

Abstract
The kidneys and the blood system mutually exert influence in maintaining homeostasis in the body. Because the kidneys control erythropoiesis by producing erythropoietin and by supporting hematopoiesis, anemia is associated with kidney diseases. Anemia is the most prevalent genetic disorder, and it is caused by a deficiency of glucose 6-phosphate dehydrogenase (G6PD), for which sulfhydryl oxidation due to an insufficient supply of NADPH is a likely direct cause. Elevated reactive oxygen species (ROS) result in the sulfhydryl oxidation and hence are another potential cause for anemia. ROS are elevated in red blood cells (RBCs) under superoxide dismutase (SOD1) deficiency in C57BL/6 mice. SOD1 deficient mice exhibit characteristics similar to autoimmune hemolytic anemia (AIHA) and systemic lupus erythematosus (SLE) at the gerontic stage. An examination of AIHA-prone New Zealand Black (NZB) mice, which have normal SOD1 and G6PD genes, indicated that ROS levels in RBCs are originally high and further elevated during aging. Transgenic overexpression of human SOD1 in erythroid cells effectively suppresses ROS elevation and ameliorates AIHA symptoms such as elevated anti-RBC antibodies and premature death in NZB mice. These results support the hypothesis that names oxidative stress as a risk factor for AIHA and other autoimmune diseases such as SLE. Herein we discuss the association between oxidative stress and SLE pathogenesis based mainly on the genetic and phenotypic characteristics of NZB and New Zealand white mice and provide insight into the mechanism of SLE pathogenesis.
Keywords: Autoimmune hemolytic anemia, Systemic lupus erythematosus, Red blood cells, New Zealand black mice, New Zealand white mice
Core tip: Superoxide dismutase (SOD1) deficient C57BL/6 mice exhibit characteristics similar to autoimmune hemolytic anemia (AIHA) and systemic lupus erythematosus (SLE) at the gerontic stage. An examination of AIHA-prone New Zealand Black (NZB) mice indicated that reactive oxygen species (ROS) levels in red blood cells are originally high and further elevated during aging. Transgenic overexpression of human SOD1 in erythroid cells effectively suppresses ROS elevation and ameliorates AIHA symptoms in NZB mice. Herein we discuss the association between oxidative stress and SLE pathogenesis based mainly on the genetic and phenotypic characteristics of NZB and New Zealand white mice.
INTRODUCTION
The kidney has multiple functions that include maintaining the functions of homeostasis such as the excretion of waste, maintenance of the electrolyte balance of body fluids, and endocrine secretion. As an endocrine organ, the kidney plays an essential role in erythropoiesis by producing erythropoietin that supports hematopoiesis in bone marrow[1]. Chronic kidney disease causes renal anemia by reducing erythropoietin production, and, hence, exogenous erythropoietin is widely used as a potent medicine for the treatment of patients with renal anemia[2]. Defected iron metabolism due to chronic inflammation and cytokine imbalance is also involved in chronic kidney disease-induced anemia[3].
A variety of contributing factors including defected hematopoiesis and accelerated hemolysis are involved in anemic pathogenesis. Glucose 6-phosphate dehydrogenase (G6PD) deficiency, which is the most common genetic defect in the human population[4], causes an insufficient supply of NADPH in RBCs and results in anemia. Although the actual mechanism of anemia due to the G6PD deficiency is not totally understood, the involvement of sulfhydryl oxidation is suspected to be a contributing factor.
Aberrant immune responses in some autoimmune diseases also cause anemia. Autoimmune hemolytic anemia (AIHA) is the pathological condition whereby antibodies attack RBCs, and it often precedes a diagnosis of systemic lupus erythematosus (SLE)[5,6]. Both genetic and environmental factors are involved in the etiology of AIHA and SLE, but molecular mechanisms for a majority of the diseases are largely ambiguous. Reactive oxygen species (ROS) are elevated and appear to be a likely underlying mechanism for these pathological conditions[7-9]. In this review article we discuss recent advances in the research on AIHA and SLE from the viewpoint of oxidative stress using animal models.
G6PD deficiency and oxidative stress
ROS are produced under various conditions such as inflammation and hypoxia-reperfusion injury, and they are involved in a variety of diseases including anemia and renal failure[10]. While reduction-oxidation (redox) reactions play essential roles in metabolic reactions, which includes oxidative phosphorylation that consumes respired oxygen, ROS are simultaneously produced as byproducts. Meanwhile, hemoglobin (Hb), which constitutes a major protein (5 mmol/L) in RBCs and contains Fe(II)-heme. When hemoglobin is oxygenated (Hb-O2), a part of Hb-O2 suffers autoxidation to methemoglobin (MetHb), which possesses Fe(III)-heme and is unable to bind oxygen, and releases superoxide[11,12]. Calculation has shown that the rate for the autoxidation of hemoglobin is 2%-3% (in humans) and 4% (in mice) of total hemoglobin per day. Thus, RBCs are under oxidative stress constitutively, and cellular components face the risk of oxidative damage (Figure 1). NADPH is the principle electron donor for most redox systems that include antioxidation by glutathione peroxidase-glutathione reductase and peroxiredoxin (Prdx)-thioredoxin reductase axes[13] and reductive carbonyl detoxification by the aldo-keto reductase family. Under healthy conditions, the resultant methemoglobin is reduced back by methemoglobin reductase in a NADPH-dependent manner and kept at low levels.
Figure 1.
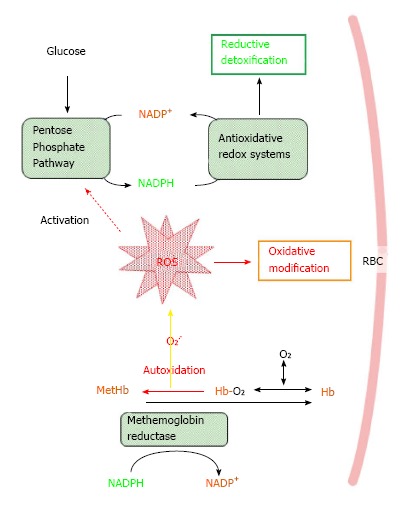
Oxidative stress and antioxidative/redox systems in red blood cells. A part of oxygenized hemoglobin (Hb-O2) is autoxidized to methemoglobin (MetHb) and releases superoxide (O2-.), which may cause oxidative damage to RBCs. MetHb can be reduced back to Hb by methemoglobin reductase in NADPH-dependent manner. G6PD, a rate-determining enzyme in the pentose phosphate pathway, is involved in the production of NADPH that supports antioxid/redox systems as well as methemoglobin reduction by donating redox potential. G6PD deficiency affects entire antioxidative/redox systems, which can consequently accelerate the destruction of RBCs and lead to anemia. RBCs: Red blood cells. NADPH: Triphosphopyridine nucleotide.
Elimination of the resultant ROS and maintaining the redox potential within cells are prerequisites for the survival for RBCs, so that antioxidative enzymes, such as superoxide dismutase (SOD), catalase, glutathione peroxidase, and Prdx, have crucial roles in keeping RBCs healthy. Antioxidants with a small molecular weight, notably glutathione and vitamin C (ascorbic acid), also play roles in redox homeostasis. Oxidative stress induced by SOD and Prdx deficiencies participate in the pathogenesis of anemia, as described below.
Approximately 60 years have passed since the discovery of G6PD deficiency, but the actual mechanism of G6PD deficiency-triggered anemia remains undefined[4]. Because G6PD is the rate-determining enzyme in the pentose phosphate pathway and is involved in the production of NADPH, a G6PD deficiency shifts the cellular redox balance to an oxidized state[14]. Meanwhile, most redox proteins, excluding the ones possessing electron-accepting prosthetic groups, consist of reactive sulfhydryl residues, which are also highly susceptible to oxidative modification. Oxidized or aged proteins undergo proteolytic degradation, and RBCs lack the cellular organelle and protein synthesis machinery that is necessary for their renewal. Thus, oxidative stress appears to cause selective decreases in redox-sensitive proteins. The production of NADPH is elevated by activated G6PD in response to oxidative stress[15], and it supports the reductive detoxification of ROS and oxidized molecules, although its activation mechanism is still unclear.
Mouse models developing anemia, AIHA, and SLE
There are animal models that are applicable to research into the etiology of anemia. For example, a direct cause for iron deficiency anemia is defective hemoglobin synthesis due to insufficient heme supply. The involvement of oxidative stress has been implicated in the pathogenesis of some types of chemically induced anemia, such as that induced by pheynylhydrazine[16].
Several strains of animals that spontaneously develop anemia have been used for pathophysiological examinations. New Zealand Black (NZB) mice constitute a strain that develops AIHA during late middle age, at around 40-50 wk. IgG bound to RBCs increases from about 3 mo of age and induces anemia from about 6 mo of age onward[5]. AIHA is exacerbated by an aberrant immune system with notably impaired CD4+CD25+ regulatory T cells[17] and a Th1 and Th2 cytokine imbalance[18]. Peripheral B-1 cells appear to be a source of autoantibody-producing cells[19]. A dominant T-cell epitope in AIHA is a major glycosylated membrane protein of RBCs, which is also known as an anion transporter band 3[20]. When the AE1 gene encoding band 3 is deleted, the congenic NZB mice still produce autoantibodies against another glycoprotein, glycophorin, and develop AIHA[21]. Thus, a defect in these glycoproteins is not a primary cause, but other latent abnormalities remain.
Defected genes have been identified in pathological model animals for SLE, MRL/lpr and MRL/gld mice[22]. Mutations in Fas and Fas ligand genes cause SLE in MRL/lpr and MRL/gld mice, respectively, via malfunctioning apoptotic removal of self-recognizing preB cells at an infant stage[23]. Although mutations in FAS/APO-1 and Fas ligand are found in human SLE patients[24-26], the incidence is not high. Thus, causal factors for SLE are still largely unknown in the human population. (NZB x NZW) F1 mice are another SLE model animal and show characteristics similar to human SLE[27]. While NZB mice spontaneously develop AIHA symptoms that are limited to the blood system, (NZB x NZW) F1 mice exhibit symptoms in a systemic fashion that include lupus nephritis and cardiovascular abnormalities[28-30]. Although NZW mice possess a larval defect in the immune system, they show virtually normal phenotypes and survival times. Genetic analysis of NZW mice has advanced in the past decade, and the latent factor responsible for the onset of SLE has been unveiled.
Anemia observed in antioxidative enzyme gene-modified mice
Because antioxidation plays an essential role in maintaining RBC function, a deficiency of antioxidative enzymes occasionally exerts severe damage to RBCs. Anemia is caused by a deficiency of antioxidative enzymes SOD1[31], SOD2[32], Prdx1[33], and Prdx2[34], but not by deficiencies of glutathione peroxidase 1[35] or catalase[36]. Phenotypic characteristics regarding anemia differ in genetically modified mice, as follows.
SOD1 DEFICIENCY
Among three SOD isozymes present in mammals, SOD1 is a sole superoxide-scavenging enzyme in mature RBCs, and its deficiency causes anemia[31]. Hemoglobin is a major protein in RBCs, and suffers autoxidation, which results in the production of superoxide[11,12]. Without SOD1, the radical chain reaction initiated by superoxide oxidatively damages RBCs, and ultimately accelerates their destruction. Thus, SOD1-deficient RBCs show a shortened life span that is approximately 60%-70% that of the RBCs of wild-type mice[31].
SOD1 deficiency accelerates hemolysis in the blood and phagocytotic removal of RBCs by liver Kupffer cells[37]. An elevation of ROS levels in RBCs, oxidation of RBC components, and augmented production of autoantibodies in RBCs have been observed in SOD1 deficient C57BL/6 (B6) mice[31]. Elevated production of antibodies against lipid peroxidation products, 4-hydroxynonenal and acrolein, occurs[15]. A general antioxidant, N-acetyl cysteine (NAC), ameliorates these phenotypes and suppresses anemia and AIHA development. Restricted expression of human SOD1 in erythroid cells suppresses oxidative stress in RBCs, which rescues aberrant phenotypes related to anemia and autoimmune responses in SOD1-deficient B6 mice. This substantial amount of evidence supports the notion that overproduced ROS due to SOD1 deficiency can trigger anemia.
Superoxide is continuously produced from oxygenized hemoglobin[11], and hence it is regarded as one of the sources for ROS. Based on theoretical calculation[12], an approximate 200-fold elevation in superoxide results from SOD1 deficiency. Superoxide would conversely result in the conversion of hemoglobin to methemoglobin and enhance the oxidative modification of RBCs. A marked reduction in glutathione peroxidase 1 protein and its activity is seen in SOD1 deficiency[38], which is caused by an irreversible inactivation via conversion of the catalytic selenocysteine to dihydroalanine by elevated ROS[39]. However, the contribution of this low glutathione peroxidase 1 activity to anemia is ambiguous because a deficiency of either glutathione peroxidase 1 or catalase does not cause hematological abnormalities in mice[35,36]. Because thioredoxin reductase is also a selenoenzyme[40], inactivation by the elevated ROS due to a SOD1 deficiency may have a role in the destruction of RBCs.
SOD2 DEFICIENCY
Mice lacking SOD2, a mitochondria-specific isoform, in the whole body show dilated cardiomyopathy, hepatic lipid accumulation and early neonatal death[41]. Hematopoietic chimeras in which all blood cells are derived from the fetal liver stem cells of SOD2-deficient mice are employed to examine the effect of SOD2 deficiency on hematopoiesis. The chimera mice are persistently anemic and characteristically similar to the human disorder sideroblastic anemia[32]. Enhanced protein oxidation and altered membrane deformation appear to reduce the life span of RBCs[42,43]. SOD2-deficient reticulocytes reveal up-regulated transferrin receptors[44] and mitochondrial proliferation and mitochondrial membrane thickening[45]. It is noteworthy that mature RBCs, which do not possess mitochondria, show an elevated production of ROS, abundant iron-stainable granules, and oxidatively damaged proteins. These observations imply that the life-span of the resultant RBCs is reduced due to oxidative damage that is experienced before final maturation of the erythroid cells.
PRDX DEFICIENCY
Among 6 Prdx family members, deficiency of either Prdx1[33] or Prdx2[34] causes hemolytic anemia. Prdx1-deficient mice show increased ROS, hemoglobin instability, Heinz body formation, and a decreased erythrocyte life span[33]. Cancers develop in some organs of Prdx1-deficient mice, but a causal connection to anemia is unknown. Prdx2 is a predominant form of Prdx family members in RBCs[46] and its function in RBCs has been thoroughly characterized. Prdx2 exists as either a stable dimer or a hyperoxidized form in RBCs[47]. Prdx2 functions in a dimer form with a head-to-tail arrangement. During the peroxidase reaction two pairs of disulfide bonds between the catalytic Cys at the N-terminus and the resolving Cys at the C-terminus in the two subunits are formed as an intermediate[13]. However, Prdx2 appears to function as a non-catalytic scavenger of peroxides in RBCs due to an insufficient thioredoxin-thioredoxin reductase system[48,49]. Sulfenic acid is a physiological intermediate of sulfhydryl groups in the catalytic Cys, but excessively produced hydrogen peroxide hyperoxidizes it to sulfinic acid and then sulfonic acid during the reaction cycle of Prdx, which results in a loss of peroxidase activity[13]. The slow turnover rate of Prdx2 increases the chance for hyperoxidization by hydrogen peroxide in RBCs. Sulfinic acid in Prdx can be converted back to sulfhydryl by sulfiredoxin in an ATP-dependent manner in many cells[50,51]. However, because of an insufficient amount of sulfiredoxin in RBCs, hyperoxidized Prdx2 would proceed to proteolytic removal. Although cyclic changes of the hyperoxidized Prdx has been shown in cultured RBCs[52], this phenomenon cannot be explained by virtue of sulfiredoxin but may be caused by the proteolytic removal of hyperoxidized Prdx2. Because Prdx2 is involved in maintaining hemoglobin stability[53], hemolytic anemia found in Prdx2-deficient mice may be related to the decrease in the life-span of hemoglobin.
Oxidative stress as a potential cause for anemia and autoimmune responses in NZB mice
SOD1-deficient mice produce anti-RBC autoantibody and ultimately develop lupus nephritis-like symptoms in the gerontic stage[15,31], so that we hypothesized that oxidative stress is one of the causal factors for some autoimmune diseases such as SLE and AIHA in C57BL/6 mice. However SOD1 deficiency is far from a physiologic condition, and we have tried to validate this hypothesis based on physiological conditions using NZB mice.
NZB mice[54] and (NZB × NZW)F1 mice[55] are the established model animals that spontaneously develop AIHA and SLE, respectively, at around 40-50 wk of age. Abnormal proteolytic cleavage of the membrane proteins of RBCs has been proposed as a likely cause because the cleaved membrane proteins, such as band 3, are highly antigenic[56-58]. However, elevated proteolytic activity in the RBCs of AIHA patients or AIHA-prone mice is unknown. Despite extensive investigation on the etiology of the mice, it remains unclear what actually triggers the autoantibody production in the NZB mice[5].
We first recognized that the ROS levels are originally high at a young age (4 wk) and increase as NZB mice age compared to control mice[38]. Increases in the autoantibodies against RBCs show a correlation with the elevated levels of ROS in RBCs. Antioxidants such as NAC suppress autoantibody production in the mice, supporting the oxidative stress theory of AIHA in mice. The onset of AIHA occurs prematurely and mortality increases in the SOD1-deficient congenic NZB mice compared with control NZB mice[59]. The transgenic expression of human SOD1 in RBCs reduces oxidative stress to RBCs and oxidative modification of lipids and proteins and consequently rescues the AIHA phenotypes in NZB mice. Figure 2 provides a schematic mechanism for the onset of AIHA based on our hypothesis regarding the oxidative damage of RBCs. Because oxidative modification elevates during aging and oxidized molecules are highly antigenic, oxidative stress would elevate the autoantibodies by increasing the autoantigens, and would ultimately cause AIHA onset.
Figure 2.
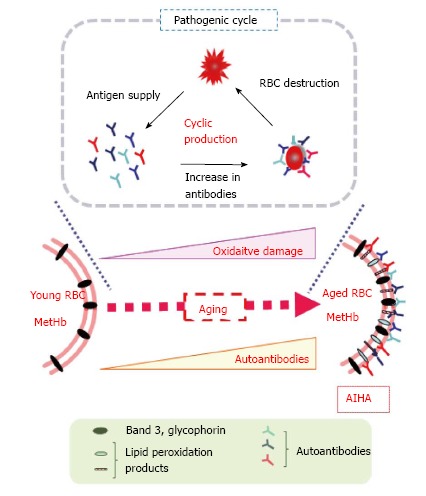
Hypothetical role of oxidative stress in triggering the autoimmune reaction against red blood cells in New Zealand Black mice. Elevated ROS trigger oxidative modification of RBC components and result in the production of oxidatively modified compounds such as 4-hydroxy 2-nonenal and acrolein that are highly antigenic. During aging, the oxidation of susceptible molecules and the production of antibodies recognizing them occurs repeatedly, which results in an accumulation of epitopes and autoantibodies. The elevated levels of autoantibodies ultimately trigger AIHA in aged NZB mice. RBCs: Red blood cells; AIHA: Autoimmune hemolytic anemia; MetHb: Methemoglobin; NZB: New Zealand Black; ROS: Reactive oxygen species.
Either suppression of the antioxidative/redox system or activation of ROS generation causes the elevated ROS in RBCs. As mentioned above, the mechanism of G6PD deficiency-induced anemia is attributed to a short supply of NADPH, which triggers the oxidation of sulfhydryls in RBCs[4]. However, no report has shown abnormalities in glucose G6PD in the RBCs of NZB mice. Regarding the antioxidative enzymes catalase, SOD, glutathione peroxidase, and glutathione reductase, nonsynonymous nucleotide polymorphisms have been identified in the genes in 10 inbred mouse strains, including NZB mice[60]. Thus, the origin of oxidative stress in NZB mice is unclear as of this writing.
Oxidative stress as a potential cause for SLE
Superoxide anion diffuses across the RBC membrane via the anion channel band 3 protein[61], which is a potent antigenic molecule in RBCs[56-58]. ROS appear to derive from inside the RBCs because lipid peroxides are high in the RBCs but about the same in plasma among the congenic mouse groups. Based on theoretical calculation, more than 100 mol/L of superoxide is released daily from hemoglobin autoxidation[12].
Involvement of oxidative stress has been implied in the pathogenesis of human SLE. For example, lipid peroxidation product 4-hydroxy 2-nonenal may modify Ro60, which is the 60-kDa autoantigen of autoimmunity in both SLE Sjögren syndrome, and differentially participate in Sjögren syndrome or SLE[62,63]. Children with SLE carry increased levels of 4-hydroxy 2-nonenal-modified proteins in plasma[64]. Plasma concentrations of 4-hydroxy 2-nonenal as well as malondialdehyde and oxidized glutathione increase during aging in human plasma and RBCs[65]. Both mitochondrial electron transport chain activity at complex I and oxygen consumption are increased in the lymphocytes of SLE patients[66]. On the contrary, NAC suppresses oxygen consumption and hydrogen peroxide levels. Other studies have shown the beneficial effects that antioxidants such as vitamin E, all-trans-retinoic acid, fish oil, and cystamine has on (NZB X NZW)F1 mice[67-69].
Oxidative modification is caused by relatively large amounts of ROS and generally causes oxidation in a non-specific manner. While lymphocytes are defective, and aberrant immune responses occur in AIHA and SLE, it is unclear how they are stimulated to produce autoantibodies. Because oxidized cells are efficiently phagocytosed by macrophages, there is more chance for the immune system to recognize the resultant oxidized molecules as antigens[70,71]. In fact, oxidatively modified albumin is well recognized by the antibodies from SLE patients[72], and oxidatively modified lipids are identified as epitopes for innate immunity and are responsible for diseases such as atherogenesis[73-75]. Lipid peroxidation products, such as 4-hydroxy 2-nonenal and acrolein, have been identified as bona fide epitopes for autoantibodies on RBC membranes[15]. Thus, oxidative stress participates in the formation of novel epitopes by oxidizing proteins and lipids. It is also noteworthy that anti-DNA antibodies, which are typically elevated in SLE patients, also recognize 4-hydroxy 2-nonenal-bound proteins[76,77].
Hypothetical mechanism for SLE onset in (NZB x NZW)F1 mice
An early genetic study suggests that three genes, one from NZB and two from NZW mice, are involved in the development of SLE in the (NZB x NZW) F1 mice and that the gene from NZB mice should function dominantly[78]. Recent genetic studies have indicated several candidate genes for AIHA and/or SLE in model mice[79]. Three major genomic intervals (Sle1, Sle2, and Sle3) have been identified on the New Zealand mouse strains and regarded as systemic autoimmune disease susceptibility loci in NZM2410 mice, which is an acute lupus-prone strain derived from a cross between NZB and NZW[28,30]. High titers of IgG autoantibodies against nuclear proteins and DNA are produced by B6 mice congenic for the Sle1 locus[80]. T cells specific for histone are present[81], implicating Sle1 in the loss of tolerance that leads to the development of antinuclear antibodies. The Sle1b sublocus contains the SLAM (signaling lymphocyte activating molecule) family (Slamf) genes derived from the lupus-prone NZW mice[82,83].
Several candidate genes for autoimmune diseases in humans have also been screened out by genome-wide association studies[84]. Those genes include HLA, STAT4, and PTPN22. Among them, an allelic variant of protein tyrosine phosphatase nonreceptor 22 (PTPN22) shows the most promise because it has been associated with multiple human autoimmune diseases, such as type 1 diabetes, rheumatoid arthritis, and SLE. PTPN22 encodes lymphoid tyrosine phosphatase (Lyp) which participates in the negative regulation of T-cell receptor (TCR) proximal signaling[85,86]. Lyp is also referred to as PEST domain-enriched tyrosine phosphatase (Pep), and it suppresses the activity of the Src family protein tyrosine kinases and inhibits T-cell activity[87]. Because PEST domain, which is rich in proline (P)-glutamate (E)-serine (S)-threonine (T), undergoes rapid degradation, Lyp is vulnerable to proteolytic cleavage. Lyp reportedly negatively regulates T cell receptor signaling[88,89], and the decreased activity would conversely activate the signaling pathway.
In the past two decades, the signaling function of ROS has attracted much attention in the research field of oxidative stress. In this aspect, ROS specifically inactivates susceptible molecules, e.g., phosphotyrosine phosphatase (PTP) families such as PTP1B, Cdc25, SHP1 and SHP2[90,91]. PTP has reactive cys-SH at its catalytic center, which is a preferred target of locally produced ROS. Multiple reports have indicated that PTP variants are linked to human hereditary disorders[92], which indicate that PTP activities play pivotal roles and hence oxidative inactivation affects a variety of cells including lymphocytes.
Because Lyp/Pep is a member of the PTP superfamily and easily oxidized by ROS such as hydrogen peroxide, it may play a role in the sustained activation of lymphocytes, and, hence, it would also play a role in the autoimmune response. A Pep variant (Pep-R619W; Rep with substitution of arginine-619 to Tryptophane-619) protein linked to autoimmune disease is more rapidly degraded and shows greater association with, and in vitro cleavage by, calpain 1 than normal allele Pep-R619[93]. Conversely, Pep overexpression in T cells attenuates autoimmune diabetes in NOD mice by preferentially modulating TCR signaling-mediated functions in diabetogenic T cells but not in regulatory T cells[94]. Lyp-R620W is also involved in the breakdown of peripheral tolerance and in the entry of autoreactive B cells into the naive B cell compartment. Moreover, lymphocytes with a variant of Pep-R619W, corresponding to human Lyp-R620W, are hyper-responsive to antigen-receptor engagement. Thus, Pep-R619W uniquely modulates T and B cell homeostasis, leading to a loss in tolerance[95].
Elevated ROS would cause inactivation of Lyp/Pep by oxidizing catalytic Cys and may accelerate its degradation via the PEST domain. If ROS inactivates Lyp/Pep, the incidence of autoimmune response would be elevated. This oxidative stress-triggered SLE onset is only hypothetical at this moment and hence requires direct demonstration. The crystallographic analysis of Lyp shows a unique disulfide bond that may play a role in protecting the enzyme from irreversible oxidation[96], and hydrogen peroxide actually inactivates the Lyp phosphatase to a lesser extent compared with CD45 phosphatase[97]. Based on the literature and our own observations, we can propose a hypothetical model to explain SLE onset in (NZB x NZW)F1 mice (Figure 3). Because the F1 mice inherit a SLAM variant from NZW mice and high levels of ROS from NZB mice, which may oxidatively inactivate the Lyp/Pep, lymphocytes are hyper-activated, leading to SLE onset in aged mice. Low CD45 phosphotyrosine phosphatase activities that have been reported by two groups[98,99] may support our hypothesis.
Figure 3.
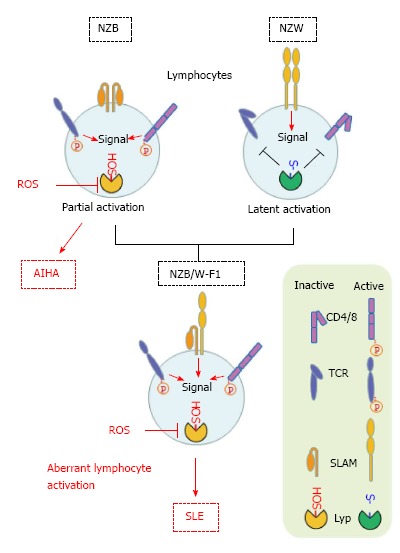
Hypothetical model for triggering systemic lupus erythematosus in (NZB x NZW) F1 mice. Lyp/pep suppressively regulates lymphocyte receptors such as CD4/8 and TCR. Production of ROS are originally high in the cells derived from NZB mice and oxidize the sulfhydryl group in the catalytic Cys to sulfenic acid (-SOH). So that Lyp/pep proteins in lymphocytes in the F1 mice are prone to oxidative inactivation. On the other hand, lymphocytes in NZW mice possess the variant form of SLAM that would be responsible for sustained activation of the lymphocytes. (NZB x NZW) F1 mice inherit this potentially pathogenic nature from parental strains. SLE: Systemic lupus erythematosus; ROS: Reactive oxygen species; NZB: New Zealand black; TCR: T-cell receptor; AIHA: Autoimmune hemolytic anemia.
Potential roles of oxidative stress in lupus nephritis
Lupus nephritis is a serious pathological condition of SLE. The incidence of SLE in women is nine times greater than in men[100], while the sex difference is not observed for the autoantibody production in SOD1-deficient mice[31]. Immune complex formation and complement activation are major causes, but other pathogenic factor is involved in lupus nephritis[101]. Despite deficiency of the gamma chain of the Fc receptor in F1 mice, ameliorated glomerulonephritis, immune complex deposition still occurs[102]. Thus, glomerular deposition of C1q as immune complexes, complement activation, and Fc gamma receptor activation together appear to be required for the renal damage[103]. As discussed above, oxidative stress is a potent risk factor for the autoantibody production by affecting immune system and hence would be involved in the kidney damage by increasing the immune complex deposition. However, since kidney is the organ considerably susceptible to oxidative damage[104], elevated ROS may directly affect the renal function and be an independent risk factor for lupus nephritis in SLE.
Perspectives
In addition to the results of studies on the supplementary administration of antioxidative compounds, observations from pathological models and genetically modified mice support the view that ROS are one of the underlying mechanisms for AIHA and/or SLE. ROS cause opposing responses; they trigger cell growth arrest and accelerate cellular senescence, but stimulate the cellular proliferation on the other hand[90,91]. In the latter case, transient elevation in ROS levels occurs when cells are stimulated by growth factor and is involved in sustaining the signal transduction. Antioxidant therapy appears to be effective, but may be potentially adverse because of a possible impairment of the ROS signaling during proliferation of hematopoietic cells. Elucidation of target molecules by oxidative modification and pathogenesis could lead to safer forms of preventive and therapeutic treatment.
Footnotes
P- Reviewer: Fujiwara N, Han C, La Montagna G, Yong D S- Editor: Tian YL L- Editor: A E- Editor: Lu YJ
Supported by The Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation from the Japan Society for the Promotion of Sciences.
Conflict-of-interest: There are no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 18, 2014
First decision: August 14, 2014
Article in press: February 9, 2015
References
- 1.Bauer C, Kurtz A. Oxygen sensing in the kidney and its relation to erythropoietin production. Annu Rev Physiol. 1989;51:845–856. doi: 10.1146/annurev.ph.51.030189.004213. [DOI] [PubMed] [Google Scholar]
- 2.Sato Y, Yanagita M. Renal anemia: from incurable to curable. Am J Physiol Renal Physiol. 2013;305:F1239–F1248. doi: 10.1152/ajprenal.00233.2013. [DOI] [PubMed] [Google Scholar]
- 3.Canavesi E, Alfieri C, Pelusi S, Valenti L. Hepcidin and HFE protein: Iron metabolism as a target for the anemia of chronic kidney disease. World J Nephrol. 2012;1:166–176. doi: 10.5527/wjn.v1.i6.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beutler E. Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111:16–24. doi: 10.1182/blood-2007-04-077412. [DOI] [PubMed] [Google Scholar]
- 5.Izui S. Autoimmune hemolytic anemia. Curr Opin Immunol. 1994;6:926–930. doi: 10.1016/0952-7915(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 6.Scatizzi JC, Haraldsson MK, Pollard KM, Theofilopoulos AN, Kono DH. The Lbw2 locus promotes autoimmune hemolytic anemia. J Immunol. 2012;188:3307–3314. doi: 10.4049/jimmunol.1103561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon C, Bradley H, Waring RH, Emery P. Abnormal sulphur oxidation in systemic lupus erythematosus. Lancet. 1992;339:25–26. doi: 10.1016/0140-6736(92)90144-r. [DOI] [PubMed] [Google Scholar]
- 8.Dhaliwal G, Cornett PA, Tierney LM. Hemolytic anemia. Am Fam Physician. 2004;69:2599–2606. [PubMed] [Google Scholar]
- 9.Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013;9:674–686. doi: 10.1038/nrrheum.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halliwell B, Gutteridge JMC. Free Radical Biology and Medicine. 4th ed. Oxford: Oxford University Press; 2007. [Google Scholar]
- 11.Winterbourn CC. Oxidative reactions of hemoglobin. Methods Enzymol. 1990;186:265–272. doi: 10.1016/0076-6879(90)86118-f. [DOI] [PubMed] [Google Scholar]
- 12.Johnson RM, Goyette G, Ravindranath Y, Ho YS. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic Biol Med. 2005;39:1407–1417. doi: 10.1016/j.freeradbiomed.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Rhee SG, Woo HA, Kil IS, Bae SH. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev. 2007;21:267–283. doi: 10.1016/j.blre.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Iuchi Y, Okada F, Takamiya R, Kibe N, Tsunoda S, Nakajima O, Toyoda K, Nagae R, Suematsu M, Soga T, et al. Rescue of anaemia and autoimmune responses in SOD1-deficient mice by transgenic expression of human SOD1 in erythrocytes. Biochem J. 2009;422:313–320. doi: 10.1042/BJ20090176. [DOI] [PubMed] [Google Scholar]
- 16.Criswell KA, Sulkanen AP, Hochbaum AF, Bleavins MR. Effects of phenylhydrazine or phlebotomy on peripheral blood, bone marrow and erythropoietin in Wistar rats. J Appl Toxicol. 2000;20:25–34. doi: 10.1002/(sici)1099-1263(200001/02)20:1<25::aid-jat624>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Mqadmi A, Zheng X, Yazdanbakhsh K. CD4+CD25+ regulatory T cells control induction of autoimmune hemolytic anemia. Blood. 2005;105:3746–3748. doi: 10.1182/blood-2004-12-4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fagiolo E, Toriani-Terenzi C. Mechanisms of immunological tolerance loss versus erythrocyte self-antigens and autoimmune hemolytic anemia. Autoimmunity. 2003;36:199–204. doi: 10.1080/0891693031000151238. [DOI] [PubMed] [Google Scholar]
- 19.Murakami M, Yoshioka H, Shirai T, Tsubata T, Honjo T. Prevention of autoimmune symptoms in autoimmune-prone mice by elimination of B-1 cells. Int Immunol. 1995;7:877–882. doi: 10.1093/intimm/7.5.877. [DOI] [PubMed] [Google Scholar]
- 20.Fossati-Jimack L, Azeredo da Silveira S, Moll T, Kina T, Kuypers FA, Oldenborg PA, Reininger L, Izui S. Selective increase of autoimmune epitope expression on aged erythrocytes in mice: implications in anti-erythrocyte autoimmune responses. J Autoimmun. 2002;18:17–25. doi: 10.1006/jaut.2001.0563. [DOI] [PubMed] [Google Scholar]
- 21.Hall AM, Ward FJ, Shen CR, Rowe C, Bowie L, Devine A, Urbaniak SJ, Elson CJ, Barker RN. Deletion of the dominant autoantigen in NZB mice with autoimmune hemolytic anemia: effects on autoantibody and T-helper responses. Blood. 2007;110:4511–4517. doi: 10.1182/blood-2007-06-094383. [DOI] [PubMed] [Google Scholar]
- 22.Santiago-Raber ML, Laporte C, Reininger L, Izui S. Genetic basis of murine lupus. Autoimmun Rev. 2004;3:33–39. doi: 10.1016/S1568-9972(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 23.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996;98:1107–1113. doi: 10.1172/JCI118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaishnaw AK, Toubi E, Ohsako S, Drappa J, Buys S, Estrada J, Sitarz A, Zemel L, Chu JL, Elkon KB. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum. 1999;42:1833–1842. doi: 10.1002/1529-0131(199909)42:9<1833::AID-ANR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 26.Kojima T, Horiuchi T, Nishizaka H, Sawabe T, Higuchi M, Harashima SI, Yoshizawa S, Tsukamoto H, Nagasawa K, Niho Y. Analysis of fas ligand gene mutation in patients with systemic lupus erythematosus. Arthritis Rheum. 2000;43:135–139. doi: 10.1002/1529-0131(200001)43:1<135::AID-ANR17>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 27.Dubois EL, Horowitz RE, Demopoulos HB, Teplitz R. NZB/NZW mice as a model of systemic lupus erythematosus. JAMA. 1966;195:285–289. [PubMed] [Google Scholar]
- 28.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 29.Morel L, Mohan C, Yu Y, Croker BP, Tian N, Deng A, Wakeland EK. Functional dissection of systemic lupus erythematosus using congenic mouse strains. J Immunol. 1997;158:6019–6028. [PubMed] [Google Scholar]
- 30.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iuchi Y, Okada F, Onuma K, Onoda T, Asao H, Kobayashi M, Fujii J. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochem J. 2007;402:219–227. doi: 10.1042/BJ20061386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin FM, Bydlon G, Friedman JS. SOD2-deficiency sideroblastic anemia and red blood cell oxidative stress. Antioxid Redox Signal. 2006;8:1217–1225. doi: 10.1089/ars.2006.8.1217. [DOI] [PubMed] [Google Scholar]
- 33.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 34.Lee TH, Kim SU, Yu SL, Kim SH, Park DS, Moon HB, Dho SH, Kwon KS, Kwon HJ, Han YH, et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101:5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 35.Johnson RM, Goyette G, Ravindranath Y, Ho YS. Red cells from glutathione peroxidase-1-deficient mice have nearly normal defenses against exogenous peroxides. Blood. 2000;96:1985–1988. [PubMed] [Google Scholar]
- 36.Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- 37.Starzyński RR, Canonne-Hergaux F, Willemetz A, Gralak MA, Woliński J, Styś A, Olszak J, Lipiński P. Haemolytic anaemia and alterations in hepatic iron metabolism in aged mice lacking Cu,Zn-superoxide dismutase. Biochem J. 2009;420:383–390. doi: 10.1042/BJ20082137. [DOI] [PubMed] [Google Scholar]
- 38.Iuchi Y, Kibe N, Tsunoda S, Suzuki S, Mikami T, Okada F, Uchida K, Fujii J. Implication of oxidative stress as a cause of autoimmune hemolytic anemia in NZB mice. Free Radic Biol Med. 2010;48:935–944. doi: 10.1016/j.freeradbiomed.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Cho CS, Lee S, Lee GT, Woo HA, Choi EJ, Rhee SG. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid Redox Signal. 2010;12:1235–1246. doi: 10.1089/ars.2009.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu J, Holmgren A. Selenoproteins. J Biol Chem. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 42.Friedman JS, Rebel VI, Derby R, Bell K, Huang TT, Kuypers FA, Epstein CJ, Burakoff SJ. Absence of mitochondrial superoxide dismutase results in a murine hemolytic anemia responsive to therapy with a catalytic antioxidant. J Exp Med. 2001;193:925–934. doi: 10.1084/jem.193.8.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohanty JG, Nagababu E, Friedman JS, Rifkind JM. SOD2 deficiency in hematopoietic cells in mice results in reduced red blood cell deformability and increased heme degradation. Exp Hematol. 2013;41:316–321. doi: 10.1016/j.exphem.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin FM, Xu X, von Löhneysen K, Gilmartin TJ, Friedman JS. SOD2 deficient erythroid cells up-regulate transferrin receptor and down-regulate mitochondrial biogenesis and metabolism. PLoS One. 2011;6:e16894. doi: 10.1371/journal.pone.0016894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedman JS, Lopez MF, Fleming MD, Rivera A, Martin FM, Welsh ML, Boyd A, Doctrow SR, Burakoff SJ. SOD2-deficiency anemia: protein oxidation and altered protein expression reveal targets of damage, stress response, and antioxidant responsiveness. Blood. 2004;104:2565–2573. doi: 10.1182/blood-2003-11-3858. [DOI] [PubMed] [Google Scholar]
- 46.Johnson RM, Ho YS, Yu DY, Kuypers FA, Ravindranath Y, Goyette GW. The effects of disruption of genes for peroxiredoxin-2, glutathione peroxidase-1, and catalase on erythrocyte oxidative metabolism. Free Radic Biol Med. 2010;48:519–525. doi: 10.1016/j.freeradbiomed.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cox AG, Winterbourn CC, Hampton MB. Measuring the redox state of cellular peroxiredoxins by immunoblotting. Methods Enzymol. 2010;474:51–66. doi: 10.1016/S0076-6879(10)74004-0. [DOI] [PubMed] [Google Scholar]
- 48.Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109:2611–2617. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- 49.Low FM, Hampton MB, Winterbourn CC. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid Redox Signal. 2008;10:1621–1630. doi: 10.1089/ars.2008.2081. [DOI] [PubMed] [Google Scholar]
- 50.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 51.Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-cys peroxiredoxins. J Biol Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 52.O'Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. 2011;469:498–503. doi: 10.1038/nature09702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han YH, Kim SU, Kwon TH, Lee DS, Ha HL, Park DS, Woo EJ, Lee SH, Kim JM, Chae HB, et al. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem Biophys Res Commun. 2012;426:427–432. doi: 10.1016/j.bbrc.2012.08.113. [DOI] [PubMed] [Google Scholar]
- 54.Helyer BJ, Howie JB. Spontaneous auto-immune disease in NZB/BL mice. Br J Haematol. 1963;9:119–131. doi: 10.1111/j.1365-2141.1963.tb05450.x. [DOI] [PubMed] [Google Scholar]
- 55.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 56.Beppu M, Mizukami A, Nagoya M, Kikugawa K. Binding of anti-band 3 autoantibody to oxidatively damaged erythrocytes. Formation of senescent antigen on erythrocyte surface by an oxidative mechanism. J Biol Chem. 1990;265:3226–3233. [PubMed] [Google Scholar]
- 57.Poole J. Red cell antigens on band 3 and glycophorin A. Blood Rev. 2000;14:31–43. doi: 10.1054/blre.1999.0124. [DOI] [PubMed] [Google Scholar]
- 58.Shen CR, Youssef AR, Devine A, Bowie L, Hall AM, Wraith DC, Elson CJ, Barker RN. Peptides containing a dominant T-cell epitope from red cell band 3 have in vivo immunomodulatory properties in NZB mice with autoimmune hemolytic anemia. Blood. 2003;102:3800–3806. doi: 10.1182/blood-2002-07-2125. [DOI] [PubMed] [Google Scholar]
- 59.Konno T, Otsuki N, Kurahashi T, Kibe N, Tsunoda S, Iuchi Y, Fujii J. Reactive oxygen species exacerbate autoimmune hemolytic anemia in New Zealand Black mice. Free Radic Biol Med. 2013;65:1378–1384. doi: 10.1016/j.freeradbiomed.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Guo Z, Higuchi K, Mori M. Spontaneous hypomorphic mutations in antioxidant enzymes of mice. Free Radic Biol Med. 2003;35:1645–1652. doi: 10.1016/j.freeradbiomed.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 61.Lynch RE, Fridovich I. Permeation of the erythrocyte stroma by superoxide radical. J Biol Chem. 1978;253:4697–4699. [PubMed] [Google Scholar]
- 62.Scofield RH, Kurien BT, Ganick S, McClain MT, Pye Q, James JA, Schneider RI, Broyles RH, Bachmann M, Hensley K. Modification of lupus-associated 60-kDa Ro protein with the lipid oxidation product 4-hydroxy-2-nonenal increases antigenicity and facilitates epitope spreading. Free Radic Biol Med. 2005;38:719–728. doi: 10.1016/j.freeradbiomed.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 63.Kurien BT, Porter A, Dorri Y, Iqbal S, D’Souza A, Singh A, Asfa S, Cartellieri M, Mathias K, Matsumoto H, et al. Degree of modification of Ro60 by the lipid peroxidation by-product 4-hydroxy-2-nonenal may differentially induce Sjögren syndrome or systemic lupus erythematosus in BALB/c mice. Free Radic Biol Med. 2011;50:1222–1233. doi: 10.1016/j.freeradbiomed.2010.10.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grune T, Michel P, Sitte N, Eggert W, Albrecht-Nebe H, Esterbauer H, Siems WG. Increased levels of 4-hydroxynonenal modified proteins in plasma of children with autoimmune diseases. Free Radic Biol Med. 1997;23:357–360. doi: 10.1016/s0891-5849(96)00586-2. [DOI] [PubMed] [Google Scholar]
- 65.Gil L, Siems W, Mazurek B, Gross J, Schroeder P, Voss P, Grune T. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radic Res. 2006;40:495–505. doi: 10.1080/10715760600592962. [DOI] [PubMed] [Google Scholar]
- 66.Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex I is regulated by N-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal. 2014;21:56–65. doi: 10.1089/ars.2013.5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, Funauchi M, Kanamaru A. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol. 2003;170:5793–5798. doi: 10.4049/jimmunol.170.11.5793. [DOI] [PubMed] [Google Scholar]
- 68.Halade GV, Rahman MM, Bhattacharya A, Barnes JL, Chandrasekar B, Fernandes G. Docosahexaenoic acid-enriched fish oil attenuates kidney disease and prolongs median and maximal life span of autoimmune lupus-prone mice. J Immunol. 2010;184:5280–5286. doi: 10.4049/jimmunol.0903282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsu TC, Chiu CC, Wang YW, Tzang BS. Effects of cystamine on antioxidant activities and regulatory T cells in lupus-prone mice. J Cell Mol Med. 2013;17:1308–1315. doi: 10.1111/jcmm.12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.López-Revuelta A, Sánchez-Gallego JI, García-Montero AC, Hernández-Hernández A, Sánchez-Yagüe J, Llanillo M. Membrane cholesterol in the regulation of aminophospholipid asymmetry and phagocytosis in oxidized erythrocytes. Free Radic Biol Med. 2007;42:1106–1118. doi: 10.1016/j.freeradbiomed.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 72.Rasheed Z, Ahmad R, Rasheed N, Ali R. Enhanced recognition of reactive oxygen species damaged human serum albumin by circulating systemic lupus erythematosus autoantibodies. Autoimmunity. 2007;40:512–520. doi: 10.1080/08916930701574331. [DOI] [PubMed] [Google Scholar]
- 73.Binder CJ, Shaw PX, Chang MK, Boullier A, Hartvigsen K, Hörkkö S, Miller YI, Woelkers DA, Corr M, Witztum JL. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–1363. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 74.Chou MY, Hartvigsen K, Hansen LF, Fogelstrand L, Shaw PX, Boullier A, Binder CJ, Witztum JL. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med. 2008;263:479–488. doi: 10.1111/j.1365-2796.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 75.Hazen SL. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J Biol Chem. 2008;283:15527–15531. doi: 10.1074/jbc.R700054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Toyoda K, Nagae R, Akagawa M, Ishino K, Shibata T, Ito S, Shibata N, Yamamoto T, Kobayashi M, Takasaki Y, et al. Protein-bound 4-hydroxy-2-nonenal: an endogenous triggering antigen of antI-DNA response. J Biol Chem. 2007;282:25769–25778. doi: 10.1074/jbc.M703039200. [DOI] [PubMed] [Google Scholar]
- 77.Otaki N, Chikazawa M, Nagae R, Shimozu Y, Shibata T, Ito S, Takasaki Y, Fujii J, Uchida K. Identification of a lipid peroxidation product as the source of oxidation-specific epitopes recognized by anti-DNA autoantibodies. J Biol Chem. 2010;285:33834–33842. doi: 10.1074/jbc.M110.165175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knight JG, Adams DD. Three genes for lupus nephritis in NZB x NZW mice. J Exp Med. 1978;147:1653–1660. doi: 10.1084/jem.147.6.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kono DH, Theofilopoulos AN. Genetics of SLE in mice. Springer Semin Immunopathol. 2006;28:83–96. doi: 10.1007/s00281-006-0030-7. [DOI] [PubMed] [Google Scholar]
- 80.Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest. 1998;101:1362–1372. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sobel ES, Satoh M, Chen Y, Wakeland EK, Morel L. The major murine systemic lupus erythematosus susceptibility locus Sle1 results in abnormal functions of both B and T cells. J Immunol. 2002;169:2694–2700. doi: 10.4049/jimmunol.169.5.2694. [DOI] [PubMed] [Google Scholar]
- 82.Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, Yim YS, Pertsemlidis A, Garner HR, Morel L, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–780. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 83.Wang A, Batteux F, Wakeland EK. The role of SLAM/CD2 polymorphisms in systemic autoimmunity. Curr Opin Immunol. 2010;22:706–714. doi: 10.1016/j.coi.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 84.Delgado-Vega A, Sánchez E, Löfgren S, Castillejo-López C, Alarcón-Riquelme ME. Recent findings on genetics of systemic autoimmune diseases. Curr Opin Immunol. 2010;22:698–705. doi: 10.1016/j.coi.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189:111–121. doi: 10.1084/jem.189.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM. Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood. 1999;93:2013–2024. [PubMed] [Google Scholar]
- 87.Veillette A, Rhee I, Souza CM, Davidson D. PEST family phosphatases in immunity, autoimmunity, and autoinflammatory disorders. Immunol Rev. 2009;228:312–324. doi: 10.1111/j.1600-065X.2008.00747.x. [DOI] [PubMed] [Google Scholar]
- 88.Gianchecchi E, Palombi M, Fierabracci A. The putative role of the C1858T polymorphism of protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun Rev. 2013;12:717–725. doi: 10.1016/j.autrev.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 89.Zheng J, Petersen F, Yu X. The role of PTPN22 in autoimmunity: learning from mice. Autoimmun Rev. 2014;13:266–271. doi: 10.1016/j.autrev.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 90.Savitsky PA, Finkel T. Redox regulation of Cdc25C. J Biol Chem. 2002;277:20535–20540. doi: 10.1074/jbc.M201589200. [DOI] [PubMed] [Google Scholar]
- 91.Weibrecht I, Böhmer SA, Dagnell M, Kappert K, Ostman A, Böhmer FD. Oxidation sensitivity of the catalytic cysteine of the protein-tyrosine phosphatases SHP-1 and SHP-2. Free Radic Biol Med. 2007;43:100–110. doi: 10.1016/j.freeradbiomed.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 92.Hendriks WJ, Pulido R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim Biophys Acta. 2013;1832:1673–1696. doi: 10.1016/j.bbadis.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 93.Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, Dong B, Xie G, Qiu F, Hao Z, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43:902–907. doi: 10.1038/ng.904. [DOI] [PubMed] [Google Scholar]
- 94.Yeh LT, Miaw SC, Lin MH, Chou FC, Shieh SJ, Chuang YP, Lin SH, Chang DM, Sytwu HK. Different modulation of Ptpn22 in effector and regulatory T cells leads to attenuation of autoimmune diabetes in transgenic nonobese diabetic mice. J Immunol. 2013;191:594–607. doi: 10.4049/jimmunol.1203380. [DOI] [PubMed] [Google Scholar]
- 95.Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, Moon RT, Liggitt D, Wolf-Yadlin A, Buckner JH, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013;123:2024–2036. doi: 10.1172/JCI66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsai SJ, Sen U, Zhao L, Greenleaf WB, Dasgupta J, Fiorillo E, Orrú V, Bottini N, Chen XS. Crystal structure of the human lymphoid tyrosine phosphatase catalytic domain: insights into redox regulation. Biochemistry. 2009;48:4838–4845. doi: 10.1021/bi900166y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bayley R, Yang P, Buckley CD, Young SP. Measuring the specific activity of the protein tyrosine phosphatase Lyp. J Immunol Methods. 2013;388:33–39. doi: 10.1016/j.jim.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 98.Takeuchi T, Pang M, Amano K, Koide J, Abe T. Reduced protein tyrosine phosphatase (PTPase) activity of CD45 on peripheral blood lymphocytes in patients with systemic lupus erythematosus (SLE) Clin Exp Immunol. 1997;109:20–26. doi: 10.1046/j.1365-2249.1997.4371334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blasini AM, Alonzo E, Chacón R, Riera R, Stekman IL, Rodriguez MA. Abnormal pattern of tyrosine phosphorylation in unstimulated peripheral blood T lymphocytes from patients with systemic lupus erythematosus. Lupus. 1998;7:515–523. doi: 10.1191/096120398678920604. [DOI] [PubMed] [Google Scholar]
- 100.Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 101.Sterner RM, Hartono SP, Grande JP. The Pathogenesis of Lupus Nephritis. J Clin Cell Immunol. 2014;5:205. doi: 10.4172/2155-9899.1000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 103.Trouw LA, Groeneveld TW, Seelen MA, Duijs JM, Bajema IM, Prins FA, Kishore U, Salant DJ, Verbeek JS, van Kooten C, et al. Anti-C1q autoantibodiesdeposit in glomeruli but are only pathogenic in combination with glomerular C1q-containing immune complexes. J Clin Invest. 2004;114:679–688. doi: 10.1172/JCI21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kooman JP, Kotanko P, Schols AM, Shiels PG, Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol. 2014;10:732–742. doi: 10.1038/nrneph.2014.185. [DOI] [PubMed] [Google Scholar]