blood viscosity, and promoting microthrombi formation and ischemic necrosis in the vasa recta, and therefore inducing structural changes and haematuria[87]. Haematuria is mainly recurrent and macroscopic, and could be asymptomatic or painful due to passage of clots through the ureter. Furthermore, haemoglobinuria is also frequent is this patients due to its recurrent haemolytic anaemia crisis.
CONCLUSION
Recent findings suggest a pathogenic role for glomerular hematuria in kidney disease. Thus, the occurrence of macroscopic hematuria-associated AKI in IgAN nephropathy is associated with subsequent persistent impairment of renal function in a significant proportion of patients. An excessive anticoagulation, as a result of warfarin therapy, may also result in macroscopic hematuria-associated AKI and may compromise long-term renal function. Finally, persistent isolated microhematuria may also induce ESRD. The intrinsic pathogenical mechanism of glomerular haematuria remains unclear. Dysmorphic urinary RBCs pointed GFB dysfunction or damage as a possible alteration associated to this pathological process. Three possible pathological mechanisms may be implicated in GFB dysfunction and subsequent haematuria onset, including genetic alteration of GFB components, aberrant deposition of toxic molecules in the GFB, and an enhanced inflammatory response. However, although it has been identified some of the mechanisms involved in haematuria-associated renal damage, it is necessary to characterize new pathogenic effects of hematuria to identify new potential therapeutic targets. Future studies, in this line, will be of great interest.
Footnotes
P- Reviewer: Landry DL S- Editor: Gong XM L- Editor: A E- Editor: Lu YJ
Supported by Grants from FIS (Programa Miguel Servet: CP10/00479, PI13/00802 and PI14/00883) and Spanish Society of Nephrology to Moreno JA; and Institute of Research Queen Sophia, FRIAT and ISCIII fund PI10/00072 to Egido J.
Conflict-of-interest: There are no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 21, 2014
First decision: September 28, 2014
Article in press: December 31, 2014
References
- 1.Kelly JD, Fawcett DP, Goldberg LC. Assessment and management of non-visible haematuria in primary care. BMJ. 2009;338:a3021. doi: 10.1136/bmj.a3021. [DOI] [PubMed] [Google Scholar]
- 2.Cohen RA, Brown RS. Clinical practice. Microscopic hematuria. N Engl J Med. 2003;348:2330–2338. doi: 10.1056/NEJMcp012694. [DOI] [PubMed] [Google Scholar]
- 3.Clarkson AR, Seymour AE, Thompson AJ, Haynes WD, Chan YL, Jackson B. IgA nephropathy: a syndrome of uniform morphology, diverse clinical features and uncertain prognosis. Clin Nephrol. 1977;8:459–471. [PubMed] [Google Scholar]
- 4.Vivante A, Afek A, Frenkel-Nir Y, Tzur D, Farfel A, Golan E, Chaiter Y, Shohat T, Skorecki K, Calderon-Margalit R. Persistent asymptomatic isolated microscopic hematuria in Israeli adolescents and young adults and risk for end-stage renal disease. JAMA. 2011;306:729–736. doi: 10.1001/jama.2011.1141. [DOI] [PubMed] [Google Scholar]
- 5.Gutiérrez E, González E, Hernández E, Morales E, Martínez MA, Usera G, Praga M. Factors that determine an incomplete recovery of renal function in macrohematuria-induced acute renal failure of IgA nephropathy. Clin J Am Soc Nephrol. 2007;2:51–57. doi: 10.2215/CJN.02670706. [DOI] [PubMed] [Google Scholar]
- 6.Kido R, Shibagaki Y, Iwadoh K, Nakajima I, Fuchinoue S, Fujita T, Teraoka S. Persistent glomerular hematuria in living kidney donors confers a risk of progressive kidney disease in donors after heminephrectomy. Am J Transplant. 2010;10:1597–1604. doi: 10.1111/j.1600-6143.2010.03077.x. [DOI] [PubMed] [Google Scholar]
- 7.Kincaid-Smith P, Bennett WM, Dowling JP, Ryan GB. Acute renal failure and tubular necrosis associated with hematuria due to glomerulonephritis. Clin Nephrol. 1983;19:206–210. [PubMed] [Google Scholar]
- 8.Praga M, Gutierrez-Millet V, Navas JJ, Ruilope LM, Morales JM, Alcazar JM, Bello I, Rodicio JL. Acute worsening of renal function during episodes of macroscopic hematuria in IgA nephropathy. Kidney Int. 1985;28:69–74. doi: 10.1038/ki.1985.120. [DOI] [PubMed] [Google Scholar]
- 9.Brodsky SV, Nadasdy T, Rovin BH, Satoskar AA, Nadasdy GM, Wu HM, Bhatt UY, Hebert LA. Warfarin-related nephropathy occurs in patients with and without chronic kidney disease and is associated with an increased mortality rate. Kidney Int. 2011;80:181–189. doi: 10.1038/ki.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deltas C, Pierides A, Voskarides K. The role of molecular genetics in diagnosing familial hematuria(s) Pediatr Nephrol. 2012;27:1221–1231. doi: 10.1007/s00467-011-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1–266. [PubMed] [Google Scholar]
- 12.KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150. doi: 10.1038/ki.2013.243. [DOI] [PubMed] [Google Scholar]
- 13.KDIGO clinical practice guideline for glomerulonephritis. Kidney Int Suppl. 2012;2:139–274. [Google Scholar]
- 14.Szeto CC, Lai FM, To KF, Wong TY, Chow KM, Choi PC, Lui SF, Li PK. The natural history of immunoglobulin a nephropathy among patients with hematuria and minimal proteinuria. Am J Med. 2001;110:434–437. doi: 10.1016/s0002-9343(01)00659-3. [DOI] [PubMed] [Google Scholar]
- 15.Nath KA. Heme oxygenase-1 and acute kidney injury. Curr Opin Nephrol Hypertens. 2014;23:17–24. doi: 10.1097/01.mnh.0000437613.88158.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nath KA, Vercellotti GM, Grande JP, Miyoshi H, Paya CV, Manivel JC, Haggard JJ, Croatt AJ, Payne WD, Alam J. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int. 2001;59:106–117. doi: 10.1046/j.1523-1755.2001.00471.x. [DOI] [PubMed] [Google Scholar]
- 17.Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol. 2007;18:414–420. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 18.Paller MS, Jacob HS. Cytochrome P-450 mediates tissue-damaging hydroxyl radical formation during reoxygenation of the kidney. Proc Natl Acad Sci USA. 1994;91:7002–7006. doi: 10.1073/pnas.91.15.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heyman SN, Brezis M. Acute renal failure in glomerular bleeding: a puzzling phenomenon. Nephrol Dial Transplant. 1995;10:591–593. [PubMed] [Google Scholar]
- 20.Fairley KF, Birch DF. Hematuria: a simple method for identifying glomerular bleeding. Kidney Int. 1982;21:105–108. doi: 10.1038/ki.1982.16. [DOI] [PubMed] [Google Scholar]
- 21.Moreno JA, Martín-Cleary C, Gutiérrez E, Rubio-Navarro A, Ortiz A, Praga M, Egido J. Haematuria: the forgotten CKD factor? Nephrol Dial Transplant. 2012;27:28–34. doi: 10.1093/ndt/gfr749. [DOI] [PubMed] [Google Scholar]
- 22.Jarad G, Miner JH. Update on the glomerular filtration barrier. Curr Opin Nephrol Hypertens. 2009;18:226–232. doi: 10.1097/mnh.0b013e3283296044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salmon AH, Neal CR, Harper SJ. New aspects of glomerular filtration barrier structure and function: five layers (at least) not three. Curr Opin Nephrol Hypertens. 2009;18:197–205. doi: 10.1097/MNH.0b013e328329f837. [DOI] [PubMed] [Google Scholar]
- 24.Menon MC, Chuang PY, He CJ. The glomerular filtration barrier: components and crosstalk. Int J Nephrol. 2012;2012:749010. doi: 10.1155/2012/749010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haraldsson BS. The endothelium as part of the integrative glomerular barrier complex. Kidney Int. 2014;85:8–11. doi: 10.1038/ki.2013.317. [DOI] [PubMed] [Google Scholar]
- 27.Fridén V, Oveland E, Tenstad O, Ebefors K, Nyström J, Nilsson UA, Haraldsson B. The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 2011;79:1322–1330. doi: 10.1038/ki.2011.58. [DOI] [PubMed] [Google Scholar]
- 28.Watts RA, Scott DG, Jayne DR, Ito-Ihara T, Muso E, Fujimoto S, Harabuchi Y, Kobayashi S, Suzuki K, Hashimoto H. Renal vasculitis in Japan and the UK--are there differences in epidemiology and clinical phenotype? Nephrol Dial Transplant. 2008;23:3928–3931. doi: 10.1093/ndt/gfn354. [DOI] [PubMed] [Google Scholar]
- 29.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–4119. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kallenberg CG. Pathophysiology of ANCA-associated small vessel vasculitis. Curr Rheumatol Rep. 2010;12:399–405. doi: 10.1007/s11926-010-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen TK, Murakami C, Manno RL, Geetha D. Hematuria duration does not predict kidney function at 1 year in ANCA-associated glomerulonephritis. Semin Arthritis Rheum. 2014;44:198–201. doi: 10.1016/j.semarthrit.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Iturbe B, Musser JM. The current state of poststreptococcal glomerulonephritis. J Am Soc Nephrol. 2008;19:1855–1864. doi: 10.1681/ASN.2008010092. [DOI] [PubMed] [Google Scholar]
- 34.Montseny JJ, Meyrier A, Kleinknecht D, Callard P. The current spectrum of infectious glomerulonephritis. Experience with 76 patients and review of the literature. Medicine (Baltimore) 1995;74:63–73. doi: 10.1097/00005792-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Sethi S, Fervenza FC, Zhang Y, Zand L, Meyer NC, Borsa N, Nasr SH, Smith RJ. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013;83:293–299. doi: 10.1038/ki.2012.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lien JW, Mathew TH, Meadows R. Acute post-streptococcal glomerulonephritis in adults: a long-term study. Q J Med. 1979;48:99–111. [PubMed] [Google Scholar]
- 37.Moroni G, Pozzi C, Quaglini S, Segagni S, Banfi G, Baroli A, Picardi L, Colzani S, Simonini P, Mihatsch MJ, et al. Long-term prognosis of diffuse proliferative glomerulonephritis associated with infection in adults. Nephrol Dial Transplant. 2002;17:1204–1211. doi: 10.1093/ndt/17.7.1204. [DOI] [PubMed] [Google Scholar]
- 38.Gale DP. How benign is hematuria? Using genetics to predict prognosis. Pediatr Nephrol. 2013;28:1183–1193. doi: 10.1007/s00467-012-2399-y. [DOI] [PubMed] [Google Scholar]
- 39.Miner JH. Developmental biology of glomerular basement membrane components. Curr Opin Nephrol Hypertens. 1998;7:13–19. doi: 10.1097/00041552-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Oka M, Nozu K, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, Morisada N, Yan K, Matsuo M, Yoshikawa N, et al. Natural history of genetically proven autosomal recessive Alport syndrome. Pediatr Nephrol. 2014;29:1535–1544. doi: 10.1007/s00467-014-2797-4. [DOI] [PubMed] [Google Scholar]
- 41.Barsotti P, Muda AO, Mazzucco G, Massella L, Basolo B, De Marchi M, Rizzoni G, Monga G, Faraggiana T. Distribution of alpha-chains of type IV collagen in glomerular basement membranes with ultrastructural alterations suggestive of Alport syndrome. Nephrol Dial Transplant. 2001;16:945–952. doi: 10.1093/ndt/16.5.945. [DOI] [PubMed] [Google Scholar]
- 42.Longo I, Scala E, Mari F, Caselli R, Pescucci C, Mencarelli MA, Speciale C, Giani M, Bresin E, Caringella DA, et al. Autosomal recessive Alport syndrome: an in-depth clinical and molecular analysis of five families. Nephrol Dial Transplant. 2006;21:665–671. doi: 10.1093/ndt/gfi312. [DOI] [PubMed] [Google Scholar]
- 43.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11:649–657. doi: 10.1681/ASN.V114649. [DOI] [PubMed] [Google Scholar]
- 44.Carasi C, Van’t Hoff WG, Rees L, Risdon RA, Trompeter RS, Dillon MJ. Childhood thin GBM disease: review of 22 children with family studies and long-term follow-up. Pediatr Nephrol. 2005;20:1098–1105. doi: 10.1007/s00467-005-1879-8. [DOI] [PubMed] [Google Scholar]
- 45.Sevillano A, Gutierrez E, Morales E, Hernandez E, Molina M, Gonzalez E, Praga M. Multiple kidney cysts in thin basement membrane disease with proteinuria and kidney function impairment. Clin Kidney J. 2014;7:251–256. doi: 10.1093/ckj/sfu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Temme J, Peters F, Lange K, Pirson Y, Heidet L, Torra R, Grunfeld JP, Weber M, Licht C, Müller GA, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012;81:779–783. doi: 10.1038/ki.2011.452. [DOI] [PubMed] [Google Scholar]
- 47.Plaisier E, Chen Z, Gekeler F, Benhassine S, Dahan K, Marro B, Alamowitch S, Paques M, Ronco P. Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am J Med Genet A. 2010;152A:2550–2555. doi: 10.1002/ajmg.a.33659. [DOI] [PubMed] [Google Scholar]
- 48.Van Agtmael T, Schlötzer-Schrehardt U, McKie L, Brownstein DG, Lee AW, Cross SH, Sado Y, Mullins JJ, Pöschl E, Jackson IJ. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet. 2005;14:3161–3168. doi: 10.1093/hmg/ddi348. [DOI] [PubMed] [Google Scholar]
- 49.Spetie DN, Nadasdy T, Nadasdy G, Agarwal G, Mauer M, Agarwal AK, Khabiri H, Nagaraja HN, Nahman NS, Hartman JA, et al. Proposed pathogenesis of idiopathic loin pain-hematuria syndrome. Am J Kidney Dis. 2006;47:419–427. doi: 10.1053/j.ajkd.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 50.Chen M, Cui Z, Zhao MH. ANCA-associated vasculitis and anti-GBM disease: the experience in China. Nephrol Dial Transplant. 2010;25:2062–2065. doi: 10.1093/ndt/gfq134. [DOI] [PubMed] [Google Scholar]
- 51.Zhou XJ, Lv JC, Zhao MH, Zhang H. Advances in the genetics of anti-glomerular basement membrane disease. Am J Nephrol. 2010;32:482–490. doi: 10.1159/000321324. [DOI] [PubMed] [Google Scholar]
- 52.Fakhouri F, Frémeaux-Bacchi V, Noël LH, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol. 2010;6:494–499. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 53.Medjeral-Thomas NR, O’Shaughnessy MM, O’Regan JA, Traynor C, Flanagan M, Wong L, Teoh CW, Awan A, Waldron M, Cairns T, et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol. 2014;9:46–53. doi: 10.2215/CJN.04700513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Medjeral-Thomas N, Malik TH, Patel MP, Toth T, Cook HT, Tomson C, Pickering MC. A novel CFHR5 fusion protein causes C3 glomerulopathy in a family without Cypriot ancestry. Kidney Int. 2014;85:933–937. doi: 10.1038/ki.2013.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, Zavros M, Maxwell PH, Cook HT, Demosthenous P, Hadjisavvas A, et al. Familial C3 glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol. 2011;6:1436–1446. doi: 10.2215/CJN.09541010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003;63:2286–2294. doi: 10.1046/j.1523-1755.63.6s.2.x. [DOI] [PubMed] [Google Scholar]
- 58.Hastings MC, Delos Santos NM, Wyatt RJ. Renal survival in pediatric patients with IgA nephropathy. Pediatr Nephrol. 2007;22:317–318. doi: 10.1007/s00467-006-0303-3. [DOI] [PubMed] [Google Scholar]
- 59.Ronkainen J, Ala-Houhala M, Autio-Harmainen H, Jahnukainen T, Koskimies O, Merenmies J, Mustonen J, Ormälä T, Turtinen J, Nuutinen M. Long-term outcome 19 years after childhood IgA nephritis: a retrospective cohort study. Pediatr Nephrol. 2006;21:1266–1273. doi: 10.1007/s00467-006-0163-x. [DOI] [PubMed] [Google Scholar]
- 60.Kusumoto Y, Takebayashi S, Taguchi T, Harada T, Naito S. Long-term prognosis and prognostic indices of IgA nephropathy in juvenile and in adult Japanese. Clin Nephrol. 1987;28:118–124. [PubMed] [Google Scholar]
- 61.Syrjänen J, Mustonen J, Pasternack A. Hypertriglyceridaemia and hyperuricaemia are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant. 2000;15:34–42. doi: 10.1093/ndt/15.1.34. [DOI] [PubMed] [Google Scholar]
- 62.Roberts IS. Pathology of IgA nephropathy. Nat Rev Nephrol. 2014;10:445–454. doi: 10.1038/nrneph.2014.92. [DOI] [PubMed] [Google Scholar]
- 63.Gutiérrez E, Zamora I, Ballarín JA, Arce Y, Jiménez S, Quereda C, Olea T, Martínez-Ara J, Segarra A, Bernis C, et al. Long-term outcomes of IgA nephropathy presenting with minimal or no proteinuria. J Am Soc Nephrol. 2012;23:1753–1760. doi: 10.1681/ASN.2012010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, Julian BA, Wyatt RJ, Mestecky J. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–513. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- 65.Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368:2402–2414. doi: 10.1056/NEJMra1206793. [DOI] [PubMed] [Google Scholar]
- 66.Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant. 2013;28:2946–2960. doi: 10.1093/ndt/gft253. [DOI] [PubMed] [Google Scholar]
- 67.Strøm EH, Banfi G, Krapf R, Abt AB, Mazzucco G, Monga G, Gloor F, Neuweiler J, Riess R, Stosiek P. Glomerulopathy associated with predominant fibronectin deposits: a newly recognized hereditary disease. Kidney Int. 1995;48:163–170. doi: 10.1038/ki.1995.280. [DOI] [PubMed] [Google Scholar]
- 68.Yoshino M, Miura N, Ohnishi T, Suzuki K, Kitagawa W, Nishikawa K, Imai H. Clinicopathological analysis of glomerulopathy with fibronectin deposits (GFND): a case of sporadic, elderly-onset GFND with codeposition of IgA, C1q, and fibrinogen. Intern Med. 2013;52:1715–1720. doi: 10.2169/internalmedicine.52.0046. [DOI] [PubMed] [Google Scholar]
- 69.Gemperle O, Neuweiler J, Reutter FW, Hildebrandt F, Krapf R. Familial glomerulopathy with giant fibrillar (fibronectin-positive) deposits: 15-year follow-up in a large kindred. Am J Kidney Dis. 1996;28:668–675. doi: 10.1016/s0272-6386(96)90247-4. [DOI] [PubMed] [Google Scholar]
- 70.Javaugue V, Karras A, Glowacki F, McGregor B, Lacombe C, Goujon JM, Ragot S, Aucouturier P, Touchard G, Bridoux F. Long-term kidney disease outcomes in fibrillary glomerulonephritis: a case series of 27 patients. Am J Kidney Dis. 2013;62:679–690. doi: 10.1053/j.ajkd.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 71.Nasr SH, Valeri AM, Cornell LD, Fidler ME, Sethi S, Leung N, Fervenza FC. Fibrillary glomerulonephritis: a report of 66 cases from a single institution. Clin J Am Soc Nephrol. 2011;6:775–784. doi: 10.2215/CJN.08300910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arrondel C, Vodovar N, Knebelmann B, Grünfeld JP, Gubler MC, Antignac C, Heidet L. Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol. 2002;13:65–74. doi: 10.1681/ASN.V13165. [DOI] [PubMed] [Google Scholar]
- 73.Kopp JB, Winkler CA, Nelson GW. MYH9 genetic variants associated with glomerular disease: what is the role for genetic testing? Semin Nephrol. 2010;30:409–417. doi: 10.1016/j.semnephrol.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Han KH, Lee H, Kang HG, Moon KC, Lee JH, Park YS, Ha IS, Ahn HS, Choi Y, Cheong HI. Renal manifestations of patients with MYH9-related disorders. Pediatr Nephrol. 2011;26:549–555. doi: 10.1007/s00467-010-1735-3. [DOI] [PubMed] [Google Scholar]
- 75.Kopp JB. Glomerular pathology in autosomal dominant MYH9 spectrum disorders: what are the clues telling us about disease mechanism? Kidney Int. 2010;78:130–133. doi: 10.1038/ki.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin HY, Chong KW, Hsu JH, Yu HC, Shih CC, Huang CH, Lin SJ, Chen CH, Chiang CC, Ho HJ, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2:450–456. doi: 10.1161/CIRCGENETICS.109.862920. [DOI] [PubMed] [Google Scholar]
- 78.Prabakaran T, Birn H, Bibby BM, Regeniter A, Sørensen SS, Feldt-Rasmussen U, Nielsen R, Christensen EI. Long-term enzyme replacement therapy is associated with reduced proteinuria and preserved proximal tubular function in women with Fabry disease. Nephrol Dial Transplant. 2014;29:619–625. doi: 10.1093/ndt/gft452. [DOI] [PubMed] [Google Scholar]
- 79.Liebau MC, Braun F, Höpker K, Weitbrecht C, Bartels V, Müller RU, Brodesser S, Saleem MA, Benzing T, Schermer B, et al. Dysregulated autophagy contributes to podocyte damage in Fabry‘s disease. PLoS One. 2013;8:e63506. doi: 10.1371/journal.pone.0063506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brodsky SV, Satoskar A, Chen J, Nadasdy G, Eagen JW, Hamirani M, Hebert L, Calomeni E, Nadasdy T. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of 9 cases. Am J Kidney Dis. 2009;54:1121–1126. doi: 10.1053/j.ajkd.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 81.Moll S, Huffman J. Cholesterol emboli associated with warfarin treatment. Am J Hematol. 2004;77:194–195. doi: 10.1002/ajh.20210. [DOI] [PubMed] [Google Scholar]
- 82.Kapoor KG, Bekaii-Saab T. Warfarin-induced allergic interstitial nephritis and leucocytoclastic vasculitis. Intern Med J. 2008;38:281–283. doi: 10.1111/j.1445-5994.2008.01646.x. [DOI] [PubMed] [Google Scholar]
- 83.Yanagita M. Gas6, warfarin, and kidney diseases. Clin Exp Nephrol. 2004;8:304–309. doi: 10.1007/s10157-004-0305-z. [DOI] [PubMed] [Google Scholar]
- 84.Brodsky SV, Collins M, Park E, Rovin BH, Satoskar AA, Nadasdy G, Wu H, Bhatt U, Nadasdy T, Hebert LA. Warfarin therapy that results in an International Normalization Ratio above the therapeutic range is associated with accelerated progression of chronic kidney disease. Nephron Clin Pract. 2010;115:c142–c146. doi: 10.1159/000312877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.An JN, Ahn SY, Yoon CH, Youn TJ, Han MK, Kim S, Chin HJ, Na KY, Chae DW. The occurrence of warfarin-related nephropathy and effects on renal and patient outcomes in korean patients. PLoS One. 2013;8:e57661. doi: 10.1371/journal.pone.0057661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ozcan A, Ware K, Calomeni E, Nadasdy T, Forbes R, Satoskar AA, Nadasdy G, Rovin BH, Hebert LA, Brodsky SV. 5/6 nephrectomy as a validated rat model mimicking human warfarin-related nephropathy. Am J Nephrol. 2012;35:356–364. doi: 10.1159/000337918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pham PT, Pham PC, Wilkinson AH, Lew SQ. Renal abnormalities in sickle cell disease. Kidney Int. 2000;57:1–8. doi: 10.1046/j.1523-1755.2000.00806.x. [DOI] [PubMed] [Google Scholar]