

Abstract
Tumour biomarker status is being used more and more frequently to guide treatment decisions in patients with metastatic colorectal cancer (mCRC). Continued cycles of hypothesis generation and biomarker testing in retrospective, prospective–retrospective and prospective analyses from studies of the epidermal growth factor (EGFR)-targeted monoclonal antibodies (mAbs), panitumumab and cetuximab, have resulted in improved patient selection in mCRC. Initial data suggested EGFR-targeted mAb treatment should be limited to patients with KRAS exon 2 wild-type (WT) tumours, but the availability of tumour samples from large phase III studies permitted evaluation of additional potential biomarkers of activity for these agents. Subsequent analyses further refined the target population to those patients whose tumours were WT for KRAS and NRAS exons 2, 3 and 4 (i.e., those with RAS WT status). Here, we review key clinical data for panitumumab in mCRC across the lines of treatment, assessing in detail the impact of more comprehensive RAS selection on patient outcomes. Panitumumab data across first- to third-line therapy consistently demonstrate that by testing tumour RAS status, it is possible to select patients more likely to benefit from treatment.
Key Points
| RAS mutations predict a lack of response to panitumumab. |
| Detecting RAS mutations improves patient selection in mCRC. |
| Panitumumab has an optimal risk/benefit profile in tumours without RAS mutations. |
Introduction
A predictive biomarker is a characteristic that can be objectively measured and evaluated as an indicator of treatment response (positively predictive) or lack of response (negatively predictive) [1]. Biomarker-guided treatment has the potential to improve clinical outcomes by allowing physicians to tailor therapy to those patients most likely to benefit, thereby sparing potential side effects in patients who are unlikely to respond to treatment. Avoiding treating patients who are unlikely to benefit improves the overall risk/benefit profile of targeted agents; it also has the advantage of being potentially cost-saving, in terms of reducing the use of ineffective drugs, the strain on hospital resources and the need for side-effect management.
Tumour biomarker status is increasingly used to guide treatment decisions in patients with cancer and has been a rapidly developing area of research in metastatic colorectal cancer (mCRC). Improved patient selection through the use of biomarkers is likely to be particularly beneficial in mCRC because of the heterogeneity of response amongst these patients and the costs and toxicities associated with the available targeted therapies [2]. Approximately 27–43 % of mCRC tumours harbour KRAS exon 2 mutations that lead to constitutive activation of downstream signalling pathways [3]. Results of several uncontrolled [4, 5] and phase II [6] studies led to the hypothesis that the presence of KRAS exon 2 mutations might be associated with a lack of response to the epidermal growth factor receptor (EGFR)-targeted monoclonal antibodies (mAbs) panitumumab and cetuximab (Fig. 1) [6–15]. Initial analyses from the 408 study comparing panitumumab + best supportive care (BSC) with BSC alone in patients with mCRC receiving predominantly third-line treatment, supported the use of KRAS as a biomarker [13].
Fig. 1.
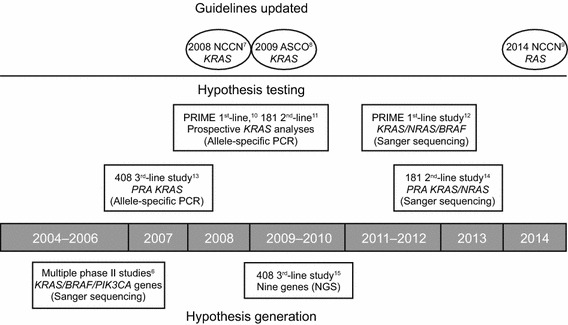
The cycle of hypothesis generation/testing to refine the target metastatic colorectal cancer population for panitumumab treatment. ASCO American Society of Clinical Oncology, NCCN National Comprehensive Cancer Network, NGS next-generation sequencing, PCR polymerase chain reaction, PRA prospective–retrospective analysis
As the availability of tumour samples for biomarker analysis was a requirement for entry into several phase III panitumumab trials [10, 11, 13], prospective–retrospective analyses were possible to further test the hypothesis that tumour KRAS status may predict response to anti-EGFR therapy in mCRC. In this context, a prospective–retrospective analysis is defined as the prospective analysis of a new biomarker hypothesis that was not prespecified at study start; these analyses are conducted on banked tumour samples from a clinical trial [16]. Wang et al. [17] defined such analyses as “a completed or post-interim analysis from a trial where biomarker samples were collected prior to treatment initiation and where the mechanistic hypothesis would be ‘prospectively specified’ prior to an approved diagnostic assay testing”. Hence, the data analysis is considered a prospective analysis of the hypothesis.
As more data were generated around the sensitivity of tumours to EGFR inhibitors, there was an ongoing cycle of hypothesis generation and testing. As a result, study protocols were developed to enable prospective analysis of new biomarkers, including prospective analyses of KRAS in the phase III PRIME [10] and 181 [11] panitumumab studies (Fig. 1; Table 1). The subsequent confirmation of KRAS exon 2 mutations as being negatively predictive of EGFR-targeted mAb efficacy led to their use being restricted to patients with KRAS exon 2 wild-type (WT) tumours [18] and meant that for the first time patients with mCRC could be specifically selected for treatment based on the molecular profile of their tumours. This was among the most important developments in personalised mCRC management in recent years.
Table 1.
Full trial names and abbreviations
| Trial acronym | Full trial name |
|---|---|
| 181 | A randomised, multicentre phase III study to compare the efficacy of panitumumab in combination with FOLFIRI to the efficacy of FOLFIRI alone in patients with previously treated metastatic colorectal cancer |
| 314 | A single arm multicentre phase II study of panitumumab in combination with irinotecan/5-fluorouracil/leucovorin in patients with metastatic colorectal cancer |
| 408 | An open-label, randomised, phase III clinical trial of panitumumab plus best supportive care versus best supportive care in subjects with metastatic colorectal cancer |
| ASPECCT | A randomised, multicentre, open-label, phase III study to compare the efficacy and safety of panitumumab and cetuximab in subjects with previously treated, wild-type KRAS, metastatic colorectal cancer |
| CRYSTAL | Cetuximab combined with irinotecan in first-line therapy for metastatic colorectal cancer |
| FIRE-3 | FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer |
| OPUS | Oxaliplatin and cetuximab in first-line treatment of metastatic colorectal cancer |
| PEAK | Panitumumab efficacy in combination with mFOLFOX6 against bevacizumab plus mFOLFOX6 in mCRC subjects with wild-type KRAS tumours |
| PLANET | Safety and efficacy study of FOLFOX4 + panitumumab vs FOLFIRI + panitumumab in subjects WT KRAS colorectal cancer and liver-only metastases |
| PRIME | The panitumumab randomised trial in combination with FOLFOX4 for metastatic colorectal cancer to determine efficacy |
| SPIRITT | Second-line panitumumab, irinotecan treatment trial |
Continued investigation of the molecular mechanisms underpinning tumour development and growth in mCRC led to the hypothesis that, even with a KRAS exon 2 WT population, other mutations might confer resistance to EGFR inhibitors. Based on the similarity between NRAS and KRAS oncogenes, and the fact that mutations in either gene at codons 12, 13, 61, 117 and 146 had similar biochemical effects [19, 20], these additional RAS mutations were hypothesised as biomarkers of resistance to EGFR-targeted mAb activity. Furthermore, in the clinic, colorectal tumours harbour mutations at these same codons of NRAS and KRAS, and these mutations tend to be mutually exclusive within tumour cells, suggesting functional redundancy [21]. As tumour samples were available for testing, this preclinical molecular hypothesis led to prospective–retrospective analyses of the impact of tumour RAS status in the PRIME and 181 trials (Fig. 1). The resulting data showed for the first time that mutations in KRAS beyond those in exon 2, as well as mutations in the related NRAS gene, also have a significant impact on the risk/benefit profile of panitumumab in mCRC [12, 14]. Patients whose tumours harbour RAS mutations do not benefit from treatment with an EGFR inhibitor, whereas those with RAS WT status appear to gain from an improved risk/benefit profile compared with those having KRAS exon 2 WT tumours. Similar results have since been seen in retrospective analyses of older cetuximab trials [22, 23].
Patients can, therefore, now be selected for panitumumab or cetuximab therapy based on their RAS status, meaning that patients whose tumours are WT for KRAS and NRAS (exons 2, 3 and 4) are eligible, but patients whose tumours harbour mutations in these exons should not receive EGFR inhibitor treatment [24, 25]. An ongoing study (NCT01412957) of panitumumab monotherapy in the third-line mCRC setting will be the first trial to prospectively analyse outcomes for patients based on their tumour RAS status. This is a rapidly moving field, with new data supporting the shift from KRAS to RAS testing being published regularly over the last 18 months and regulatory bodies in Europe responding quickly to these advances, updating the labels of both panitumumab and cetuximab to reflect the need for RAS testing in mCRC [24, 25]. Here we review the key clinical data for panitumumab in mCRC across the lines of treatment, and assess in detail the impact of more comprehensive RAS selection on patient outcomes.
First-Line Setting
There have been four key first-line panitumumab trials in patients with mCRC, including a total of 1699 patients. Prospective and prospective–retrospective analyses of tumour samples from these studies have allowed RAS status to be determined for 1500 (88 %) of the included patients. PRIME, the largest of these trials, was conducted in an unselected patient population, as was the 314 study, whereas PEAK and PLANET only included patients with known KRAS exon 2 WT status.
Phase III Data—The PRIME trial
PRIME (NCT00364013) was a randomised (1:1) phase III study comparing the efficacy and safety of first-line panitumumab 6.0 mg/kg once every 2 weeks (Q2W) + FOLFOX4 versus FOLFOX4 alone in patients with previously untreated mCRC [10]. PRIME was the first randomised, first-line study in which results were prospectively analysed by tumour KRAS exon 2 status. A key inclusion criterion was that paraffin-embedded tumour tissue from the primary tumour or metastasis had to be available for central biomarker analyses. Importantly, sufficient tumour tissue was collected to enable additional hypothesis-driven biomarker analyses after the primary efficacy analysis had been conducted.
In PRIME, a prespecified mutational analysis was conducted on banked tumour samples that were previously characterised as KRAS exon 2 WT to test for mutations in NRAS exon 2 (codons 12 and 13), and KRAS and NRAS exons 3 (codon 61) and 4 (codons 117 and 146). Assessment of tumour BRAF status was also prespecified (exon 15 codon 600). Gene alterations in KRAS and NRAS exon 3 (codon 59) were investigated as an exploratory endpoint. The RAS WT population, therefore, included patients whose tumours harboured no mutations in any of the following: KRAS and NRAS exons 2 (codons 12 and 13), 3 (codons 59 [exploratory analysis set only] and 61), and 4 (codons 117 and 146).
Impact of KRAS Exon 2 Status
Overall, 1183 patients were randomised in PRIME. In total, 1096 patients (93 %) had tumour KRAS results, of whom 656 (60 %) were found to have KRAS exon 2 WT tumours. The primary analysis from this study was prespecified to be performed when >50 % of patients with KRAS exon 2 WT mCRC had died from any cause. In the primary analysis (54 % of patients had died at this point), panitumumab + FOLFOX4 significantly improved progression-free survival (PFS) compared with FOLFOX4 alone in patients with KRAS exon 2 WT tumours (Table 2) [10]. Overall survival (OS) and objective response rate (ORR) were numerically higher in the panitumumab + FOLFOX4 versus FOLFOX4 group, but these between-treatment differences were not statistically significant. In KRAS exon 2 WT patients, surgical resection was attempted in 10.5 % of those receiving panitumumab + FOLFOX4 and 9.4 % of those receiving FOLFOX4 alone; complete resections were performed in 8.3 versus 7.0 % of patients, respectively [10]. In contrast, in patients with KRAS exon 2 mutant (MT) tumours, PFS was significantly reduced in the panitumumab + FOLFOX4 group versus the FOLFOX4 group, and median OS was numerically lower (Table 2).
Table 2.
Impact of KRAS exon 2 [10] and RAS [12, 24] status on outcomes in the PRIME trial (primary analysis data)
| KRAS exon 2 WT [10] | RAS WTa [12, 24] | |||
|---|---|---|---|---|
| Panitumumab + FOLFOX4 (n = 325) | FOLFOX4 (n = 331) | Panitumumab + FOLFOX4 (n = 259) | FOLFOX4 (n = 253) | |
| Median PFS, months | 9.6 | 8.0 | 10.1 | 7.9 |
| HR [95 % CI]; p value | 0.80 [0.66–0.97]; 0.02 | 0.72 [0.58–0.90]; 0.004 | ||
| Median OS, months | 23.9 | 19.7 | 26.0 | 20.2 |
| HR [95 % CI]; p value | 0.83 [0.67–1.02]; 0.072 | 0.78 [0.62–0.99]; 0.043 | ||
| ORR,a % [95 % CI] |
n = 317 55 [50–61] |
n = 323 48 [42–53] |
n = 149 59 [52–65] |
n = 114 46 [40–53] |
| OR [95 % CI]; p value | 1.35 [NA]; 0.068 | 1.63 [1.13–2.38]; 0.009 | ||
| KRAS exon 2 MT [10] | RAS MTb [12, 24] | |||
|---|---|---|---|---|
| Panitumumab + FOLFOX4 (n = 221) | FOLFOX4 (n = 219) | Panitumumab + FOLFOX4 (n = 272) | FOLFOX4 (n = 276) | |
| Median PFS, months | 7.3 | 8.8 | 7.3 | 8.7 |
| HR [95 % CI]; p value | 1.29 [1.04–1.62]; 0.02 | 1.31 [1.07–1.60]; 0.008 | ||
| Median OS, months | 15.5 | 19.3 | 15.6 | 19.2 |
| HR [95 % CI]; p value | 1.24 [0.98–1.57]; 0.068 | 1.25 [1.02–1.55]; 0.034 | ||
| ORR,c % [95 % CI] |
n = 215 40 [33–46] |
n = 211 40 [34–47] |
NA | NA |
| OR [95 % CI]; p value | NA | NA | ||
CI confidence interval, HR hazard ratio, MT mutant, NA not available, OR odds ratio, ORR objective response rate, OS overall survival, PFS progression-free survival, WT wild type
aWild type at KRAS and NRAS exons 2 (codons 12 and 13), 3 (codon 61) and 4 (codons 117 and 146)
bMutations at any of KRAS and NRAS exons 2 (codons 12 and 13), 3 (codon 61) and 4 (codons 117 and 146). Of 620 patients with KRAS exon 2 WT tumours and RAS data, 108 (17 %) had other RAS mutations (RAS status determined using bidirectional Sanger sequencing and WAVE-based Surveyor® Scan Kits (Transgenomic)
cIncluded only patients with baseline measurable disease per central review
Significant PFS improvements (median 10.0 vs 8.6 months; hazard ratio (HR) 0.80 [95 % confidence intervals {CI} 0.67–0.95]; p = 0.01) were maintained in the KRAS exon 2 WT panitumumab + FOLFOX4 group in a prespecified ‘final’ analysis scheduled to occur ~30 months after the last patient enrolled (68 % of patients had died at this point) [26]. Median OS (23.9 vs 19.7 months; HR 0.88 [95 % CI 0.73–1.06]; p = 0.17) was also numerically higher for the panitumumab + FOLFOX4 group versus FOLFOX4 in the KRAS exon 2 WT population in this analysis. An exploratory analysis of updated survival (>80 % OS events, providing the most up-to-date estimation of OS) was also carried out, which demonstrated a significant OS improvement for KRAS exon 2 WT patients receiving panitumumab + FOLFOX4 versus FOLFOX4 (HR 0.83 [95 % CI 0.70–0.98]; p = 0.03) [26].
Impact of RAS Mutations on Efficacy
RAS ascertainment rates in PRIME were high, with tumour RAS status determined for 1060 of the 1183 (90 %) patients randomised in this study; among those 1060 patients, 512 (48 %) were found to have RAS WT mCRC [12]. Of the 620 patients with KRAS exon 2 WT tumours who could be evaluated for tumour RAS status, 108 (17 %) had mutations elsewhere in either the KRAS or NRAS genes. Sites of the RAS mutations found in the PRIME study are shown in Fig. 2a. A further seven patients had mutations in codon 59 of KRAS or NRAS exon 3, which were not originally prespecified for analysis—these patients were excluded from the exploratory analysis population (n = 505). Tumour RAS/BRAF ascertainment rate was also high, with evaluable samples available for 89 % (1047/1183) of patients. Of the 619 patients with KRAS exon 2 WT mCRC who were evaluated for tumour BRAF status, 53 (9 %) were found to have BRAF V600E mutations.
Fig. 2.
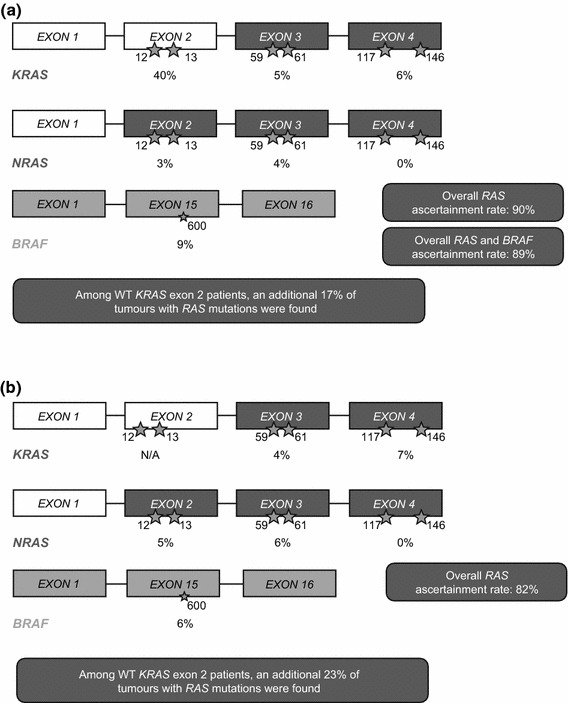
RAS mutation hotspots in the first-line a PRIME [12] and b PEAK [31, 32] studies. Stars denote codon position; percentages denote the proportion of patients with available data who had a mutation within the specified gene exon. N/A not applicable as KRAS exon 2 WT status was defined in the trial eligibility criteria, WT wild type
In the primary RAS analysis, PFS, OS and ORR were significantly improved in patients with RAS WT tumours receiving panitumumab + FOLFOX4 versus FOLFOX4 alone (Table 2) [12]. A clinically significant 5.8-month improvement in median OS was observed in RAS WT patients treated with panitumumab + FOLFOX4 compared with those receiving FOLFOX4 alone.
In patients with RAS WT tumours, the PFS and OS benefits observed in favour of panitumumab + FOLFOX4 versus FOLFOX4 were observed across all subpopulations predefined according to baseline covariates, with the exception of the Eastern Cooperative Oncology Group (ECOG) performance status 2 subgroup (Fig. 3). Consistent OS benefits were seen in the panitumumab + FOLFOX4 group versus the FOLFOX4 group in an exploratory, updated OS analysis performed when >80 % of all patients in PRIME had died from any cause. In this analysis, median OS was 25.8 versus 20.2 months, respectively (HR 0.77 [95 % CI 0.64–0.94]; p = 0.009), supporting the robustness of the primary analysis. In an additional exploratory analysis excluding the seven patients with KRAS or NRAS exon 3 codon 59 mutations, PFS (median 10.4 vs 7.9 months; HR 0.71 [95 % CI 0.57–0.89]; p = 0.002) and OS (median 26.0 vs 20.2 months; HR 0.77 [95 % CI 0.60–0.98]; p = 0.032) outcomes were further improved in the panitumumab + FOLFOX4 versus FOLFOX4 group.
Fig. 3.
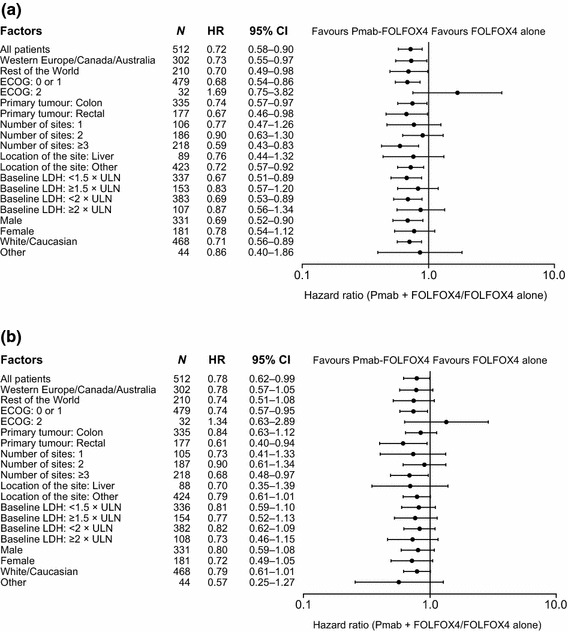
PRIME: Hazard ratios (95 % confidence intervals) for a progression-free survival; b overall survival (RAS wild-type primary analysis population) [12]. From Douillard et al. [12]. Copyright© 2013 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. CI confidence intervals, ECOG Eastern Cooperative Oncology Group, HR hazard ratio, LDH lactate dehydrogenase, Pmab panitumumab, ULN upper limit of normal
In the 108 patients without KRAS exon 2 mutations but with mutations elsewhere in KRAS or NRAS, PFS (median 7.3 vs 8.0 months; HR 1.28 [95 % CI 0.79–2.07]; p = 0.33) and OS (median 17.1 vs 18.3 months; HR 1.29 [95 % CI 0.79–2.10]; p = 0.31) were numerically shorter in the panitumumab + FOLFOX4 versus FOLFOX4 group. In the RAS MT population overall, median PFS and OS were shorter for patients receiving panitumumab + FOLFOX4 versus FOLFOX4 (Table 2), highlighting the importance of limiting the use of panitumumab + FOLFOX4 to patients with RAS WT tumours.
In an additional analysis, BRAF MT status was found to be a negative prognostic factor. In the 446 patients with neither RAS nor BRAF mutations, panitumumab + FOLFOX4 was associated with a 1.6-month improvement in PFS (HR 0.68 [95 % CI 0.54–0.87]; p = 0.002) and a 7.4-month improvement in OS (HR 0.74 [95 % CI 0.57–0.96]; p = 0.02), versus FOLFOX4 alone. In patients with no RAS mutations but with BRAF mutations (n = 53), small absolute PFS (median 6.1 vs 5.4 months; HR 0.58 [95 % CI 0.29–1.15]; p = 0.12) and OS (median 10.5 vs 9.2 months; HR 0.90 [95 % CI 0.46–1.76]; p = 0.76) differences observed in favour of the panitumumab + FOLFOX4 versus FOLFOX4 group were not statistically significant, likely due to the small sample size.
Tumour shrinkage is an important treatment aim for patients with mCRC as it increases the possibility of potentially curative surgical resection [27] and would also be expected to be accompanied with symptom relief. The presence of early tumour shrinkage of ≥30 % at week 8 has also been associated with improved PFS and OS outcomes versus shrinkage <30 % at this time point [28–30]. By week 8, significantly more RAS WT patients in the panitumumab + FOLFOX4 versus FOLFOX4 group had achieved ≥30 % tumour shrinkage (59 vs 38 %; p < 0.001) [28]. Surgical resections were performed in 14 versus 12 % of RAS WT patients (exploratory analysis population; n = 505) and complete resections were performed in 10 versus 8 % of panitumumab + FOLFOX4 versus FOLFOX4-treated patients, respectively [28].
Phase II Data—The PEAK Trial
PEAK (NCT00819780) was a 1:1 randomised, open-label, phase II study comparing modified (m)FOLFOX6 + either panitumumab (6 mg/kg Q2W) or bevacizumab (5 mg/kg Q2W) as first-line treatments in patients with KRAS exon 2 WT mCRC and unresectable metastatic disease [31]. PEAK was the first randomised trial to compare the treatment effect of an EGFR-targeted mAb with that of an anti-vascular endothelial growth factor (VEGF) therapy in combination with oxaliplatin-containing chemotherapy in the first-line treatment of patients with KRAS exon 2 WT mCRC. In this study, KRAS exon 2 WT status (codons 12 and 13 were assessed) of paraffin-embedded tumour tissue had to be determined using a validated test. A prespecified extended RAS analysis was also performed, with mutations assessed in NRAS exon 2 (codons 12 and 13), and KRAS and NRAS exons 3 (codons 59 and 61) and 4 (codons 117 and 146). BRAF exon 15 (codon 600) mutations were also assessed [32].
Two clinical snapshots were reported in PEAK—an event-driven prespecified primary analysis (performed when ~168 PFS events had occurred) and an exploratory analysis performed ~1 year after the last patient enrolled.
Efficacy in the KRAS Exon 2 WT Population
Overall, 285 patients with KRAS exon 2 WT mCRC were randomised and comprised the primary analysis population [31]. PFS was similar between the panitumumab + mFOLFOX6 and bevacizumab + mFOLFOX6 arms in the primary analysis (Fig. 4a), despite the relative dose intensity being 8 % lower in the panitumumab arm. OS outcomes were immature at the time of this analysis, with 87 deaths (31 %) reported in the KRAS exon 2 WT population overall. At the time of the exploratory analysis, 130 deaths (46 %) had been reported. In this analysis, a significant OS benefit was observed for panitumumab + mFOLFOX6 versus bevacizumab + mFOLFOX6 (Fig. 5a). Post-study treatment included an EGFR-targeted mAb in 21 and 38 % of patients and included anti-VEGF therapy in 40 and 24 % of patients in the panitumumab and bevacizumab arms, respectively.
Fig. 4.
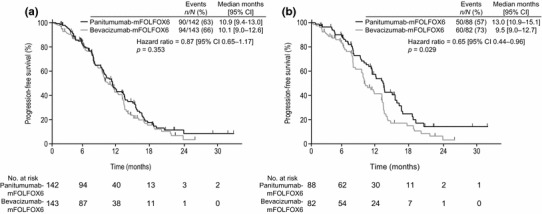
PEAK: progression-free survival in the a Wild-type KRAS exon 2 population (primary analysis, data cut-off 30 May 2012); b Wild-type RAS population (prespecified analysis, data cut-off Jan 3 2013) [31]. From Schwartzberg et al. [31]. Reprinted with permission © 2014 American Society of Clinical Oncology. All rights reserved. CI confidence intervals
Fig. 5.
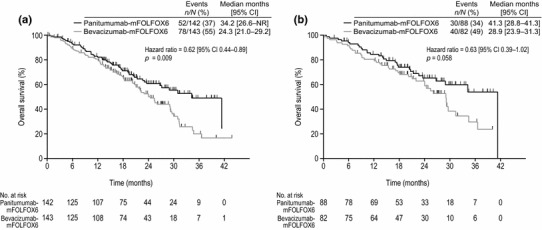
PEAK: overall survival in the a Wild-type KRAS exon 2 population; b Wild-type RAS population (primary analysis, data cut-off 3 January 2013) [31]. From Schwartzberg et al. [31]. Reprinted with permission © 2014 American Society of Clinical Oncology. All rights reserved. CI confidence intervals, NR not recorded
In PEAK, ORRs were similar between treatments (58 % [95 % CI 49–66] vs 54 % [95 % CI 45–62]) for the panitumumab vs bevacizumab arms, respectively).
Efficacy in the RAS WT Population
In PEAK, tumour samples from 250/285 (88 %) patients underwent a prespecified extended RAS analysis and 233 (82 %) results were obtained; 170/221 (77 %) patients with KRAS exon 2 WT mCRC had RAS WT tumours and 51/221 (23 %) had other RAS mutations [31]. The sites of mutations found in the PEAK study are shown in Fig. 2b.
PFS was significantly improved in patients with RAS WT tumours receiving panitumumab + mFOLFOX6 versus bevacizumab + mFOLFOX6 (Fig. 4b). The OS benefits seen in KRAS exon 2 WT patients receiving panitumumab + mFOLFOX6 versus bevacizumab + mFOLFOX6 were greater in the RAS WT population (Fig. 5b). Median OS in the KRAS exon 2 WT population was 34.2 versus 24.3 months in the panitumumab versus bevacizumab arms, respectively (HR 0.62 [95 % CI 0.44–0.89]; p = 0.009) in comparison to 41.3 versus 28.9 months (HR 0.63 [95 % CI 0.39–1.02]; p = 0.058) for these agents, respectively, in the RAS WT population. Post-study treatment in the RAS WT population included EGFR-targeted mAbs in 22 and 37 % of patients and anti-VEGF therapy in 40 versus 33 % of patients in the panitumumab and bevacizumab arms, respectively, suggesting that second-line treatment was unlikely to be driving differences in outcome.
In PEAK, ORRs in the RAS WT group were 64 % [95 % CI 53–74] versus 60 % [95 % CI 49–71] for panitumumab + mFOLFOX6 versus bevacizumab + mFOLFOX6, respectively. ORR results for patients with KRAS exon 2 WT/other RAS MT tumours were consistent with those reported for the KRAS exon 2 WT population.
Phase II Data—The PLANET Trial
PLANET (NCT00885885) was a phase II, open-label, randomised, parallel-group study including patients with KRAS exon 2 WT mCRC and unresectable liver-limited metastases, who had recurrence after prior adjuvant and/or surgical treatment [33]. Patients were randomised to receive preoperative panitumumab (6 mg/kg Q2W) with either FOLFOX4 or FOLFIRI for 4–8 cycles. Patients with stable disease or who remained unresectable received additional cycles until disease progression (PD) or unacceptable toxicity. Six cycles of adjuvant treatment were also administered after surgery. The impact of RAS mutations in exons 2, 3 and 4 of KRAS and NRAS on treatment efficacy was also determined in an exploratory analysis of this study.
Efficacy in the KRAS Exon 2 WT Population
Overall, 77 patients with KRAS exon 2 WT mCRC were randomised and comprised the primary analysis set in PLANET [33]. Efficacy results were generally similar between treatments. Overall, the ORR was 70 % and was 74 % for panitumumab + FOLFOX4 versus 67 % for panitumumab + FOLFIRI. Liver resection was reported in 52 % of patients (45 % for panitumumab + FOLFOX4 vs 59 % for panitumumab + FOLFIRI) after preoperative treatment. Median PFS was 12.6 months for both treatments (p = 0.943) and median OS was 32.5 versus 42.4 months for patients receiving panitumumab + FOLFOX4 versus panitumumab + FOLFIRI, respectively (p = 0.848).
Impact of RAS Mutations Beyond KRAS Exon 2
RAS status was determined for 64 patients (83 %) in PLANET, of whom 53 (83 %) were found to have RAS WT and 11 (17 %) were found to have RAS MT mCRC [33]. Overall, in RAS WT patients, the ORR increased to 76 % and was 78 % for panitumumab + FOLFOX4 versus 73 % for panitumumab + FOLFIRI. In RAS MT patients, the ORR was 55 % overall and was 50 % versus 57 % for panitumumab + FOLFOX4 versus panitumumab + FOLFIRI. In the RAS WT population, median PFS was 12.8 versus 14.8 months (p = 0.621) and median OS was 39.0 versus 45.8 months (p = 0.935) for panitumumab + FOLFOX4 versus panitumumab + FOLFIRI groups, respectively.
Phase II Data—The 314 Trial
The 314 trial (NCT00508404) was a phase II, single-arm study in which panitumumab (6 mg/kg Q2W) + FOLFIRI was administered to patients with previously untreated mCRC [34]. After the importance of KRAS as a biomarker was demonstrated in patients receiving anti-EGFR therapies [5, 13], the protocol of this study was amended to enable prospective evaluation of outcomes by tumour KRAS exon 2 status. Subsequently, an exploratory analysis of panitumumab + FOLFIRI activity by tumour RAS/BRAF status was performed using tumour samples from participating patients with known KRAS exon 2 WT status. In this analysis, mutations were assessed in NRAS exon 2 (codons 12 and 13), KRAS and NRAS exons 3 (codons 59 and 61) and 4 (codons 117 and 146) and BRAF exon 15 (codon 600) using bidirectional Sanger sequencing.
Impact of KRAS Exon 2 Status
Overall, 145/154 (94 %) patients included in the study had KRAS evaluable samples, of whom 86 (59 %) had KRAS exon 2 WT tumours and 59 (41 %) had KRAS exon 2 MT tumours [34]. A higher proportion of patients in the KRAS exon 2 WT group (56 % [95 % CI 45–67]) than the MT group (38 % [95 % CI 26–52]) had an objective response (difference 18 % [95 % CI 1–35]; odds ratio [OR] 2.1 [95 % CI 1.0–4.4]). Median duration of response (DoR; 13.0 vs 7.4 months; HR 0.3 [95 % CI 0.1–0.6]), PFS (8.9 vs 7.2 months; HR 0.5 [95 % CI 0.3–0.7]) and time to progression (TTP; 11.2 vs 7.3 months; HR 0.4 [95 % CI 0.3–0.6]) also favoured the KRAS exon 2 WT versus MT group. Seven patients (8 %) in the KRAS exon 2 WT group had a complete resection, compared with three (5 %) in the KRAS MT group. Most patients undergoing a complete resection had a complete resection of liver metastases (KRAS exon 2 WT: n = 6 vs MT: n = 1).
Impact of RAS/BRAF Mutations on Activity
RAS/BRAF status was retrospectively determined in 143/154 (93 %) patients from the 314 study. Of these, 69 patients (48 %) had RAS WT and 74 (52 %) had RAS MT tumours; 60 patients (42 %) had RAS WT/BRAF WT tumours, and 83 (58 %) had RAS MT or BRAF MT tumours [35].
The ORR was higher in patients with RAS WT versus RAS MT mCRC (59 vs 41 %; OR 2.0 [95 % CI 1.0–4.2]). Longer median DoR (13.0 vs 5.8 months; HR 0.16 [95 % CI 0.07–0.37]) and PFS (11.2 vs 7.3 months; HR 0.37 [95 % CI 0.24–0.58]) and numerically longer TTP (13.2 vs 7.3 months) were also observed in the RAS WT versus MT groups. Patients with RAS WT/BRAF WT versus RAS MT or BRAF MT status also had improved ORR (68 vs 37 %; OR 3.6 [95 % CI 1.7–7.9]), median DoR (13.0 vs 5.8 months; HR 0.16 [95 % CI 0.07–0.37]) and PFS (13.2 vs 6.9 months; HR 0.25 [95 % CI 0.15–0.41]) and numerically longer TTP (13.3 vs 7.2 months). Resection rates were 13 versus 9 % in RAS WT versus RAS MT populations and 15 versus 8 % for RAS WT/BRAF WT versus RAS MT or BRAF MT populations, respectively.
Second-Line Setting
There has been one large second-line panitumumab trial, conducted in an unselected patient population (n = 1186); RAS data were available for 1014 patients (85 %). There has also been a smaller (n = 182) second-line trial conducted in patients with KRAS exon 2 WT mCRC; however, RAS analyses are not available from this study.
Phase III Data—The 181 Trial
20050181 (NCT00339183) was an open-label, phase III, randomised (1:1) study comparing panitumumab 6 mg/kg Q2W + FOLFIRI versus FOLFIRI alone as second-line treatments for patients who had progressed on one prior fluoropyrimidine-based mCRC therapy [11]. As the impact of KRAS mutations on EGFR inhibitor therapy became apparent, the study protocol was amended to permit data to be prospectively analysed by tumour KRAS exon 2 status.
Two clinical snapshots were reported for the KRAS exon 2 WT population in this study. The primary analyses of PFS and OS were conducted after 381 PFS events had occurred and after 407 deaths, respectively [11]. A further prespecified final analysis of PFS and OS was conducted ~30 months after the last patient enrolled [36].
Mutations were subsequently assessed in NRAS exon 2 (codons 12 and 13), KRAS and NRAS exons 3 (codons 59 and 61) and 4 (codons 117 and 146) and BRAF exon 15 (codon 600). Results of this prospective–retrospective analysis have recently been reported [14].
Efficacy in the KRAS Exon 2 WT Population
Overall, 1083/1186 patients (91 %) in the 181 study had tumour KRAS data available, of whom 597 patients (55 %) had KRAS WT tumours [11]. In the primary analysis, a statistically significant PFS benefit was observed in the panitumumab + FOLFIRI versus FOLFIRI group (Table 3). There was no significant difference in OS between panitumumab + FOLFIRI versus FOLFIRI groups, however, significantly more patients receiving panitumumab + FOLFIRI versus FOLFIRI alone had an objective response (Table 3).
Table 3.
Impact of KRAS exon 2 [36] and RAS [14, 30] status on outcomes in the 181 trial (primary analysis data)
| KRAS exon 2 WT [11] | RAS WTa [14, 30] | |||
|---|---|---|---|---|
| Panitumumab + FOLFIRI (n = 303) | FOLFIRI (n = 294) | Panitumumab + FOLFIRI (n = 208) | FOLFIRI (n = 213) | |
| Median PFS, months | 5.9 | 3.9 | 6.4 | 4.6 |
| HR [95 % CI]; p value | 0.73 [0.59–0.90]; 0.004 | 0.70 [0.54–0.91]; 0.007 | ||
| Median OS, months | 14.5 | 12.5 | 16.2 | 13.9 |
| HR [95 % CI]; p value | 0.85 [0.70–1.04]; 0.12 | 0.81 [0.63–1.03]; 0.08 | ||
| ORR,c % [95 % CI] | 35 [30–41] | 10 [7–14] | 41 [34–48] | 10 [6–15] |
| OR [95 % CI]; p value | NA [NA]; 0.001 | NA | ||
| KRAS exon 2 MT [11] | RAS MTb [14] | |||
|---|---|---|---|---|
| Panitumumab + FOLFOX4 (n = 238) | FOLFOX4 (n = 248) | Panitumumab + FOLFOX4 (n = 299) | FOLFOX4 (n = 294) | |
| Median PFS, months | 5.0 | 4.9 | 4.8 | 4.0 |
| HR [95 % CI]; p value | 0.85 [0.68–1.06]; 0.14 | 0.86 [0.71–1.05]; 0.14 | ||
| Median OS, months | 11.8 | 11.1 | 11.8 | 11.1 |
| HR [95 % CI]; p value | 0.94 [0.76–1.15]; ND | 0.91 [0.76–1.10]; 0.34 | ||
| ORR,c % [95 % CI] | 13 [9–18] | 14 [10–19] | NA | NA |
| OR [95 % CI]; p value | NA [NA]; 1.0 | NA | ||
CI confidence interval, HR hazard ratio, MT mutant, NA not available, ND not done, OR odds ratio, ORR objective response rate, OS overall survival, PFS progression-free survival, WT wild type
aWild type at KRAS and NRAS exons 2 (codons 12 and 13), 3 (codon 61) and 4 (codons 117 and 146)
bMutations at any of KRAS and NRAS exons 2 (codons 12 and 13), 3 (codon 61) and 4 (codons 117 and 146). Of 597 patients with KRAS exon 2 WT tumours and RAS data, 107 (18 %) had other RAS mutations (RAS status determined using bidirectional Sanger sequencing and WAVE-based Surveyor® Scan Kits (Transgenomic)
cIncluded only patients with baseline measurable disease per central review
Results of the final analysis were consistent with the primary analysis. The addition of panitumumab to FOLFIRI significantly improved PFS (median 6.7 vs 4.9 months; HR 0.82 [95 % CI 0.69–0.97]; p = 0.023) and ORR (35 vs 10 %; OR 5.5 [95 % CI 3.32–8.87]; p < 0.0001) but not OS (median 14.5 vs 12.5 months; HR 0.92 [0.78–1.10]; p = 0.37) in patients with KRAS exon 2 WT mCRC [36].
Efficacy in the RAS WT Population
Tumour RAS/BRAF status was determined for 1014/1186 patients (85 %) in the 181 study; 107/597 KRAS exon 2 WT patients (18 %) were found to harbour additional RAS mutations. BRAF mutations were found in 45/541 patients (8 %) [14].
In the RAS WT population (n = 421), patients receiving panitumumab + FOLFIRI versus FOLFIRI had significantly improved PFS and a trend towards improved OS (Table 3). Analysis of prespecified subgroups showed that the treatment benefit of panitumumab was generally consistent across subgroups, although the HR did not favour panitumumab in patients who had longer time (>6 months) to PD. RAS WT patients receiving panitumumab + FOLFIRI had significantly longer PFS if they were aged <65 years (HR 0.64 [95 % CI 0.45–0.89]; p = 0.008), had an ECOG performance status of 0 or 1 (HR 0.68 [95 % CI 0.52–0.88]; p = 0.0042) or had BRAF WT mCRC (HR 0.68 [95 % CI 0.51–0.90]; p = 0.0063) [37]. Those who had received prior oxaliplatin (HR 0.64 [95 % CI 0.47–0.86]; p = 0.0035) or no prior bevacizumab (HR 0.72 [95 % CI 0.54–0.95]; p = 0.0204) also showed significant PFS benefits on addition of panitumumab to FOLFIRI. Overall, patients with BRAF mutations had worse prognosis, irrespective of treatment arm. Finally, more RAS WT patients in the panitumumab + FOLFIRI versus FOLFIRI group achieved tumour shrinkage ≥30 % at week 8 (37 vs 7 %; difference: 30 % [95 % CI 22–38]) and the ORR was also higher in this group (41 % [95 % CI 34–48] vs 10 % [95 % CI 6–15]; difference 31 % [95 % CI 23–38]) [30].
Patients with RAS MT tumours had similar PFS and OS in the panitumumab + FOLFIRI versus FOLFIRI groups (Table 3). BRAF mutations were associated with numerically worse PFS (median 2.5 vs 1.8 months; HR 0.69 [95 % CI 0.32–1.49]; p = 0.34) and OS (median 4.7 vs 5.7 months; HR 0.64 [95 % CI 0.32–1.28]; p = 0.20) in patients with RAS WT tumours, irrespective of treatment received [14].
Phase II Data—The SPIRITT trial
SPIRITT (NCT00418938) was a randomised, open-label, phase II study of panitumumab 6 mg/kg Q2W + FOLFIRI versus bevacizumab 5 or 10 mg/kg Q2W + FOLFIRI as second-line treatments for patients with KRAS exon 2 WT mCRC previously treated with first-line bevacizumab + oxaliplatin-based therapy. No prior irinotecan or anti-EGFR therapy was permitted [38].
Efficacy in the KRAS Exon 2 WT Population
One hundred and eighty-two patients with KRAS exon 2 WT mCRC were included in the initial analysis (n = 91 in each treatment arm). Median PFS (primary endpoint: 7.7 vs 9.2 months; HR 1.01 [95 % CI 0.68–1.50]) and OS (18.0 vs 21.4 months; HR 1.06 [95 % CI 0.75–1.49]) did not differ significantly between panitumumab + FOLFIRI and bevacizumab + FOLFIRI groups [38]. Objective responses were reported in 32 % [95 % CI 23–43] versus 19 % [95 % CI 11–29] of patients in the panitumumab versus bevacizumab groups, respectively. Currently, no RAS data have been reported from this study.
Third-Line and Beyond
RAS data have been reported from one large panitumumab trial conducted in an unselected mCRC population (n = 463) who had PD following at least two chemotherapy regimens. RAS data were available for 288 patients (62 %). A second large trial (ASPECCT) compared panitumumab and cetuximab specifically in patients (n = 999) with KRAS exon 2 WT mCRC. RAS analyses are not yet available from this study.
Phase III Data—The 408 Trial
The 20020408 trial (NCT00113763) was a randomised, open-label, phase III study of panitumumab 6 mg/kg Q2W + BSC versus BSC alone as treatment for patients with EGFR-expressing mCRC after failure of fluoropyrimidines and oxaliplatin- and irinotecan-containing regimens [39]. To ensure adequate exposure to prior chemotherapy, average dose intensity of irinotecan (≥65 mg/m2 per week) and of oxaliplatin (≥30 mg/m2 per week) were required. All but one patient in this study had received at least two prior chemotherapy lines; 37 % had received three prior lines. This study also incorporated an optional open-label extension study in which BSC patients experiencing PD in the pivotal phase III study could cross over to receive panitumumab monotherapy [40].
A prospective–retrospective analysis from the 408 study [13] first tested the clinical hypothesis that a larger treatment effect of EGFR-targeted mAbs would be observed in patients with KRAS exon 2 WT tumours compared with patients with KRAS exon 2 MT tumours. An exploratory analysis was also subsequently performed in patients with KRAS exon 2 WT tumours to assess the impact of mutations in NRAS exon 2 (codons 12 and 13), and KRAS and NRAS exon 3 (codon 61), using massively parallel sequencing [15].
Impact of KRAS Exon 2 Status
Of the 231 patients included in the panitumumab + BSC arm, 208 (90 %) had biomarker data; of these, 124 (60 %) had KRAS exon 2 WT mCRC [13]. Of the 232 patients included in the BSC arm, 219 (94 %) had biomarker data; of these, 119 (54 %) had KRAS exon 2 WT mCRC. Overall, 176 BSC patients subsequently received panitumumab in the crossover study. Biomarker data were available for 167 (95 %) of the patients who underwent crossover; 90 (54 %) had KRAS WT mCRC.
In the pivotal phase III study, the treatment effect on PFS was significantly greater in patients with KRAS exon 2 WT tumours than in patients with KRAS exon 2 MT tumours [13]. Median PFS was significantly longer for panitumumab + BSC versus BSC alone (median 12.3 weeks vs 7.3 weeks; HR 0.45 [95 % CI 0.34–0.59]; p < 0.0001) and ORRs were 17 % [95 % CI 11–25 %] versus 0 %, respectively. Median PFS was similar between treatments for patients with KRAS exon 2 MT tumours (median 7.4 vs 7.3 weeks; HR 0.99 [95 % CI 0.73–1.36]). PFS was also significantly longer in patients with KRAS exon 2 WT versus MT tumours receiving panitumumab treatment in the crossover study (median 16.4 vs 7.9 weeks; HR 0.32 [95 % CI 0.22–0.45]).
OS was similar between treatments among patients with KRAS exon 2 WT (HR 0.99 [95 % CI 0.75–1.29]) or KRAS exon 2 MT tumours (HR 1.02 [95 % CI 0.75–1.39]). However, OS analysis was confounded by the high proportion of patients who crossed over from the BSC arm to receive panitumumab therapy following PD.
Impact of RAS Mutations on Efficacy
Overall, 320 archival tumour samples were available from the 463 patients originally included in the 408 study, 288 of whom (90 %) provided RAS data [15]. In an exploratory analysis, panitumumab was associated with longer PFS versus BSC in patients with KRAS WT mCRC (codons 12, 13 and 61 assessed; HR 0.39 [95 % CI 0.28–0.56]) [15]. Among KRAS WT (n = 153) patients, a treatment effect for PFS favouring panitumumab occurred in patients with NRAS WT mCRC (n = 138; HR 0.39 [95 % CI 0.27–0.56]) and BRAF WT mCRC (n = 115; HR 0.37 [95 % CI 0.24–0.55]), but not in patients with NRAS mutations (n = 11; HR 1.94 [95 % CI 0.44–8.44]).
Phase III Data—The ASPECCT Trial
ASPECCT (NCT01001377) was a randomised, open-label, phase III non-inferiority study of panitumumab (6.0 mg/kg Q2W) versus cetuximab (initial dose of 400 mg/m2, followed by 250 mg/m2 once a week [QW]) in patients with KRAS exon 2 WT, chemorefractory mCRC [41]. Patients must have failed prior regimens containing both irinotecan and oxaliplatin for metastatic disease and have previously received a thymidylate synthase inhibitor (fluorouracil, capecitabine, raltitrexed, or fluorouracil-uracil). Patients in the cetuximab group received premedication with an H1 antagonist before infusion; premedication for infusion reactions was not required in the panitumumab group.
Efficacy in the KRAS Exon 2 WT Population
At the time of primary analysis (n = 999), 77 % (383/499) versus 78 % (392/500) of patients in the panitumumab and cetuximab arms had died and 477 patients in each group had progressed or died [41]. Median PFS was similar between panitumumab and cetuximab arms (4.1 vs 4.4 months, respectively; HR 1.00 [95 % CI 0.88–1.14]). For the primary endpoint of OS, panitumumab was found to be non-inferior to cetuximab (Z-score: −3.19; p = 0.0007; median OS 10.4 vs 10.0 months, respectively; HR 0.97 [95 % CI 0.84–1.11]). Similar proportions of patients also experienced an objective response (22 % [95 % CI 18–26] vs 20 % [95 % CI 16–24] for panitumumab and cetuximab groups, respectively; OR 1.15 [95 % CI 0.83–1.58]). No RAS data have been reported from this study.
Safety and Tolerability
Across the KRAS exon 2 WT patients included in these studies, treatment-related grade 3/4 adverse events (AEs) occurred in 82 % of patients receiving panitumumab + FOLFOX4 [10], 68–78 % of patients receiving panitumumab + FOLFIRI [11, 38] and 25 % of those receiving panitumumab + BSC [13]. The most common grade 3/4 AEs of interest (AEs known to be associated with EGFR-targeted mAbs and/or relevant chemotherapy regimen) were integument toxicity (36 % in patients receiving panitumumab + FOLFIRI [34], 25 % in patients receiving panitumumab + BSC [13]), skin toxicity (32–36 % in patients receiving panitumumab + oxaliplatin-based regimens [10, 31], 31–37 % in patients receiving panitumumab + FOLFIRI [11, 34]) and diarrhoea (18 % in patients receiving panitumumab + FOLFOX4 [10], 14–24 % in patients receiving panitumumab + FOLFIRI [11, 34], 2 % in patients receiving panitumumab + BSC [13]). Discontinuations due to AEs occurred in 24 % of patients receiving panitumumab + mFOLFOX6 [31], 29 % of patients receiving panitumumab + FOLFIRI [38] and 7 % of those receiving panitumumab + BSC [13]. In ASPECCT, AE profiles were generally similar for the two EGFR-targeted mAbs. However, despite the use of premedication in the cetuximab group, there was a numeric difference in the incidence of grade 3/4 infusion reactions for the panitumumab versus cetuximab group (<0.5 vs 2 %, respectively). In contrast, grade 3/4 hypomagnesaemia was numerically more frequent in the panitumumab versus cetuximab arm (7 vs 3 %) [41].
In general, the type, incidence and severity of AEs and overall safety profile of panitumumab were similar in patients with RAS WT tumours [12, 31, 35, 42] to those previously reported for those with KRAS exon 2 WT tumours receiving panitumumab treatment [10, 13, 31, 34]. No new safety signals were identified in these extended RAS analyses. Safety data specifically in the RAS WT population have not yet been reported in the PLANET and 181 studies. Selection of patients according to tumour RAS status does not, therefore, appear to be associated with an increase in EGFR-related toxicities.
Discussion
Preclinical and clinical studies of panitumumab support the concept that hypothesis-generating molecular analyses can be used to develop retrospective studies that inform prospective trials on the use of tumour biomarkers, to predict response to targeted anti-cancer treatment. The ‘RAS story’ represents a breakthrough in personalised medicine for patients with mCRC, and illustrates well how molecular selection can be developed to define patient populations for targeted oncology treatment. To date, six key panitumumab trials including a total of 3348 patients have reported extended RAS analyses, with RAS data currently available from 2802 patients (84 %), of whom 1378 (49 %) had RAS WT tumours. In the trials in which collection of tumour samples was mandatory, RAS ascertainment rates were ~90 % [12, 14, 31].
In the first- [10, 12, 31, 33], second- [11, 14, 36] and third-line [13, 15, 42] settings overall, the efficacy benefits of panitumumab treatment were generally more pronounced in RAS WT patients than in the corresponding KRAS exon 2 WT populations, demonstrating the benefit of further refining the target patient population for this agent. Notably, median OS for RAS WT patients in the panitumumab + mFOLFOX6 group from PEAK was 41.3 months [31]; however, patient numbers and the number of deaths at last analysis were relatively low in this phase II study.
The impact of RAS mutations in KRAS exon 3 and 4 and NRAS exons 2, 3 and 4 appears to be similar to that of KRAS exon 2 mutations, with all mutations being predictive of a lack of response to panitumumab therapy. The impact of tumour BRAF mutations was also assessed in retrospective and prospective–retrospective analyses from some of these panitumumab studies, with data generally suggesting that these mutations are indicative of poor prognosis, irrespective of treatment received [12, 35]. No new safety signals were noted for the RAS WT populations [12, 31, 35, 42], with the overall safety profile being similar to that observed in patients with KRAS WT tumours [10, 13, 31, 34]. Safety profiles in all of these studies were as expected based on the mechanisms of action and previously reported data for the study treatments. In general, these studies have consistently reported negative outcomes for patients with RAS MT tumours receiving an EGFR-targeted mAb combined with oxaliplatin-containing chemotherapy [10, 12, 31, 43]. Retrospective data from clinical trials of cetuximab generally appear to support these observations [22, 23].
Efficacy benefits have been noted on addition of cetuximab to FOLFOX4 in RAS WT patients from the first-line OPUS trial (RAS ascertainment rate: 66 % [n = 118/179]) [22] and on addition of cetuximab to FOLFIRI in the corresponding patient population in the first-line CRYSTAL trial (RAS ascertainment rate: 65 % [n = 430/666]) [23]. These data confirm the impact of tumour RAS status on efficacy of all EGFR-targeted mAbs in patients with mCRC. The efficacy and safety of these two EGFR-targeted mAbs has recently been compared in ASPECCT, the first head-to-head monotherapy study of Q2W panitumumab versus QW cetuximab in patients with KRAS exon 2 WT, chemorefractory mCRC [41]. In the primary analysis, panitumumab was non-inferior to cetuximab for OS in the KRAS exon 2 WT population; no RAS analyses have been performed to date. Safety was similar for these two agents with the exception of grade 3/4 infusion reactions (less common with panitumumab: <0.5 vs 2 %, respectively) and grade 3/4 hypomagnesaemia (less common with cetuximab 7 vs 3 %) [41]. Because of the key role of EGFR signalling in skin, skin toxicity is a common side effect of all EGFR-targeted therapies [44]. Proactive treatment with skin moisturizers, sunscreen, topical steroid and/or doxycycline is recommended to help limit the impact of these AEs on quality of life. In a recent panitumumab study comparing pre-emptive or reactive treatment (after skin toxicity developed) of skin toxicity, pre-emptive treatment reduced the incidence of grade 2 + skin toxicities by more than 50 % and patients in this group experienced less quality-of-life impairment compared with those receiving reactive treatment [45].
There are currently no studies investigating optimal treatment sequence of biologics in mCRC; nonetheless, head-to-head trials support the use of EGFR-targeted agents as first-line therapy, as they suggest an OS benefit for these agents relative to anti-VEGF therapy in patients with KRAS exon 2 WT mCRC, with patients with RAS WT tumours even more likely to benefit from anti-EGFR therapy [31, 46, 47]. For example, in the phase III FIRE-3 trial, improved OS was seen with cetuximab + FOLFIRI versus bevacizumab + FOLFIRI in both KRAS exon 2 WT (n = 592; 28.7 vs 25.0 months, HR 0.77 [95 % CI 0.62–0.96]; p = 0.017) and RAS WT patients (n = 342; 33.1 vs 25.6 months, HR 0.70 [95 % CI 0.53–0.92]; p = 0.011) [46]. However, ORR was the primary endpoint in this study and no differences were found between treatments for this measure in either the KRAS exon 2 WT (62 % [95 % CI 56–68] vs 58 % [95 % CI 52–64]; OR 1.18 [95 % CI 0.85–1.64]; p = 0.18) or RAS WT (66 % [95 % CI 58–73] vs 60 % [95 % CI 52–67]; OR 1.28 [95 % CI 0.83–1.99]; p = 0.32) intent-to-treat populations. However, in a predefined per-protocol analysis (n = 526), ORR was higher in the cetuximab versus bevacizumab arms (72 vs 63 %, respectively; p = 0.017) [46]. Furthermore, in a subsequent analysis in which response data were independently, centrally reviewed, significant benefits in favour of the cetuximab + FOLFIRI versus bevacizumab + FOLFIRI group were noted. In this analysis, ORRs were 67 % [95 % CI 60–73] versus 54 % [95 % CI 48–61] in the KRAS exon 2 WT population, respectively (OR 1.68; p = 0.0076) and 71 % [95 % CI 63–79] versus 56 % [95 % CI 48–65] in the RAS WT population (OR 1.93; p = 0.015), respectively [47]. The RAS analyses from FIRE-3 included 407 of the 592 patients (69 %) with KRAS exon 2 WT tumours, who had tumour tissue available [46].
Preliminary results from the randomised, phase III Cancer and Leukaemia Group B (CALGB) 80405 trial comparing first-line bevacizumab or cetuximab combined with chemotherapy in 1137 patients with mCRC have also recently been reported [48]. In this study, 73 % of patients overall received mFOLFOX6 and 27 % received FOLFIRI as their chemotherapy backbone. Although this trial enrolled patients over a 10-year period, only 24-month follow-up data for the 11th interim analysis have so far been presented. After 24 months’ follow up, PFS (10.8 vs 10.4 months; HR 1.04 [95 % CI 0.91–1.17]; p = 0.55) and OS (29.0 vs 29.9 months; HR 0.92 [95 % CI 0.78–1.09]; p = 0.34) were similar in the bevacizumab and cetuximab arms, respectively. Median OS was numerically higher specifically in cetuximab patients receiving the mFOLFOX6 backbone; median OS was 26.9 versus 30.1 months in the bevacizumab versus cetuximab groups, respectively (HR 0.9 [95 % CI 0.7–1.0]; p = 0.09). Overall, 27 % of those receiving bevacizumab versus 32 % of those receiving cetuximab discontinued treatment due to PD. The proportions withdrawing from treatment due to an AE were also similar (56 vs 55 %, respectively). This study included patients with KRAS exon 2 WT tumours, and data from a retrospective analysis of outcomes according to RAS status have recently been reported. In this analysis, median OS was 31.2 versus 32.0 months for RAS WT patients receiving bevacizumab or cetuximab combined with chemotherapy, respectively (HR 0.9 [95 % CI 0.7–1.1]; p = 0.40) [49].
Retrospective analyses assessing the impact of post-PD therapy on efficacy can also give insights into optimal treatment sequencing. In an analysis from the PRIME trial, RAS WT patients receiving first-line panitumumab + FOLFOX4 followed by anti-VEGF therapy had the longest median OS. This sequence was associated with a median OS of 40 months versus 36 months for FOLFOX4 alone followed by anti-VEGF treatment (HR 0.64 [95 % CI 0.41–1.00]; p = 0.0494) and an OS of 26 months for panitumumab + FOLFOX4 versus 21 months for FOLFOX4 followed by treatment not containing an anti-VEGF agent (HR 0.69 [95 % CI 0.53–0.92]; p = 0.0096) [50]. Similarly, in PEAK, median OS was numerically longer for RAS WT patients receiving first-line therapy with panitumumab + mFOLFOX6 (41.3 months, 40 % received subsequent anti-VEGF therapy) than in those who received bevacizumab + mFOLFOX6 (28.9 months, 38 % subsequently received an EGFR-targeted mAb; HR 0.63 [95 % CI 0.39–1.02]; p = 0.058) [31].
Given the importance of tumour RAS testing, particularly for individuals in whom oxaliplatin-based chemotherapy is being planned, it is essential that RAS testing is of a sufficiently high quality to give robust results. The summary of product characteristics for panitumumab acknowledge this point and state that “Mutational status should be determined by an experienced laboratory using validated test methods for detection of KRAS (exons 2, 3 and 4) and NRAS (exons 2, 3 and 4) mutations”. External quality assurance (EQA) programmes have a critical role in ensuring that RAS testing is carried out to a high standard [51]. For example, the European Society of Pathology EQA programme has provided recommendations and an overview of validated laboratory methods, standardised operating procedures, and accreditation criteria relevant for RAS mutation testing [52, 53]. The cost of incorporating tumour RAS testing into clinical practice is important to consider. However, these upfront costs are likely to be offset by subsequent savings in patient care (e.g., by avoiding unnecessary treatment and hospital stays in patients unlikely to respond), as was seen with the introduction of KRAS [54–58] and also BRAF [59] testing.
Although RAS WT status is clearly important for activity of EGFR-targeted mAbs, a proportion of patients with RAS WT tumours still do not respond to these agents, suggesting that other mutations/alterations may also impact on their activity. The PI3K pathway is partly modulated by KRAS activation during EGFR signalling and alterations in this pathway, such as PI3KCA mutations and loss of PTEN expression/activity, may also lead to lack of response to EGFR-targeted mAbs [1, 60–64]. Furthermore, other studies have suggested that overexpression of EGFR ligands such as amphiregulin and epiregulin may predict response to cetuximab [65–67]. However, these potential biomarkers need to be evaluated/validated in properly designed studies to assess their potential usefulness in clinical practice.
To enable biomarker analysis to continue to optimise the treatment of patients with mCRC, collection of tumour samples should be recommended for all future mCRC trials. If biomarker analyses are contemplated, then banked tumour samples may be used to perform prospective analyses prior to the primary trial analysis, potentially providing more robust data. Ideally, sufficient sample should be taken to allow biomarker analyses to continue for several years after initial trial results are published, as new biomarkers are continually being proposed that require validation in a clinical trial setting.
Currently, biopsy in solid tumours has several disadvantages, including its invasive nature and the limitation that it provides a single ‘snapshot’ of the tumour before treatment with biologics. In the future, liquid biopsies of circulating plasma DNA may allow for a less invasive approach to assessing biomarkers in mCRC patients: mutations detected in circulating plasma DNA show good concordance with tumour mutations, particularly when samples are paired [68–70]. When combined with sensitive molecular testing, this technique has the potential to improve patient selection and aid the early detection of PD and the emergence of resistance. For example, KRAS mutations have been detected in the sera of previously KRAS WT patients during treatment with panitumumab [71] or cetuximab [70], in some cases up to 10 months ahead of radiographic progression [70]. Data suggest that EGFR-targeted mAb treatment causes amplification of rare, pre-existing cells harbouring these mutations in ostensibly KRAS WT tumours [71]. Recently, liquid biopsies from patients in the ASPECCT study detected EGFR S492R mutations, which appear to confer resistance to cetuximab but not panitumumab, emerging in 16 % versus 1 % of patients during cetuximab versus panitumumab treatment, respectively [72], supporting the idea that this technique may be particularly useful for assessing biomarkers of resistance and progression. Nonetheless, further clinical studies are required to progress and refine this approach to molecular monitoring.
Most of the current evidence regarding the use of RAS as a biomarker comes from retrospective or prospective–retrospective analyses, using banked samples from completed phase III trials. Debate continues around how much evidence is sufficient when assessing a potential new biomarker using retrospective analyses. Nonetheless, with consistent observations noted in all of these analyses, the weight of evidence from the studies reviewed here suggests that RAS mutations are a robust biomarker for a lack of EGFR-targeted mAb activity. In future studies, there may be sufficient supporting evidence to prospectively test biomarker hypotheses by randomising patients based on their biomarker status to further define the optimum patient population for EGFR-targeted mAbs. Future studies will likely prospectively evaluate the use of these agents specifically in RAS WT patients, while others will exploit the improved molecular understanding of processes underpinning tumour growth and development to assess new targeted combination treatments. Biomarker research in mCRC continues to rapidly evolve and promises to further optimise outcomes via better patient selection and improved tailoring of anti-cancer treatment to the molecular profile of individual patients’ tumours.
In conclusion, data across first- to third-line therapy consistently demonstrate that by testing tumour RAS status, it is possible to select patients more likely to benefit from panitumumab therapy. Consistent effects have also been noted in trials that have reported RAS data for cetuximab in patients with mCRC. EGFR-targeted mAbs have not shown a positive risk/benefit ratio in patients whose tumours harbour mutations in any of KRAS and NRAS exons 2 (codons 12 and 13), 3 (codons 59 and 61) and 4 (codons 117 and 146); accordingly, their licensed indications in Europe now state that treatment should be confined to patients with RAS WT tumours [24, 25]. The risk/benefit profile of panitumumab is thereby improved by restricting treatment to patients whose tumours do not harbour RAS mutations. Notably, EGFR-targeted mAbs may even cause harm when combined with oxaliplatin-containing regimens in patients with RAS MT tumours, highlighting the importance of RAS testing in mCRC. Biomarker studies are ongoing in mCRC and will help to further define the optimum patient population for EGFR-targeted mAbs.
Acknowledgments
This review covers clinical data predominantly from Amgen-sponsored trials of panitumumab; Amgen was therefore involved in design, execution, data collection and analysis of these studies. Medical writing support (funded by Amgen [Europe] GmbH) was provided by Dawn Batty PhD of Bioscript Medical Ltd. MP has received grants and consulting fees from Amgen. MK has received financial support from Amgen in the form of consulting fees/honoraria for participating in the PEAK trial steering committee, a travel grant and payment for lectures. FR has received grants, consulting fees, support for travel and payment for lectures from Amgen, Merck, Roche, Sanofi, Celgene and Bayer. JHT is an employee of Amgen (Europe) GmbH and holds stocks for Amgen Inc. JYD has received financial support from Amgen in the form of consulting fees for providing expertise/advice on files submitted to regulatory agencies, travel to meetings, fees for participation in Amgen steering committee and payment for lectures at Amgen Expert meetings, symposia and educational events. He has also received a research grant (paid to institute), support for travel to meetings/manuscript preparation and fees for participating in Advisory boards from Merck Serono. JYD has also received support for travel to meetings/manuscript preparation and fees for participating in an Advisory board and IDMC from Bayer.
Ethical Standards
All panitumumab studies mentioned in this review article were approved by the appropriate ethics committee and were therefore performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All patients included in these studies gave their informed consent prior to their inclusion.
References
- 1.Heinemann V, Douillard JY, Ducreux M, Peeters M. Targeted therapy in metastatic colorectal cancer—an example of personalised medicine in action. Cancer Treat Rev. 2013;39(6):592–601. doi: 10.1016/j.ctrv.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Pritchard CC, Grady WM. Colorectal cancer molecular biology moves into clinical practice. Gut. 2011;60(1):116–129. doi: 10.1136/gut.2009.206250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqui AD, Piperdi B. KRAS mutation in colon cancer: a marker of resistance to EGFR-I therapy. Ann Surg Oncol. 2010;17(4):1168–1176. doi: 10.1245/s10434-009-0811-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67(6):2643–2648. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 5.Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer. 2007;96(8):1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freeman DJ, Juan T, Reiner M, Hecht JR, Meropol NJ, Berlin J, et al. Association of K-ras mutational status and clinical outcomes in patients with metastatic colorectal cancer receiving panitumumab alone. Clin Colorectal Cancer. 2008;7(3):184–190. doi: 10.3816/CCC.2008.n.024. [DOI] [PubMed] [Google Scholar]
- 7.National Comprehensive Cancer Network®: NCCN Guidelines™ Version 1.2008 Colon Cancer. http://www.nccn.org/. Accessed 7 Apr 2015.
- 8.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27(12):2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 9.National Comprehensive Cancer Network®: NCCN Guidelines™ Version 1.2014 Colon Cancer. http://www.nccn.org/. Accessed 7 Apr 2015.
- 10.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 11.Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28(31):4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 12.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 13.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 14.Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, et al. Updated analysis of KRAS/NRAS and BRAF mutations in study 20050181 of panitumumab (pmab) + FOLFIRI for 2nd-line treatment (tx) of metastatic colorectal cancer (mCRC). J Clin Oncol 2014;32(Suppl.). Abstract 3568.
- 15.Peeters M, Oliner K, Parker A, Siena S, Van Cutsem E, Huang J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase 3 study of metastatic colorectal cancer. Clin Cancer Res. 2013;19(7):1902–1912. doi: 10.1158/1078-0432.CCR-12-1913. [DOI] [PubMed] [Google Scholar]
- 16.Patterson SD, Cohen N, Karnoub M, Truter SL, Emison E, Khambata-Ford S, et al. Prospective-retrospective biomarker analysis for regulatory consideration: white paper from the industry pharmacogenomics working group. Pharmacogenomics. 2011;12(7):939–951. doi: 10.2217/pgs.11.52. [DOI] [PubMed] [Google Scholar]
- 17.Wang SJ, Cohen N, Katz DA, Ruano G, Shaw PM, Spear B. Retrospective validation of genomic biomarkers—what are the questions, challenges and strategies for developing useful relationships to clinical outcomes—workshop summary. Pharmacogenomics J. 2006;6(2):82–88. doi: 10.1038/sj.tpj.6500363. [DOI] [PubMed] [Google Scholar]
- 18.European Medicines Agency: committee for medicinal products for human use (CHMP) summary of opinion (post authorisation) for Vectibix (panitumumab). 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/000741/WC500144827.pdf. Accessed 7 Apr 2015.
- 19.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70(14):5901–5911. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2(3):344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bokemeyer C, Köhne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, et al. Treatment outcome according to tumor RAS mutation status in OPUS study patients with metastatic colorectal cancer (mCRC) randomized to FOLFOX4 with/without cetuximab. J Clin Oncol 2014;32(Suppl.). Abstract 3505.
- 23.Van Cutsem E, Lenz HJ, Köhne CH, Heinemann V, Tejpar S, Melezinek I, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015;33(7):692–700. doi: 10.1200/JCO.2014.59.4812. [DOI] [PubMed] [Google Scholar]
- 24.European Medicines Agency: Vectibix® (panitumumab) prescribing information. 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000741/WC500047710.pdf. Accessed 7 Apr 2015.
- 25.European Medicines Agency: Erbitux® (cetuximab) prescribing information. 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000558/WC500029119.pdf. Accessed 7 Apr 2015.
- 26.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25(7):1346–1355. doi: 10.1093/annonc/mdu141. [DOI] [PubMed] [Google Scholar]
- 27.Chiappa A, Makuuchi M, Lygidakis NJ, Zbar AP, Chong G, Bertani E, et al. The management of colorectal liver metastases: expanding the role of hepatic resection in the age of multimodal therapy. Crit Rev Oncol Hematol. 2009;72(1):65–75. doi: 10.1016/j.critrevonc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Douillard JY, Siena S, Peeters M, Koukakis R, Terwey JH, Tabernero J. Impact of early tumour shrinkage and resection on outcomes in patients with wild-type RAS metastatic colorectal cancer. Eur J Cancer. 2015. (in press). [DOI] [PubMed]
- 29.Rivera F, Karthaus M, Hecht JR, Fasola G, Canon JL, Koukakis R, et al. First-line treatment with modified FOLFOX6 (mFOLFOX6) + panitumumab (pmab) or bevacizumab (bev) in wild-type (WT) RAS metastatic colorectal carcinoma (mCRC): tumor response outcomes beyond RECIST. In: Gastrointestinal cancers symposium 2015, San Francisco. Abstract 660.
- 30.Peeters M, Price T, Cervantes A, Sobrero A, Ducreux M, André T, et al. Tumour shrinkage and response outcomes during second-line panitumumab (pmab) + FOLFIRI vs FOLFIRI treatment. Ann Oncol 2014;25(Suppl. 4). Abstract 7739.
- 31.Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, et al. PEAK: A randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32(21):2240–2247. doi: 10.1200/JCO.2013.53.2473. [DOI] [PubMed] [Google Scholar]
- 32. Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Yu H, et al. Analysis of KRAS/NRAS mutations in PEAK: a randomized phase II study of FOLFOX6 plus panitumumab (pmab) or bevacizumab (bev) as first-line treatment (tx) for wild-type (WT) KRAS (exon 2) metastatic colorectal cancer (mCRC). J Clin Oncol 2013;31(Suppl.). Abstract 3631.
- 33.Abad A, Massuti B, Grávalos C, Escudero P, Guillén-Ponce C, Manzano JL, et al, On behalf of the Spanish Cooperative Group for the Treatment of Digestive Tumors (TTD). Phase II trial of panitumumab plus FOLFOX4 or FOLFIRI in subjects with KRAS wild-type colorectal cancer and liver-limited disease: the PLANET study. J Clin Oncol 2014;32(Suppl.). Abstract 3560.
- 34.Köhne CH, Hofheinz R, Mineur L, Letocha H, Greil R, Thaler J, et al. First-line panitumumab plus irinotecan/5-fluorouracil/leucovorin treatment in patients with metastatic colorectal cancer. J Cancer Res Clin Oncol. 2012;138(1):65–72. doi: 10.1007/s00432-011-1061-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karthaus M, Hofheinz R, Mineur L, Greil R, Thaler J, Fernebro E, et al. Impact of tumour RAS/BRAF status on efficacy of first-line panitumumab + FOLFIRI in patients (pts) with metastatic colorectal cancer (mCRC). Ann Oncol 2014;25(Suppl. 4). Abstract 5672.
- 36.Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, et al. Final results from a randomized phase 3 study of FOLFIRI ± panitumumab for second-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25(1):107–116. doi: 10.1093/annonc/mdt523. [DOI] [PubMed] [Google Scholar]
- 37.Peeters M, Price T, Cervantes A, Sobrero A, Ducreux M, André T, et al. Impact of baseline covariates and prior therapy on the efficacy of second-line panitumumab (pmab) + FOLFIRI vs FOLFIRI treatment. Ann Oncol 2014;25(Suppl. 4). Abstract 7798.
- 38.Hecht JR, Cohn AL, Dakhil SR, Saleh MN, Piperdi B, Cline-Burkhardt VJM, et al. SPIRITT (study 20060141): a randomized phase II study of FOLFIRI with either panitumumab (pmab) or bevacizumab (bev) as second-line treatment (tx) in patients (pts) with wild-type (WT) KRAS metastatic colorectal cancer (mCRC). J Clin Oncol 2013;31(Suppl. 4). Abstract 454.
- 39.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 40.Van Cutsem E, Siena S, Humblet Y, Canon JL, Maurel J, Bajetta E, et al. An open-label, single-arm study assessing safety and efficacy of panitumumab in patients with metastatic colorectal cancer refractory to standard chemotherapy. Ann Oncol. 2008;19(1):92–98. doi: 10.1093/annonc/mdm399. [DOI] [PubMed] [Google Scholar]
- 41.Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014;15(6):569–579. doi: 10.1016/S1470-2045(14)70118-4. [DOI] [PubMed] [Google Scholar]
- 42.Patterson SD, Peeters M, Siena S, Van Cutsem E, Humblet Y, Van Laethem JL, et al. Comprehensive analysis of KRAS and NRAS mutations as predictive biomarkers for single agent panitumumab (pmab) response in a randomized, phase III metastatic colorectal cancer (mCRC) study (20020408). J Clin Oncol 31(Suppl.). Abstract 3617.
- 43.Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22(7):1535–1546. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 44.Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer. 2006;6(10):803–812. doi: 10.1038/nrc1970. [DOI] [PubMed] [Google Scholar]
- 45.Lacouture ME, Mitchell EP, Piperdi B, Pillai MV, Shearer H, Iannotti N, et al. Skin toxicity evaluation protocol with panitumumab (STEPP), a phase II, open-label, randomized trial evaluating the impact of a pre-emptive skin treatment regimen on skin toxicities and quality of life in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28(8):1351–1357. doi: 10.1200/JCO.2008.21.7828. [DOI] [PubMed] [Google Scholar]
- 46.Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 47.Heinemann V, Modest DP, Fischer von Weikersthal L, Decker T, Kiani A, Vehling-Kaiser U, et al. Independent radiological evaluation of objective response, early tumour shrinkage, and depth of response in FIRE-3 (AIO KRK-0306) Ann Oncol. 2014;25(Suppl. 2):ii117. doi: 10.1093/annonc/mdu193.30. [DOI] [Google Scholar]
- 48.Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Mahoney MR, O’Neil BH, et al. CALGB/SWOG 80405: Phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the rectum or colon (MCRC). J Clin Oncol 2014;32(Suppl.):5S. (Abstract LBA3).
- 49.Lenz H, Niedzwiecki D, Innocenti F, Blanke C, Mahoney MR, O’Neil BH, et al. CALGB/SWOG 80405: Phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with expanded ras analyses untreated metastatic adenocarcinoma of the colon or rectum (mCRC). Ann Oncol 25(Suppl. 2). Abstract 501O.
- 50.Peeters M, Douillard JY, Siena S, Price T, Tabernero J, Sidhu R, et al. Impact of post-progression anti-vascular endothelial growth factor-containing therapy on survival in patients with metastatic colorectal cancer: data from the PRIME study. Eur J Cancer. 2013;49(Suppl. 4):S18. doi: 10.1016/S0959-8049(13)70139-4. [DOI] [Google Scholar]
- 51.van Krieken H, Tol J. Setting future standards for KRAS testing in colorectal cancer. Pharmacogenomics. 2009;10(1):1–3. doi: 10.2217/14622416.10.1.1. [DOI] [PubMed] [Google Scholar]
- 52.European Society of Pathology: colon external quality assessment scheme 2014. http://kras.eqascheme.org/. Accessed 7 Apr 2015.
- 53.van Krieken JH, Siebers AG, Normanno N. European consensus conference for external quality assessment in molecular pathology. Ann Oncol. 2013;24(8):1958–1963. doi: 10.1093/annonc/mdt153. [DOI] [PubMed] [Google Scholar]
- 54.Shankaran V, Bentrem DJ, Mulcahy MF, Bennett CL, Benson A, III. Economic implications of KRAS testing in metastatic colorectal cancer (mCRC). In: Gastrointestinal cancers symposium 2009, San Francisco. Abstract 298.
- 55.Mancl EE, Kolesar JM, Vermeulen LC. Clinical and economic value of screening for Kras mutations as predictors of response to epidermal growth factor receptor inhibitors. Am J Health Syst Pharm. 2009;66(23):2105–2112. doi: 10.2146/ajhp090036. [DOI] [PubMed] [Google Scholar]
- 56.Vijayaraghavan A, Efrusy MB, Goke B, Kirchner T, Santas CC, Goldberg RM. Cost-effectiveness of KRAS testing in metastatic colorectal cancer patients in the United States and Germany. Int J Cancer. 2012;131(2):438–445. doi: 10.1002/ijc.26400. [DOI] [PubMed] [Google Scholar]
- 57.Gallagher J, BBC News. Cancer cost ‘crisis’ warning from oncologists. 2011. http://www.bbc.co.uk/news/health-15032862. Accessed 7 Apr 2015.
- 58.Königsberg R, Hulla W, Klimpfinger M, Reiner-Concin A, Steininger T, Buchler W, et al. Clinical and economic aspects of KRAS mutational status as predictor for epidermal growth factor receptor inhibitor therapy in metastatic colorectal cancer patients. Oncology. 2011;81(5–6):359–364. doi: 10.1159/000334919. [DOI] [PubMed] [Google Scholar]
- 59.Blank PR, Moch H, Szucs TD, Schwenkglenks M. KRAS and BRAF mutation analysis in metastatic colorectal cancer: a cost-effectiveness analysis from a Swiss perspective. Clin Cancer Res. 2011;17(19):6338–6346. doi: 10.1158/1078-0432.CCR-10-2267. [DOI] [PubMed] [Google Scholar]
- 60.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 61.Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- 62.Sood A, McClain D, Maitra R, Basu-Mallick A, Seetharam R, Kaubisch A, et al. PTEN gene expression and mutations in the PIK3CA gene as predictors of clinical benefit to anti-epidermal growth factor receptor antibody therapy in patients with KRAS wild-type metastatic colorectal cancer. Clin Colorectal Cancer. 2012;11(2):143–150. doi: 10.1016/j.clcc.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mao C, Liao RY, Chen Q. Loss of PTEN expression predicts resistance to EGFR-targeted monoclonal antibodies in patients with metastatic colorectal cancer. Br J Cancer. 2010;102(5):940. doi: 10.1038/sj.bjc.6605575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Razis E, Briasoulis E, Vrettou E, Skarlos DV, Papamichael D, Kostopoulos I, et al. Potential value of PTEN in predicting cetuximab response in colorectal cancer: an exploratory study. BMC Cancer. 2008;8:234. doi: 10.1186/1471-2407-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 66.Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–5074. doi: 10.1200/JCO.2008.21.3744. [DOI] [PubMed] [Google Scholar]
- 67.Baker JB, Dutta D, Watson D, Maddala T, Munneke BM, Shak S, et al. Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2011;104(3):488–495. doi: 10.1038/sj.bjc.6606054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011;17(24):7808–7815. doi: 10.1158/1078-0432.CCR-11-1712. [DOI] [PubMed] [Google Scholar]
- 69.Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18(12):3462–3469. doi: 10.1158/1078-0432.CCR-11-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Newhall K, Price T, Peeters M, Kim TW, Li J, Cascinu S, et al. Frequency of S492R mutations in the epidermal growth factor receptor: analysis of plasma DNA from metastatic colorectal cancer patients treated with panitumumab or cetuximab monotherapy; 16th World Congress on Gastrointestinal Cancer 2014. Ann Oncol 25(Suppl. 2): ii109. (Abstract O-0011). [DOI] [PMC free article] [PubMed]