

Abstract
Hyperoxaluria is characterized by an increased urinary excretion of oxalate. Primary and secondary hyperoxaluria are two distinct clinical expressions of hyperoxaluria. Primary hyperoxaluria is an inherited error of metabolism due to defective enzyme activity. In contrast, secondary hyperoxaluria is caused by increased dietary ingestion of oxalate, precursors of oxalate or alteration in intestinal microflora. The disease spectrum extends from recurrent kidney stones, nephrocalcinosis and urinary tract infections to chronic kidney disease and end stage renal disease. When calcium oxalate burden exceeds the renal excretory ability, calcium oxalate starts to deposit in various organ systems in a process called systemic oxalosis. Increased urinary oxalate levels help to make the diagnosis while plasma oxalate levels are likely to be more accurate when patients develop chronic kidney disease. Definitive diagnosis of primary hyperoxaluria is achieved by genetic studies and if genetic studies prove inconclusive, liver biopsy is undertaken to establish diagnosis. Diagnostic clues pointing towards secondary hyperoxaluria are a supportive dietary history and tests to detect increased intestinal absorption of oxalate. Conservative treatment for both types of hyperoxaluria includes vigorous hydration and crystallization inhibitors to decrease calcium oxalate precipitation. Pyridoxine is also found to be helpful in approximately 30% patients with primary hyperoxaluria type 1. Liver-kidney and isolated kidney transplantation are the treatment of choice in primary hyperoxaluria type 1 and type 2 respectively. Data is scarce on role of transplantation in primary hyperoxaluria type 3 where there are no reports of end stage renal disease so far. There are ongoing investigations into newer modalities of diagnosis and treatment of hyperoxaluria. Clinical differentiation between primary and secondary hyperoxaluria and further between the types of primary hyperoxaluria is very important because of implications in treatment and diagnosis. Hyperoxaluria continues to be a challenging disease and a high index of clinical suspicion is often the first step on the path to accurate diagnosis and management.
Keywords: Primary hyperoxaluria, Transplantation, Renal stones, Secondary hyperoxaluria, Renal failure
Core tip: Hyperoxaluria is a disorder characterized by increased urinary oxalate excretion. Primary hyperoxaluria is an inherited defect of oxalate metabolism while secondary hyperoxaluria is seen in states of increased ingestion of oxalate, its precursors or altered gut flora. These disorders can lead to recurrent renal stones, nephrocalcinosis and eventually end stage renal disease. Despite these common features, the sub types of hyperoxaluria differ in their pathogenesis, severity of clinical presentation and treatment plan. Prompt clinical recognition and distinction between these disorders is essential not only for timely intervention but also impacts prognosis in patients with hyperoxaluria.
INTRODUCTION
Oxalate is the ionic form of oxalic acid and is derived from various animal and plant sources. Oxalate is excreted mainly through the kidneys. Hyperoxaluria is a state of disordered metabolism characterized by an increased urinary excretion of oxalate. The normal daily oxalate excretion in healthy individuals ranges between 10-40 mg per 24 h. Concentrations exceeding 40-45 mg per 24 h are considered as clinical hyperoxaluria[1-3]. This may result from increased endogenous production of oxalate in primary hyperoxaluria (PH) or from increased intestinal absorption or increased intake of oxalate precursors in secondary hyperoxaluria (SH).
Hyperoxaluria has the potential to cause devastating consequences which can present as early as infancy or in the sixth decade of life and if not addressed appropriately, can cause significant morbidity and mortality including End Stage Renal Disease (ESRD)[4]. Elevated plasma oxalate levels lead to oxalate deposition in various organ systems. Systemic oxalosis should be prevented but the diagnosis is often delayed in more than 40% of patients. In a survey by Hoppe et al[5], 30% of the patients were diagnosed only when they had already reached ESRD. In some cases, the diagnosis may first be made when the disease recurs following renal transplant[6]. Hyperoxaluria continues to be a challenging disease and appropriate treatment requires a high index of suspicion and a timely diagnosis.
This review highlights the mechanisms underlying both primary and secondary hyperoxaluria, clinical manifestations, important elements in screening and diagnosis, and our current knowledge of modalities of treatment.
SOURCES OF OXALATE
Oxalate is obtained from exogenous sources as well as endogenous synthesis. Oxalate is abundantly found in plant and animal sources. Dietary sources richest in oxalate include nuts, plums, chocolate, beetroot, strawberries, rhubarb, tofu and spinach[1,7]. Juicing is a recent popular trend where a diet based mainly on fruits and vegetable juices is consumed and may supply a very high amount of daily oxalate[8,9]. Studies have demonstrated that as the dietary intake of oxalate increases, so does the urinary concentration of oxalate[10]. Endogenous synthesis of oxalate occurs in the liver[11] through a pathway that generates glyoxalate as an intermediate molecule[12]. Glyoxalate is synthesized from oxidation of glycolate through enzymatic action of glycolate oxidase or from metabolism of hydroxyproline which is found in collagen or dietary sources. Increased glyoxalate is converted to oxalate by action of lactate dehydrogenase in the absence of enzymatic activity as is seen in the various types of PH[12,13]. This pathway is depicted in Figure 1.
Figure 1.
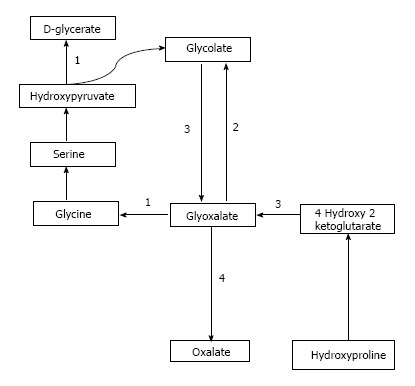
Pathway of oxalate synthesis and enzymatic defects in PH. A: PH1, alanine glyoxalate aminotransferase; B: PH 2, glycolate reductase hydroxy pyruvate reductase; C: PH 3, 4-hydroxy 2-ketoglutarate aldolase; D: Lactate dehydrogenase.
RENAL HANDLING OF OXALATE
Renal oxalate handling comprises glomerular filtration, tubular secretion and tubular reabsorption[14,15]. Glomerular filtration depends on the plasma oxalate levels while tubular transport is mediated by SLC26 family of transport proteins. SLC26A1 mediates oxalate uptake into the cell across the basolateral membrane in exchange for sulfate[16,17]. On the apical side of the tubular cells, SLC26A6 is the dominant chloride-oxalate exchanger which promotes chloride reabsorption in exchange for oxalate secretion and has been implicated in the development of renal stones. This exchanger also mediates intestinal secretion of oxalate and loss of this exchanger has been shown to promote increased intestinal absorption of oxalate in the small intestine[18,19]. In rat kidney, tubular reabsorption has been demonstrated in the S1 and S2 segments of the proximal tubule[14] which may help decrease the tendency for calcium oxalate supersaturation in the earlier parts of the nephron[3].
Overall, the contribution of tubular secretion in addition to glomerular filtration is critical in regulating plasma oxalate levels as a strong correlation has been demonstrated between high plasma oxalate levels and oxalate secretion[20]. It has also been noted that tubular oxalate secretion is increased in PH patients possibly in an attempt to mitigate the life threatening consequences of systemic oxalosis[21]. Increased tubular secretion has also been noted in patients with hyperoxaluria following intestinal bypass[22].
GENETIC AND BIOCHEMICAL BASIS OF DISEASE
Primary hyperoxaluria
Primary hyperoxaluria type 1 (PH1) is the most common and severe form of PH. It accounts for approximately 80% of the cases of PH and is caused by defect in the Vitamin B6 dependent hepatic peroxisomal enzyme, Alanine Glyoxalate Aminotransferase (AGT). This enzyme catalyzes the transamination of L-alanine and glyoxalate to pyruvate and glycine. The enzyme defect has been attributed to a mutation in the AGXT gene located on chromosome 2[23,24].
Primary hyperoxaluria type 2 (PH2) represents about 10% of the patients with PH. Dysfunction of the enzyme glyoxalate/hydroxypyruvate reductase (GRHPR) occurs secondary to a mutation in the GRHPR gene located on chromosome 10[25-27]. Consequently, there is increased urinary excretion of L-glyceric acid and oxalate.
Primary hyperoxaluria type 3 (PH 3) is a recently described entity and it occurs in 10% PH cases. The genetic defect in PH3 has been localized to the HOGA1 gene located on chromosome 9 which codes for the mitochondrial 4-hydroxy 2-oxoglutarate aldolase[28]. This enzyme breaks down 4-hydroxy 2-oxoglutarate into pyruvate and glyoxalate which in turn is converted into oxalate.
SECONDARY HYPEROXALURIA
The causes of SH are increase in dietary and intestinal absorption (enteric hyperoxaluria), excessive intake of oxalate precursors and alteration in intestinal microflora.
Increased dietary intake of oxalate
Oxalate rich dietary sources include rhubarb and spinach and daily intake may be in excess of 1000 mg/d[29]. Increased dietary absorption may occur in “juicing” which is being propagated as a health fad for clearing toxins from the body and also for weight loss. Previously dietary oxalate was thought to make only a minimal (10%-20%) contribution to the amount of oxalate excreted in urine but studies have shown that this is not correct. In a study by Holmes et al[10], dietary intake contributed to about 50% of the oxalate secretion proving that dietary ingestion is an important determinant in total oxalate excretion. Bioavailability of oxalate from food and, thus, urinary oxalate, is also influenced by the forms of oxalate in the food, techniques of food processing and cooking and other constituents in the meal[30]. Dietary ingestion of oxalate is reduced by concurrent ingestion of calcium or magnesium which complex with oxalate and form insoluble salts[10,31].
Hyperoxaluria associated with fat malabsorption
Fat malabsorption increases the intestinal absorption of oxalate due to increased intestinal permeability to oxalate and formation of calcium and fatty acid complexes leading to increased amounts of soluble oxalate. An intact colon is required for increased oxalate absorption via this mechanism[32]. This form of hyperoxaluria is seen in partial gastrectomy, bariatric surgery, jejunoileal bypass, and inflammatory bowel disease[7,33].
Role of oxalobacter formigenes
Oxalobacter formigenes (O. formigenes) is an aerobic gram negative bacterium that uses oxalate as its energy source and decreases intestinal absorption of oxalate and thus reduces urinary oxalate excretion[34,35]. This has been well documented in both human and animal experiments[36,37]. Loss of this bacterium occurs after the use of antibiotics[38] and its restoration may have a role in treatment of hyperoxaluria.
Excess intake of oxalate precursors
Ascorbic acid (Vitamin C) is a precursor of oxalate and intake of excessive quantities of vitamin C may result in precipitation of calcium oxalate[39,40]. Oxalate is a product of ethylene glycol causing calcium oxalate deposition and renal failure[41,42]. Hyperoxaluria has also been reported following renal transplantation due to mobilization of oxalate and deposition within the renal allograft[43]. Increased intestinal absorption of oxalate and tubular secretion has also been reported in patients with cystic fibrosis leading to hyperoxaluria[3,44,45].
“Juicing” deserves a special mention as it supplies a high amount of daily oxalate. The increased amount of fluid intake in the juices increases the paracellular absorption of oxalate in the intestines. This may overwhelm the ability of the kidney to excrete the increased dietary load especially in patients with chronic kidney disease. Oxalate is ingested in the fruits and vegetables used to make the juices such as kiwi, spinach and beetroot. Low calcium intake and ingestion of excess of vitamin C is also noted which together with the oxalate intake heighten the risk of acute kidney injury[8,9].
CLINICAL PRESENTATION
The prevalence of PH1 is approximately 1-3 cases per million population[46,47]. At least 1% of the ESRD seen in the pediatric population is attributable to PH1 in European and Japanese studies[48,49]. It is more frequently seen in Kuwaiti and Tunisian populations where consanguineous marriages are practiced[50,51]. PH1 is the most severe type of PH although there is significant variability in its clinical presentation. Patients may present early in life during infancy with life threatening oxalosis and failure to thrive or in adulthood after passing an occasional stone. Overall, the disease is characterized by recurrent nephrolithiasis and progressive nephrocalcinosis leading to renal damage and as a result, the majority of the patients reach ESRD during 3rd-5th decade of life[52,53].
PH2 is a less aggressive form of PH with better preservation of renal function and lower incidence of end stage renal disease and less severe nephrocalcinosis compared to PH1. The differences are accounted for by the higher oxalate excretion in PH1 and altered urine composition with reduced urinary levels of citrate and magnesium in PH1 compared to PH2[54].
PH3 generally presents with recurrent nephrolithiasis in the early decades of life. It is also characterized by the increase in urinary calcium levels and genetic defects in the HOGA1 gene have also been implicated in cases of idiopathic calcium oxalate urolithiasis[55]. The disease course is more benign compared to other forms and although limited clinical data is available, no cases of ESRD have been reported to date with PH3[56,57].
Patients with secondary hyperoxaluria have a predisposition to developing recurrent calcium oxalate stones due to the underlying disorder. This leads to worsening renal damage and progression to ESRD. Systemic oxalosis is less common in secondary hyperoxaluria but reported in some severe cases of Crohn’s disease[58].
SYSTEMIC OXALOSIS
Calcium oxalate salts are poorly soluble in body fluids. Calcium oxalate deposits within renal tissue as nephrocalcinosis and also forms renal stones (nephrolithiasis). This leads to progressive renal injury and inflammation and tubular obstruction leading to interstitial fibrosis, declining renal function and eventually ESRD[52,59].
When glomerular filtration rate (GFR) drops below 30-40 mL/min per 1.73 m2, renal capacity to excrete calcium oxalate is significantly impaired. At this stage, calcium oxalate starts to deposit in extra renal tissues in a process called systemic oxalosis. Calcium oxalate deposits have been reported in the myocardium, cardiac conduction system, kidneys, bones and bone marrow. This leads to cardiomyopathy, heart block and other cardiac conduction defects, vascular disease, retinopathy, synovitis, oxalate osteopathy and anemia that is noted to be resistant to treatment[52,60,61].
SCREENING FOR HYPEROXALURIA
Screening for hyperoxaluria must be undertaken in every child with the first episode of renal stone and all adults who present with recurrent calcium oxalate stones. Screening should also be done at first presentation of nephrocalcinosis or family history of stone disease at any age. Furthermore, screening must be offered to relatives of an index case. PH1 should be strongly considered in the differential in any patient with renal failure of unknown etiology, particularly when there is nephrocalcinosis with reduced renal function or a high occurrence of renal stones. Presence of monohydrate calcium oxalate crystals in biological fluids or tissues is also a strong pointer towards primary hyperoxaluria and should be followed up with additional testing[62].
DIAGNOSIS
Diagnosis of hyperoxaluria is established using a combination of clinical, radiological, biochemical, histopathological and genetic studies in primary hyperoxaluria. Precise diagnosis is of paramount importance for prognostic and treatment implications and also for prenatal screening in appropriate cases where PH is suspected.
In patients with a clinical suspicion for hyperoxaluria, the diagnostic workup should begin with ultrasound or other radiological imaging of the kidneys and the rest of the urinary tract to confirm the presence of nephrocalcinosis and urolithiasis[2,53]. Stone analysis should be done and may yield the initial diagnostic clues for PH. Stones in PH are composed of monohydrate calcium oxalate (whewellite) which assume a dumbbell shaped form[63].
The initial biochemical tests include urinary oxalate excretion preferably measured in 24 h urine collection and adjustment of the oxalate excretion per 1.73 m2 of the body surface area is recommended[2]. Urinary oxalate: urinary creatinine ratios can be used but age specific normal values must be known. These values however should be interpreted with caution as the ratios decline in early life and are also subject to variability based on nutritional intake. Oxaluria must be confirmed using two urine samples. PH is characterized by urinary oxalate excretion > 1.0 mmol/1.73 m2 per 24 h in majority and in some cases may exceed 2.0 mmol/1.73 m2/ 24 h in contrast to the normal urinary excretion which is typically < 0.45 mmol/1.73 m2 per 24 h. In patients with hyperoxaluria > 0.8 mmol/1.73 m2 per 24 h, urinary glycolate and glycerate levels should be measured. About two thirds of PH1 patients have elevated urinary glycolate levels but it is important to remember that normal glycolate levels do not exclude the diagnosis. Urinary glycerate levels are noted to be high in PH2 patients[2,53].
As GFR declines, urinary excretion of oxalate decreases and the urinary oxalate estimation may no longer be accurate. Plasma oxalate should be measured in these circumstances. In PH patients with ESRD, plasma oxalate levels are typically higher than 80 μmol/L while in non PH hyperoxaluric patients, the plasma oxalate level may range between 30-80 μmol/L[64-66]. This is in contrast to plasma oxalate levels of 1-5 μmol/L in normal subjects[1].
Non-invasive, definitive diagnosis of PH is provided by testing of AGXT, GRHPR and HOGA1 genes. There are 150 known mutations for AGXT[67], 16 for GRHP[26] and 15 for HOGA1[28,55-57,68]. Williams et al[69] showed that targeted analysis of the three most common mutations in AGXT (c.33_34insC, c.508G>A, and c.731T>C) provides the diagnosis in 34.5% PH1 patients while exon sequencing of exon 1, 4 and 7 increases the yield and allows diagnosis in 50% PH1 patients. Prenatal diagnosis can be done by testing chorionic villi. In patients with one or no known mutation, intragenic and extragenic linkage analysis is recommended for diagnosis[70,71] .When DNA screening is non diagnostic but clinical suspicion is high, liver biopsy is undertaken for establishing the diagnosis. However, this is an invasive method and carries a high risk of complications like bleeding[53].
In SH, stones are usually mixed (whewellite and weddellite) in contrast to PH. The excretion of urinary oxalate is increased in SH and may be > 0.7 mmol/1.73 m2 per 24 h but in some cases may exceed 1.0 mmol/1.73 m2 per 24 h[2,72,73]. Other available diagnostic tests include use of PCR in stool samples to identify oxalobacter formigenes[74,75]. Also, Increased intestinal oxalate absorption can be assessed by an absorption test using (13C2) oxalate[76]. This test can help identify hyperabsorbers who would benefit from dietary interventions focusing on lowering oxalate and increasing calcium in the diet. This diagnostic test also helps to differentiate between primary and secondary forms of hyperoxaluria[33].
Radiological imaging may aid in diagnosis of multisystem involvement. Renal involvement, apart from urolithiasis, may show two distinct patterns: medullary nephrocalcinosis which is evaluated well on ultrasound while CT scan is a better modality for diagnosis of cortical nephrocalcinosis. CT may also be helpful in detection of calcium oxalate deposition in various other organ systems like bowel wall, muscle and arteries. The effects on the heart can be evaluated by electrocardiography and echocardiography. Skin biopsy may be necessary for skin lesions secondary to calcium oxalate deposition which can resemble the lesions of calciphylaxis[62]. On histopathological examination, calcium oxalate crystals demonstrate a characteristic birefringence when examined under polarized light. Figures 2 and 3 demonstrate calcium oxalate deposition in renal tissue.
Figure 2.
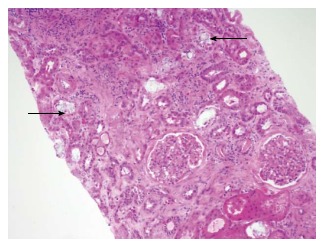
Calcium oxalate deposition in the renal tubules (black arrows).
Figure 3.
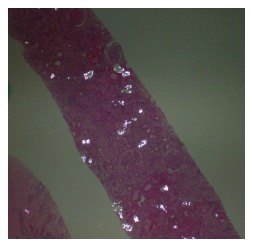
Examination of renal biopsy specimen under polarized light. Calcium oxalate crystals depict a characteristic birefringence.
TREATMENT
Conservative measures
Conservative measures are recommended soon after the diagnosis is made. High fluid intake is vital in preventing stone formation[77]. Patients with hyperoxaluria should be advised to increase their fluid intake to 3-4 L/d[53,60]. In infants and children, a gastrostomy tube may have to be placed to achieve this and special attention should be given to fluid intake in states of fluids losses like vomiting and diarrhea[57,62].
Dietary interventions do not play a major role in the management of primary hyperoxaluria as absorption of oxalate from the intestine is very small. In a study by Sikora et al[78], intestinal absorption of oxalate in patients with PH was noted to be less than 7%. This was attributed to less absorption and translocation of the SLC26A6 transporters favoring oxalate secretion over absorption. On the other hand, diet modification is a very important element in the treatment of secondary hyperoxaluria where efforts should be made to reduce oxalate intake in the diet. Calcium intake should not be restricted as it complexes with oxalate and prevents its absorption[10]. However, excessive intake of Vitamin C should be avoided.
Role of pyridoxine
Pyridoxine supplementation has been shown to be beneficial in patients with PH1. Pyridoxine functions as a cofactor for the enzyme AGT which is defective in PH1. Administration of supraphysiological doses of pyridoxine may stabilize this enzyme and also enhance its enzymatic activity[57]. The recommended initial dose of pyridoxine is 5 mg/kg with a maximum dose of 20 mg/kg[79]. Pyridoxine has been demonstrated to be effective in only 30% of the patients[80,81] and therapeutic success is noted by a approximately 30% reduction in urine oxalate excretion after 3 mo of pyridoxine supplementation at the maximal dose[53,60]. Certain genotypes (508G>A (Gly170Arg) and 454T>A (Phe153Ile) are known to be more responsive to pyridoxine treatment than others[82,83] although pyridoxine therapy should be tested in all patients with PH1. Early initiation of pyridoxine treatment and compliance with the treatment regimen in pyridoxine responsive patients may help to prevent renal failure in PH1[57].
Urinary alkalinization
Alkalinization of the urine is well known to prevent stone formation as citrate complexes with calcium and thus decreases the amount of calcium oxalate available for precipitation. This same principle can be used in patients with hyperoxaluria. Potassium citrate can be used at a dose of 0.1-0.15 g/kg body weight[84]. Urinary pH must be maintained between 6.2 and 6.8[7]. In patients with renal failure, potassium salt can be replaced by sodium citrate[85]. Other inhibitors of crystallization are orthophosphate[86] and magnesium[7] though there is no conclusive evidence that magnesium therapy alone inhibits stone formation.
Probiotics (O. formigenes)
Despite our knowledge of O. formigenes and its use of oxalate as an energy source, the use of probiotics to reduce urinary oxalate excretion has not been demonstrated in human studies[57,87]. The results in animal studies however have been encouraging[88,89].
Management of renal stones
For management of renal stones, endoscopy is currently the procedure of choice as it allows direct visualization of the stones. Extracorporeal shock wave lithotripsy (ESWL) has been the standard of treatment for many years. However, with use of this technique, the shock waves may be mistakenly used on areas of nephrocalcinosis instead of stones due to lack of direct visual assessment which is achieved with endoscopy[90]. Further, gravel in the urinary tract following the ESWL procedure may form a nidus for calcium oxalate deposition and recurrent stone formation in patients with hyperoxaluria. In contrast, endoscopy allows complete retrieval of stones and their fragments and yields excellent results[62].
Renal replacement therapy
Patients reaching ESRD need optimization of renal replacement therapy to ensure adequate oxalate removal. Oxalate deposition occurs when the oxalate levels reach the threshold for supersaturation which is estimated to be 30-45 μmol/L. Hemodialysis (HD) removes oxalate more efficiently than peritoneal dialysis (PD)[66]. However, there is significant oxalate rebound following hemodialysis and levels can reach 80% of the pre-hemodialysis levels[91]. The weekly removal of oxalate by hemodialysis or peritoneal dialysis has been calculated to be 6-10 mmol/1.73 m2[92,93] which leaves patients in a positive oxalate balance and at high risk for systemic deposition. Illies et al[94] studied 6 patients with PH1 who were on dialysis and awaiting liver transplant. Based on their observations, they made recommendations for improvement of the dialysis prescription. Dialysis should be initiated early (around GFR of 20-30 mL/min per 1.73 m2) before ESRD is reached. Dialysis should be done with high flux dialyzers and maximum possible blood flow rate. To improve efficiency of oxalate removal by HD, additional sessions per week are preferable as compared to more time per session. Combination of HD and PD may be used to further enhance oxalate elimination. The timing of HD and PD should be coordinated as PD may be more efficient in removing oxalate in the later phases of the interdialytic period when rebound is much higher than in the earlier interdialytic phase. Efforts should be made to keep the oxalate level below 50 μmol/L[94]. The intensification of dialysis may pose a burden on the patient and family and it is important to keep this in mind while designing an individualized dialysis plan.
Transplantation
Transplantation must be planned when GFR falls between 15-30 mL/min per 1.73 m2. As the defective enzyme is liver specific in PH1, these patients require preemptive liver, sequential liver- kidney, or combined liver-kidney transplantation. Transplantation strategy is decided based on individual presentation and clinical course as disease expression may vary among patients with PH1. Preemptive liver transplantation can be considered in patients who have progressive renal disease and approach a GFR of 50 mL/min per 1.73 m2. Sequential liver-kidney transplantation can be performed in children who are small for a combined liver-kidney transplant[95]. In contrast, combined liver-kidney transplant is best suited for patients who are on chronic renal replacement therapy and not responsive to pyridoxine[57]. Isolated kidney transplantation may be the procedure of choice for adult patients who are sensitive to pyridoxine[96]. However, in isolated renal transplant, allograft survival rates have been reported to be inferior in patients with primary hyperoxaluria compared to patients who received renal transplant for a non PH1 cause of ESRD[48]. Thus, caution should be exercised while advocating this approach.
For patients with PH2, isolated kidney transplantation is the preferred treatment of choice[53,57] as the defective enzyme is found in various body tissues[97]. For patients with PH3, there are no reports of ESRD to date and as a result, no recommendations for renal transplantation have been made in this subset of PH patients[57].
In secondary hyperoxaluria, there is a paucity of data regarding renal transplantation in those who develop ESRD. There is an increased risk for allograft dysfunction by the rapid release of oxalate from systemic deposits leading to recurrent nephrocalcinosis. Ceulemans et al[98] performed combined intestinal and kidney transplants in a patient with hyperoxaluria due to short bowel syndrome which may be a promising approach in patients with enteric hyperoxaluria but this needs to be evaluated in larger studies. The differences between primary and secondary hyperoxaluria are depicted in Table 1.
Table 1.
Comparison between primary and secondary hyperoxaluria
| Clinical feature | Primary hyperoxaluria | Secondary hyperoxaluria |
| Etiology | Inborn error of metabolism with specific enzymatic defects | Increased dietary intake of oxalate or precursors |
| PH 1: Alanine glyoxalate aminotransferase | Increased intestinal absorption | |
| PH 2: Glyoxalate/hydroxypyruvate reductase | Altered intestinal microflora | |
| PH 3: 4-hydroxy 2-oxoglutarate aldolase | ||
| Clinical presentation | PH 1: Recurrent stones, nephrocalcinosis, ESRD common | Recurrent renal stones, nephrocalcinosis, CKD and ESRD |
| Clinical heterogeneity in presentation, varies from an infantile to an adult onset form | ||
| PH 2: Recurrent stones, nephrocalcinosis less common, ESRD has been reported (approximately 20% cases) | ||
| PH 3: Hypercalciuria with hyperoxaluria is reported, no reports to date of ESRD | ||
| Systemic oxalosis | Frequent part of the presentation | Less common but may occur in severe cases of inflammatory bowel disease or short bowel syndrome |
| Diagnosis: History | Family history is often suggestive with other affected relatives | Dietary history may be an important pointer towards the diagnosis |
| Urinary excretion | > 1.0 mmol/1.73 m2 BSA | Usually < 1.0 mmol/1.73 m2 BSA but in some cases of enteric hyperoxaluria may extend into the primary range |
| Composition of renal stones | 95% calcium oxalate monohydrate (whewellite) | Mixed stones (whewellite and weddellite) |
| Other diagnostic points | Plasma oxalate levels in ESRD are > 60-80 mmol/L as compared from non-PH causes of ESRD | 14C test can be used to assess for increased intestinal absorption |
| Treatment: | ||
| General measures: | Daily fluid intake > 3.0 L/d | Hydration and urinary alkalinization |
| Pyridoxine in PH1 | Renal replacement therapy when ESRD occurs | |
| Urinary alkalinization | ||
| Thiazides for PH3 | ||
| Renal replacement therapy when ESRD occurs | ||
| Specific measures: | No role as dietary absorption is < 5% | Important role as dietary absorption is > 40% |
| Dietary management | ||
| O. formigenes | No role in management | No role demonstrated in human studies |
| Transplantation | PH1: Liver kidney transplant (combined or sequential) | Limited data available regarding transplants for treatment of SH |
| Isolated kidney transplant in pyridoxine sensitive adult patients | ||
| PH2: Isolated kidney transplant | ||
| PH3: No role of kidney transplant |
PH: Primary hyperoxaluria; SH: Secondary hyperoxaluria; CKD: Chronic kidney disease; ESRD: End stage renal disease; BSA: Body surface area.
Future directions
Gene therapy, chaperone treatment, liver cell transplantation and proteomic analysis of urine for diagnosis are amongst the new approaches being evaluated for management of patients with primary hyperoxaluria[7,57,62].
Additional resources
There are numerous online resources for physicians and patients to obtain more information about hyperoxaluria. The resources and their web address are outlined in Table 2.
Table 2.
Additional resources for information on hyperoxaluria
| Resource | Web address |
| Oxalosis and Hyperoxaluria Foundation | http://www.ohf.org/ |
| Rare Disease Initiative of the Renal Association | http://rarerenal.org/ |
| Rare Diseases Clinical Research Network (links to the Rare Kidney Stone Consortium) | http://www.rarediseasesnetwork.org/ |
| Children Living with Inherited Metabolic Diseases | http://www.climb.org.uk/ |
| Genetics Home Reference | http://ghr.nlm.nih.gov/ |
| Office of Rare Diseases Research | http://rarediseases.info.nih.gov/ |
| National Organization for Rare Disorders | http://www.rarediseases.org/ |
Footnotes
P- Reviewer: Sakhaee K, Thamilselvan S, Yilmaz E S- Editor: Tian YL L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: The authors declare that they have no competing interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 24, 2014
First decision: August 14, 2014
Article in press: February 9, 2015
References
- 1.Asplin JR. Hyperoxaluric calcium nephrolithiasis. Endocrinol Metab Clin North Am. 2002;31:927–949. doi: 10.1016/s0889-8529(02)00030-0. [DOI] [PubMed] [Google Scholar]
- 2.Milliner DS. The primary hyperoxalurias: an algorithm for diagnosis. Am J Nephrol. 2005;25:154–160. doi: 10.1159/000085407. [DOI] [PubMed] [Google Scholar]
- 3.Robijn S, Hoppe B, Vervaet BA, D’Haese PC, Verhulst A. Hyperoxaluria: a gut-kidney axis? Kidney Int. 2011;80:1146–1158. doi: 10.1038/ki.2011.287. [DOI] [PubMed] [Google Scholar]
- 4.Arena R, Cahalin LP. Evaluation of cardiorespiratory fitness and respiratory muscle function in the obese population. Prog Cardiovasc Dis. 2014;56:457–464. doi: 10.1016/j.pcad.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18:986–991. doi: 10.1007/s00467-003-1234-x. [DOI] [PubMed] [Google Scholar]
- 6.Spasovski G, Beck BB, Blau N, Hoppe B, Tasic V. Late diagnosis of primary hyperoxaluria after failed kidney transplantation. Int Urol Nephrol. 2010;42:825–829. doi: 10.1007/s11255-009-9690-2. [DOI] [PubMed] [Google Scholar]
- 7.Lorenzo V, Torres A, Salido E. Primary hyperoxaluria. Nefrologia. 2014;34:398–412. doi: 10.3265/Nefrologia.pre2014.Jan.12335. [DOI] [PubMed] [Google Scholar]
- 8.Lien YH. Juicing is not all juicy. Am J Med. 2013;126:755–756. doi: 10.1016/j.amjmed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Getting JE, Gregoire JR, Phul A, Kasten MJ. Oxalate nephropathy due to ‘juicing’: case report and review. Am J Med. 2013;126:768–772. doi: 10.1016/j.amjmed.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 10.Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int. 2001;59:270–276. doi: 10.1046/j.1523-1755.2001.00488.x. [DOI] [PubMed] [Google Scholar]
- 11.Farinelli MP, Richardson KE. Oxalate synthesis from [14C1]glycollate and [14C1]glyoxylate in the hepatectomized rat. Biochim Biophys Acta. 1983;757:8–14. doi: 10.1016/0304-4165(83)90146-0. [DOI] [PubMed] [Google Scholar]
- 12.Holmes RP, Assimos DG. Glyoxylate synthesis, and its modulation and influence on oxalate synthesis. J Urol. 1998;160:1617–1624. [PubMed] [Google Scholar]
- 13.Marengo SR, Romani AM. Oxalate in renal stone disease: the terminal metabolite that just won’t go away. Nat Clin Pract Nephrol. 2008;4:368–377. doi: 10.1038/ncpneph0845. [DOI] [PubMed] [Google Scholar]
- 14.Knight TF, Sansom SC, Senekjian HO, Weinman EJ. Oxalate secretion in the rat proximal tubule. Am J Physiol. 1981;240:F295–F298. doi: 10.1152/ajprenal.1981.240.4.F295. [DOI] [PubMed] [Google Scholar]
- 15.Osswald H, Hautmann R. Renal elimination kinetics and plasma half-life of oxalate in man. Urol Int. 1979;34:440–450. doi: 10.1159/000280294. [DOI] [PubMed] [Google Scholar]
- 16.Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004;447:710–721. doi: 10.1007/s00424-003-1090-3. [DOI] [PubMed] [Google Scholar]
- 17.Sindić A, Chang MH, Mount DB, Romero MF. Renal physiology of SLC26 anion exchangers. Curr Opin Nephrol Hypertens. 2007;16:484–490. doi: 10.1097/MNH.0b013e3282e7d7d0. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Z, Asplin JR, Evan AP, Rajendran VM, Velazquez H, Nottoli TP, Binder HJ, Aronson PS. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat Genet. 2006;38:474–478. doi: 10.1038/ng1762. [DOI] [PubMed] [Google Scholar]
- 19.Freel RW, Hatch M, Green M, Soleimani M. Ileal oxalate absorption and urinary oxalate excretion are enhanced in Slc26a6 null mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G719–G728. doi: 10.1152/ajpgi.00481.2005. [DOI] [PubMed] [Google Scholar]
- 20.Bergsland KJ, Zisman AL, Asplin JR, Worcester EM, Coe FL. Evidence for net renal tubule oxalate secretion in patients with calcium kidney stones. Am J Physiol Renal Physiol. 2011;300:F311–F318. doi: 10.1152/ajprenal.00411.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasidas GP, Nemat S, Rose GA. Plasma oxalate and creatinine and oxalate/creatinine clearance ratios in normal subjects and in primary hyperoxaluria. Evidence for renal hyperoxaluria. Clin Chim Acta. 1990;191:67–77. doi: 10.1016/0009-8981(90)90059-2. [DOI] [PubMed] [Google Scholar]
- 22.Lindsjö M, Fellström B, Danielson BG, Kasidas GP, Rose GA, Ljunghall S. Hyperoxaluria or hypercalciuria in nephrolithiasis: the importance of renal tubular functions. Eur J Clin Invest. 1990;20:546–554. doi: 10.1111/j.1365-2362.1990.tb01900.x. [DOI] [PubMed] [Google Scholar]
- 23.Danpure CJ, Jennings PR. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–24. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- 24.Purdue PE, Takada Y, Danpure CJ. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J Cell Biol. 1990;111:2341–2351. doi: 10.1083/jcb.111.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet. 1999;8:2063–2069. doi: 10.1093/hmg/8.11.2063. [DOI] [PubMed] [Google Scholar]
- 26.Cregeen DP, Williams EL, Hulton S, Rumsby G. Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat. 2003;22:497. doi: 10.1002/humu.9200. [DOI] [PubMed] [Google Scholar]
- 27.Williams HE, Smith LH. Hyperoxaluria in L-glyceric aciduria: possible pathogenic mechanism. Science. 1971;171:390–391. doi: 10.1126/science.171.3969.390. [DOI] [PubMed] [Google Scholar]
- 28.Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, Monico CG, Feinstein S, Ben-Shalom E, Magen D, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet. 2010;87:392–399. doi: 10.1016/j.ajhg.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes RP, Kennedy M. Estimation of the oxalate content of foods and daily oxalate intake. Kidney Int. 2000;57:1662–1667. doi: 10.1046/j.1523-1755.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 30.Massey LK. Food oxalate: factors affecting measurement, biological variation, and bioavailability. J Am Diet Assoc. 2007;107:1191–1194; quiz 1195-1196. doi: 10.1016/j.jada.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Lemann J, Pleuss JA, Worcester EM, Hornick L, Schrab D, Hoffmann RG. Urinary oxalate excretion increases with body size and decreases with increasing dietary calcium intake among healthy adults. Kidney Int. 1996;49:200–208. doi: 10.1038/ki.1996.27. [DOI] [PubMed] [Google Scholar]
- 32.Worcester EM. Stones from bowel disease. Endocrinol Metab Clin North Am. 2002;31:979–999. doi: 10.1016/s0889-8529(02)00035-x. [DOI] [PubMed] [Google Scholar]
- 33.Karaolanis G, Lionaki S, Moris D, Palla VV, Vernadakis S. Secondary hyperoxaluria: a risk factor for kidney stone formation and renal failure in native kidneys and renal grafts. Transplant Rev (Orlando) 2014;28:182–187. doi: 10.1016/j.trre.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Mittal RD, Kumar R. Gut-inhabiting bacterium Oxalobacter formigenes: role in calcium oxalate urolithiasis. J Endourol. 2004;18:418–424. doi: 10.1089/0892779041271706. [DOI] [PubMed] [Google Scholar]
- 35.Hatch M, Cornelius J, Allison M, Sidhu H, Peck A, Freel RW. Oxalobacter sp. reduces urinary oxalate excretion by promoting enteric oxalate secretion. Kidney Int. 2006;69:691–698. doi: 10.1038/sj.ki.5000162. [DOI] [PubMed] [Google Scholar]
- 36.Hoppe B, Beck B, Gatter N, von Unruh G, Tischer A, Hesse A, Laube N, Kaul P, Sidhu H. Oxalobacter formigenes: a potential tool for the treatment of primary hyperoxaluria type 1. Kidney Int. 2006;70:1305–1311. doi: 10.1038/sj.ki.5001707. [DOI] [PubMed] [Google Scholar]
- 37.Sidhu H, Schmidt ME, Cornelius JG, Thamilselvan S, Khan SR, Hesse A, Peck AB. Direct correlation between hyperoxaluria/oxalate stone disease and the absence of the gastrointestinal tract-dwelling bacterium Oxalobacter formigenes: possible prevention by gut recolonization or enzyme replacement therapy. J Am Soc Nephrol. 1999;10 Suppl 14:S334–S340. [PubMed] [Google Scholar]
- 38.Stewart CS, Duncan SH, Cave DR. Oxalobacter formigenes and its role in oxalate metabolism in the human gut. FEMS Microbiol Lett. 2004;230:1–7. doi: 10.1016/S0378-1097(03)00864-4. [DOI] [PubMed] [Google Scholar]
- 39.Canavese C, Petrarulo M, Massarenti P, Berutti S, Fenoglio R, Pauletto D, Lanfranco G, Bergamo D, Sandri L, Marangella M. Long-term, low-dose, intravenous vitamin C leads to plasma calcium oxalate supersaturation in hemodialysis patients. Am J Kidney Dis. 2005;45:540–549. doi: 10.1053/j.ajkd.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 40.Nasr SH, Kashtanova Y, Levchuk V, Markowitz GS. Secondary oxalosis due to excess vitamin C intake. Kidney Int. 2006;70:1672. doi: 10.1038/sj.ki.5001724. [DOI] [PubMed] [Google Scholar]
- 41.Stapenhorst L, Hesse A, Hoppe B. Hyperoxaluria after ethylene glycol poisoning. Pediatr Nephrol. 2008;23:2277–2279. doi: 10.1007/s00467-008-0917-8. [DOI] [PubMed] [Google Scholar]
- 42.Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. Clin J Am Soc Nephrol. 2008;3:208–225. doi: 10.2215/CJN.03220807. [DOI] [PubMed] [Google Scholar]
- 43.Worcester EM, Fellner SK, Nakagawa Y, Coe FL. Effect of renal transplantation on serum oxalate and urinary oxalate excretion. Nephron. 1994;67:414–418. doi: 10.1159/000188014. [DOI] [PubMed] [Google Scholar]
- 44.Terribile M, Capuano M, Cangiano G, Carnovale V, Ferrara P, Petrarulo M, Marangella M. Factors increasing the risk for stone formation in adult patients with cystic fibrosis. Nephrol Dial Transplant. 2006;21:1870–1875. doi: 10.1093/ndt/gfl067. [DOI] [PubMed] [Google Scholar]
- 45.Hoppe B, von Unruh GE, Blank G, Rietschel E, Sidhu H, Laube N, Hesse A. Absorptive hyperoxaluria leads to an increased risk for urolithiasis or nephrocalcinosis in cystic fibrosis. Am J Kidney Dis. 2005;46:440–445. doi: 10.1053/j.ajkd.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N. Epidemiology of primary hyperoxaluria type 1. Société de Néphrologie and the Société de Néphrologie Pédiatrique. Nephrol Dial Transplant. 1995;10 Suppl 8:3–7. doi: 10.1093/ndt/10.supp8.3. [DOI] [PubMed] [Google Scholar]
- 47.van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant. 2003;18:273–279. doi: 10.1093/ndt/18.2.273. [DOI] [PubMed] [Google Scholar]
- 48.Harambat J, van Stralen KJ, Espinosa L, Groothoff JW, Hulton SA, Cerkauskiene R, Schaefer F, Verrina E, Jager KJ, Cochat P. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol. 2012;7:458–465. doi: 10.2215/CJN.07430711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hattori S, Yosioka K, Honda M, Ito H. The 1998 report of the Japanese National Registry data on pediatric end-stage renal disease patients. Pediatr Nephrol. 2002;17:456–461. doi: 10.1007/s00467-002-0848-8. [DOI] [PubMed] [Google Scholar]
- 50.Al-Eisa AA, Samhan M, Naseef M. End-stage renal disease in Kuwaiti children: an 8-year experience. Transplant Proc. 2004;36:1788–1791. doi: 10.1016/j.transproceed.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 51.Kamoun A, Lakhoua R. End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. 1996;10:479–482. doi: 10.1007/s004670050143. [DOI] [PubMed] [Google Scholar]
- 52.Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol. 2006;21:1075–1081. doi: 10.1007/s00467-006-0124-4. [DOI] [PubMed] [Google Scholar]
- 53.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int. 2001;59:31–36. doi: 10.1046/j.1523-1755.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- 55.Monico CG, Rossetti S, Belostotsky R, Cogal AG, Herges RM, Seide BM, Olson JB, Bergstrahl EJ, Williams HJ, Haley WE, et al. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol. 2011;6:2289–2295. doi: 10.2215/CJN.02760311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams EL, Bockenhauer D, van’t Hoff WG, Johri N, Laing C, Sinha MD, Unwin R, Viljoen A, Rumsby G. The enzyme 4-hydroxy-2-oxoglutarate aldolase is deficient in primary hyperoxaluria type 3. Nephrol Dial Transplant. 2012;27:3191–3195. doi: 10.1093/ndt/gfs039. [DOI] [PubMed] [Google Scholar]
- 57.Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8:467–475. doi: 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]
- 58.Hueppelshaeuser R, von Unruh GE, Habbig S, Beck BB, Buderus S, Hesse A, Hoppe B. Enteric hyperoxaluria, recurrent urolithiasis, and systemic oxalosis in patients with Crohn’s disease. Pediatr Nephrol. 2012;27:1103–1109. doi: 10.1007/s00467-012-2126-8. [DOI] [PubMed] [Google Scholar]
- 59.Rumsby G, Cochat P. Primary hyperoxaluria. N Engl J Med. 2013;369:2163. doi: 10.1056/NEJMc1311606. [DOI] [PubMed] [Google Scholar]
- 60.Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001;12:1986–1993. doi: 10.1681/ASN.V1291986. [DOI] [PubMed] [Google Scholar]
- 61.Bobrowski AE, Langman CB. The primary hyperoxalurias. Semin Nephrol. 2008;28:152–162. doi: 10.1016/j.semnephrol.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 62.Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, Fargue S, Groothoff J, Harambat J, Hoppe B, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27:1729–1736. doi: 10.1093/ndt/gfs078. [DOI] [PubMed] [Google Scholar]
- 63.Daudon M, Jungers P, Bazin D. Peculiar morphology of stones in primary hyperoxaluria. N Engl J Med. 2008;359:100–102. doi: 10.1056/NEJMc0800990. [DOI] [PubMed] [Google Scholar]
- 64.Marangella M, Petrarulo M, Mandolfo S, Vitale C, Cosseddu D, Linari F. Plasma profiles and dialysis kinetics of oxalate in patients receiving hemodialysis. Nephron. 1992;60:74–80. doi: 10.1159/000186708. [DOI] [PubMed] [Google Scholar]
- 65.Marangella M, Petrarulo M, Vitale C, Cosseddu D, Linari F. Plasma and urine glycolate assays for differentiating the hyperoxaluria syndromes. J Urol. 1992;148:986–989. doi: 10.1016/s0022-5347(17)36796-4. [DOI] [PubMed] [Google Scholar]
- 66.Hoppe B, Kemper MJ, Bökenkamp A, Portale AA, Cohn RA, Langman CB. Plasma calcium oxalate supersaturation in children with primary hyperoxaluria and end-stage renal failure. Kidney Int. 1999;56:268–274. doi: 10.1046/j.1523-1755.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- 67.Williams EL, Acquaviva C, Amoroso A, Chevalier F, Coulter-Mackie M, Monico CG, Giachino D, Owen T, Robbiano A, Salido E, et al. Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mutat. 2009;30:910–917. doi: 10.1002/humu.21021. [DOI] [PubMed] [Google Scholar]
- 68.Riedel TJ, Knight J, Murray MS, Milliner DS, Holmes RP, Lowther WT. 4-Hydroxy-2-oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. Biochim Biophys Acta. 2012;1822:1544–1552. doi: 10.1016/j.bbadis.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams E, Rumsby G. Selected exonic sequencing of the AGXT gene provides a genetic diagnosis in 50% of patients with primary hyperoxaluria type 1. Clin Chem. 2007;53:1216–1221. doi: 10.1373/clinchem.2006.084434. [DOI] [PubMed] [Google Scholar]
- 70.Rumsby G. Experience in prenatal diagnosis of primary hyperoxaluria type 1. J Nephrol. 1998;11 Suppl 1:13–14. [PubMed] [Google Scholar]
- 71.Rumsby G, Uttley WS, Kirk JM. First trimester diagnosis of primary hyperoxaluria type I. Lancet. 1994;344:1018. doi: 10.1016/s0140-6736(94)91675-6. [DOI] [PubMed] [Google Scholar]
- 72.Rifkin SI. Transplantation for renal failure secondary to enteric hyperoxaluria: a case report. J Med Case Rep. 2007;1:31. doi: 10.1186/1752-1947-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoppe B, Hesse A, Brömme S, Rietschel E, Michalk D. Urinary excretion substances in patients with cystic fibrosis: risk of urolithiasis? Pediatr Nephrol. 1998;12:275–279. doi: 10.1007/s004670050452. [DOI] [PubMed] [Google Scholar]
- 74.Kwak C, Jeong BC, Lee JH, Kim HK, Kim EC, Kim HH. Molecular identification of Oxalobacter formigenes with the polymerase chain reaction in fresh or frozen fecal samples. BJU Int. 2001;88:627–632. doi: 10.1046/j.1464-4096.2001.02395.x. [DOI] [PubMed] [Google Scholar]
- 75.Kodama T, Akakura K, Mikami K, Ito H. Detection and identification of oxalate-degrading bacteria in human feces. Int J Urol. 2002;9:392–397. doi: 10.1046/j.1442-2042.2002.00488.x. [DOI] [PubMed] [Google Scholar]
- 76.Hesse A, Schneeberger W, Engfeld S, Von Unruh GE, Sauerbruch T. Intestinal hyperabsorption of oxalate in calcium oxalate stone formers: application of a new test with [13C2]oxalate. J Am Soc Nephrol. 1999;10 Suppl 14:S329–S333. [PubMed] [Google Scholar]
- 77.Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol. 1996;155:839–843. [PubMed] [Google Scholar]
- 78.Sikora P, von Unruh GE, Beck B, Feldkötter M, Zajaczkowska M, Hesse A, Hoppe B. [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int. 2008;73:1181–1186. doi: 10.1038/ki.2008.63. [DOI] [PubMed] [Google Scholar]
- 79.Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria--the German experience. Am J Nephrol. 2005;25:276–281. doi: 10.1159/000086358. [DOI] [PubMed] [Google Scholar]
- 80.Gibbs DA, Watts RW. The action of pyridoxine in primary hyperoxaluria. Clin Sci. 1970;38:277–286. doi: 10.1042/cs0380277. [DOI] [PubMed] [Google Scholar]
- 81.Watts RW, Veall N, Purkiss P, Mansell MA, Haywood EF. The effect of pyridoxine on oxalate dynamics in three cases of primary hyperoxaluria (with glycollic aciduria) Clin Sci (Lond) 1985;69:87–90. doi: 10.1042/cs0690087. [DOI] [PubMed] [Google Scholar]
- 82.van Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJ, Waterham HR. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int. 2004;66:746–752. doi: 10.1111/j.1523-1755.2004.00796.x. [DOI] [PubMed] [Google Scholar]
- 83.Monico CG, Rossetti S, Olson JB, Milliner DS. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005;67:1704–1709. doi: 10.1111/j.1523-1755.2005.00267.x. [DOI] [PubMed] [Google Scholar]
- 84.Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol. 1993;7:207–211. doi: 10.1007/BF00864405. [DOI] [PubMed] [Google Scholar]
- 85.Marangella M, Bagnis C, Bruno M, Vitale C, Petrarulo M, Ramello A. Crystallization inhibitors in the pathophysiology and treatment of nephrolithiasis. Urol Int. 2004;72 Suppl 1:6–10. doi: 10.1159/000076583. [DOI] [PubMed] [Google Scholar]
- 86.Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 1994;331:1553–1558. doi: 10.1056/NEJM199412083312304. [DOI] [PubMed] [Google Scholar]
- 87.Hoppe B, Groothoff JW, Hulton SA, Cochat P, Niaudet P, Kemper MJ, Deschênes G, Unwin R, Milliner D. Efficacy and safety of Oxalobacter formigenes to reduce urinary oxalate in primary hyperoxaluria. Nephrol Dial Transplant. 2011;26:3609–3615. doi: 10.1093/ndt/gfr107. [DOI] [PubMed] [Google Scholar]
- 88.Grujic D, Salido EC, Shenoy BC, Langman CB, McGrath ME, Patel RJ, Rashid A, Mandapati S, Jung CW, Margolin AL. Hyperoxaluria is reduced and nephrocalcinosis prevented with an oxalate-degrading enzyme in mice with hyperoxaluria. Am J Nephrol. 2009;29:86–93. doi: 10.1159/000151395. [DOI] [PubMed] [Google Scholar]
- 89.Hatch M, Gjymishka A, Salido EC, Allison MJ, Freel RW. Enteric oxalate elimination is induced and oxalate is normalized in a mouse model of primary hyperoxaluria following intestinal colonization with Oxalobacter. Am J Physiol Gastrointest Liver Physiol. 2011;300:G461–G469. doi: 10.1152/ajpgi.00434.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kerbl K, Clayman RV. Endourologic treatment of nephrocalcinosis. Urology. 2000;56:508. doi: 10.1016/s0090-4295(00)00675-0. [DOI] [PubMed] [Google Scholar]
- 91.Yamauchi T, Quillard M, Takahashi S, Nguyen-Khoa M. Oxalate removal by daily dialysis in a patient with primary hyperoxaluria type 1. Nephrol Dial Transplant. 2001;16:2407–2411. doi: 10.1093/ndt/16.12.2407. [DOI] [PubMed] [Google Scholar]
- 92.Hoppe B, Graf D, Offner G, Latta K, Byrd DJ, Michalk D, Brodehl J. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol. 1996;10:488–492. doi: 10.1007/s004670050145. [DOI] [PubMed] [Google Scholar]
- 93.Watts RW, Veall N, Purkiss P. Oxalate dynamics and removal rates during haemodialysis and peritoneal dialysis in patients with primary hyperoxaluria and severe renal failure. Clin Sci (Lond) 1984;66:591–597. doi: 10.1042/cs0660591. [DOI] [PubMed] [Google Scholar]
- 94.Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF. Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int. 2006;70:1642–1648. doi: 10.1038/sj.ki.5001806. [DOI] [PubMed] [Google Scholar]
- 95.Brinkert F, Ganschow R, Helmke K, Harps E, Fischer L, Nashan B, Hoppe B, Kulke S, Müller-Wiefel DE, Kemper MJ. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation. 2009;87:1415–1421. doi: 10.1097/TP.0b013e3181a27939. [DOI] [PubMed] [Google Scholar]
- 96.Saborio P, Scheinman JI. Transplantation for primary hyperoxaluria in the United States. Kidney Int. 1999;56:1094–1100. doi: 10.1046/j.1523-1755.1999.00619.x. [DOI] [PubMed] [Google Scholar]
- 97.Giafi CF, Rumsby G. Primary hyperoxaluria type 2: enzymology. J Nephrol. 1998;11 Suppl 1:29–31. [PubMed] [Google Scholar]
- 98.Ceulemans LJ, Nijs Y, Nuytens F, De Hertogh G, Claes K, Bammens B, Naesens M, Evenepoel P, Kuypers D, Vanrenterghem Y, et al. Combined kidney and intestinal transplantation in patients with enteric hyperoxaluria secondary to short bowel syndrome. Am J Transplant. 2013;13:1910–1914. doi: 10.1111/ajt.12305. [DOI] [PubMed] [Google Scholar]