

Abstract
The integrity of the lung alveolar epithelial barrier is required for the gas exchange and is important for immune regulation. Alveolar epithelial barrier is composed of flat type I cells, which make up approximately 95% of the gas-exchange surface, and cuboidal type II cells, which secrete surfactants and modulate lung immunity. p120-catenin (p120; gene symbol CTNND1) is an important component of adherens junctions of epithelial cells; however, its function in lung alveolar epithelial barrier has not been addressed in genetic models. Here, we created an inducible type II cell–specific p120-knockout mouse (p120EKO). The mutant lungs showed chronic inflammation, and the alveolar epithelial barrier was leaky to 125I-albumin tracer compared to wild type. The mutant lungs also demonstrated marked infiltration of inflammatory cells and activation of NF-κB. Intracellular adhesion molecule 1, Toll-like receptor 4, and macrophage inflammatory protein 2 were all up-regulated. p120EKO lungs showed increased expression of the surfactant proteins Sp-B, Sp-C, and Sp-D, and displayed severe inflammation after pneumonia caused by Pseudomonas aeruginosa compared with wild type. In p120-deficient type II cell monolayers, we observed reduced transepithelial resistance compared to control, consistent with formation of defective adherens junctions. Thus, although type II cells constitute only 5% of the alveolar surface area, p120 expressed in these cells plays a critical role in regulating the innate immunity of the entire lung.
Lungs are constantly exposed to pathogens; therefore, a highly restrictive alveolar epithelial barrier and finely tuned host defense mechanisms are indispensable for their protection.1,2 Unchecked inflammation is linked to various acute and chronic diseases, including edema, acute respiratory distress syndrome, and fibrosis.3,4 Although it is abundantly clear that the alveolar epithelial barrier regulates the transport of gases, liquid, and ions,5,6 the role of the barrier in the regulation of the innate immune function of lungs remains poorly understood.
The restrictiveness of the alveolar epithelial barrier is dependent on a series of interacting proteins comprising the adherens junctions (AJs) and tight junctions (TJs).7,8 The core of the epithelial AJs is composed of E-cadherin, which links cells to one another in the monolayer.9 The cytoplasmic domain of E-cadherin associates with α-catenin, β-catenin, and p120-catenin (p120, official name catenin delta 1; CTNND1).9 The α- and β-catenins can recruit proteins that link E-cadherin to the actin cytoskeleton,9 and together, these interactions maintain the tension landscape in the epithelial monolayer.10 β-Catenin also plays an essential role in the Wnt signaling pathway and thereby contributes to cell proliferation and differentiation.11 However, p120 has received comparatively less attention, although recent studies have shown that p120 has important functions in regulating cadherin stability and turnover12 and innate immunity.13
Here, we focused on the role of p120 expressed in alveolar epithelial type II cells in regulating the innate immune function of lungs. Although alveolar type II cells cover only 5% of the alveolar surface area, these cells are metabolically active.14 They produce surfactants, serve as facultative progenitor cells to repair alveolar injury, and regulate innate immune function of the lung.14 These cells express Toll-like receptors (TLRs) and tumor necrosis factor receptors.15 Interactions with pathogens or endotoxins activate these receptors to initiate NF-κB signaling to produce tumor necrosis factor,16 IL-1 and IL-6,16 regulated on activation normal T cell expressed and secreted,17 and chemokine C-X-C motif ligand 1.18 These factors play key roles in recruiting inflammatory cells.19–21 Alveolar type II cells also secrete the surfactant proteins (Sp)-A, -B, -C, and -D,22 which regulate innate and adaptive immunity by binding to antigen through interactions with surface receptors on inflammatory cell membranes.23 Here, we studied the function of p120 through disrupting the p120 gene in alveolar type II cells in mice using the rtTA/TetO system coupled with a type II cell–specific SPC promoter. In these mice, we observed unchecked chronic lung inflammation associated with increased NF-κB activity and a persistently leaky alveolar epithelial barrier. These results provide the first genetic evidence that p120 in type II cells is a central regulator of innate immunity of lungs.
Materials and Methods
Mouse Strains
The animal experiments were approved by the Animal Care Committee and Institutional Biosafety Committee of the University of Illinois at Chicago. The SPC-rtTA/TetO-Cre/p120loxP mouse line is homozygous for the p120loxP locus and harbors the transgenes SPC-rtTA and TetO-Cre.24–26 This line is designated here as p120EKO and was maintained on a mixed C57/FVB genetic background. For doxycycline (Dox)-induced type II alveolar cell–specific disruption of the p120 gene, mice were fed with a Dox diet (Teklad, Dox concentration 625 mg/kg; Harlan Laboratories, Inc./Harlan Teklad, Madison, WI) for 15 days. In our study, mutant genotype refers to SPC-rtTA/TetO-Cre/p120loxP, whereas the control genotype [wild-type (WT)] refers to +/TetO-Cre/p120loxP or SPC-rtTA/TetO-Cre/+. Control and mutant mice were fed with the Dox diet for 15 days and then fed normal food for 3 to 7 weeks before experiments as indicated later. Each experiment was made with paired mutant and control mice of the same age and sex. Genotype was determined by standard PCR. Primers used for genotyping are listed in Table 1.
Table 1.
Sequences of the Primers Used in This Study
| Primer name | Forward primer | Reverse primer |
|---|---|---|
| p120 3′loxP | 5′-TTTTAGAGCCTCCCACATACAAGC-3′ | 5′-TCAGCACCCACACAAAGGTTG-3′ |
| p120 5'loxp | 5′-TTGAACTCAGGACCGTCAGAGGAG-3′ | 5′-AAAGCAAGCCACCACCAACC-3′ |
| TetO-Cre | 5′-GCGGCATGGTGCAAGTTGAAT-3′ | 5′-CGTTCACCGGCATCAACGTTT-3′ |
| Spc-rtTA | 5′-GACACATATAAGACCCTGGTCA-3′ | 5′-AAAATCTTGCCAGCTTTCCCC-3′ |
| MIP2 | 5′-GAACATCCAGAGCTTGAGTGTGA-3′ | 5′-CTTGAGAGTGGCTATGACTTCTGTC-3′ |
| ICAM | 5′-TCACCAGGAATGTGTACCTGAC-3′ | 5′-GGCTTGTCCCTTGAGTTTTATG-3′ |
| SPA | 5′-AGGCAGACATCCACACAGCTT-3′ | 5′-ACCAGTGGTTTCTCCCAATCAC-3′ |
| SPB | 5′-GGAACACCAGTGAACAGGCTATG-3′ | 5′-AAACTGTTCACACTTTTGCCTGTCTA-3′ |
| SPC | 5′-GCCTTCTCATCGTGGTTGT-3′ | 5′-CCAGTATCATGCCCTTCCT-3′ |
| SPD | 5′-CCAACAAGGAAGCAATCTGACAT-3′ | 5′-CAAGACAAGCATGGAGAGAAAGG-3′ |
| CLO | 5′-CTTGTCCATGGCAAATGCTG-3′ | 5′-TGATCTTCTTGCTGGTCTTGC-3′ |
| TLR4 | 5′-AGTGGGTCAAGGAACAGAAGCA-3′ | 5′-CTTTACCAGCTCATTTCTCACC-3′ |
| AQP5 | 5′-GCCACCCTCATCTTCGTCTTCTTT-3′ | 5′-TGGTTGCCTATTAAGAGGGCCAGA-3′ |
Western Blotting
Freshly isolated lungs or cells were homogenized in lysis buffer [50 mmol/L phosphate buffer (pH 7.4); 2 mmol/L EDTA; 5 mmol/L EGTA; 1% Triton X-100] supplemented with leupeptin, pepstatin, aprotinin, sodium orthovanadate, and phenylmethylsulfonyl fluoride. Proteins were extracted, and the lysate protein concentration was determined by the Bradford method using Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA). Proteins (50 μg) were separated by electrophoresis and transferred to a polyvinylidene difluoride membrane. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered saline solution with 1% Tween-20 for 1 hour at room temperature. Membranes were incubated with specific primary antibodies. After incubation with appropriate secondary antibodies (peroxidase-conjugated), signals were revealed with chemiluminescence (Bio-Rad Laboratories), visualized by autoradiography, and quantified densitometrically by ImageJ version 1.48 (NIH, Bethesda, MD). Values were normalized for expression of β-actin, and results are expressed as percentages of control samples.
Immunohistochemistry
Lungs were isolated as previously described,26 fixed with 4% paraformaldehyde in PBS at 4°C overnight, and forwarded for paraffin embedding and sectioning in the Research Histology and Tissue Imaging Core (University of Illinois at Chicago). Sections were deparaffinized by xylene and rehydrated by serial dilution of ethanol, blocked with 10% donkey or goat normal serum in PBS before antibody staining. Antibodies were used as follow: anti–Sp-A (Chemicon, Billerica, MA), anti–Sp-B (Millipore, Billerica, MA), anti–Sp-C (Santa Cruz Biotechnology, Dallas, TX), anti–Sp-D (Chemicon), anti–β-actin (Santa Cruz Biotechnology), anti–phospho-p65 (Cell Signaling Technology, Danvers, MA), anti-p65 (Santa Cruz Biotechnology), anti–E-cadherin (Millipore), anti-p120 (kindly provided by Dr. Albert B. Reynolds, Vanderbilt University Medical Center, Nashville, TN), anti-zona occludens-1 (ZO-1; Invitrogen, Carlsbad, CA), and anti-T1α (Developmental Studies Hybridoma Bank under the auspices of the National Institute of Child Health and Human Development). Concentrations were used as recommended by the manufacturers.
RT-qPCR
Real-time-PCR was performed as described.26 RNA was extracted by TRIZOL (Invitrogen), treated by DNase I (Qiagen, Valencia, CA), and purified with an RNeasy kit (Qiagen). Real-time RT-PCR was performed using a QuantiTect SYBR Green RT-PCR kit (Qiagen) and the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Carlsbad, CA). Primers were obtained from Integrated DNA Technologies (Coralville, IA) and are listed in Table 1. Data were analyzed using the comparative cycle threshold (CT) method. Relative quantification to the control sample calibrator was calculated using the formula 2(−ΔΔCT). Gene expression was normalized to the level of cyclophilin, which was used as an internal control.26,27 Data are presented as relative expression levels compared with WT.
Alveolar Type II Cell Isolation and Culture
Alveolar type II cells were isolated as previously described.26,28 Animals were anesthetized with ketamine/xylazine mixture and exsanguinated. Lungs were washed with 20 mL of PBS through the pulmonary artery. Dispase (2 mL; BD Biosciences, San Jose, CA) was injected intratracheally, followed by 0.5 mL of 2% melted agarose in PBS at approximately 50°C. Ice was placed on top of the lungs immediately after agarose injection to allow its polymerization. Lungs were then isolated and incubated with dispase for 1 hour at room temperature. Lungs were mechanically dissected in DMEM containing 100 U/mL DNase (Sigma-Aldrich, St. Louis, MO). Dissociated cells were collected by centrifugation (6 minutes, 150 × g, room temperature) and incubated for 1 hour (37°C, 5% CO2) in plates coated with anti-CD32 and anti-CD45 antibodies (eBioscience, San Diego, CA). Cells were panned and collected by centrifugation (6 minutes, 150 × g, room temperature). The purity of type II cells was assessed by a modified Papanicolaou stain,28 and only preparations with >90% purity were used for subsequent experiments.
BAL Fluid Analysis
Bronchoalveolar lavage (BAL) fluid was collected, and protein and cell concentrations were analyzed as previously described.26 Approximately 1 mL of BAL was collected for each mouse, and protein concentration was determined by the Bradford method using the Bio-Rad Protein Assay Kit. Cells in BAL were quantified using a hemocytometer (Fisher Scientific, Hanover Park, IL). Cells were immobilized on Superfrost Plus microscope slides (Fisher Scientific) by cytospin centrifugation (5 minutes, 50 × g, room temperature) and stained to visualize the shape of nucleus using a Hema 3 staining kit (Fisher Scientific) as previously described.26 Concentration of albumin in the BAL was assessed using Albumin Blue Fluorescent Assay Kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions.
TEER Measurement
Transepithelial electrical resistance (TEER) was determined using the Electric Cell Substrate Impedance Sensing system29 (Applied Biophysics, Troy, NY). Alveolar type II cells were isolated and cultured to confluence on gelatin-coated culture chambers with electrodes (8W10E+ electric cell-substrate impedance sensing [ECIS] plates; Applied Biophysics). The resistance of the cultured cells was measured at an AC frequency of 4000 Hz and deducted by the naked electrode resistance. A 4000-Hz frequency was used to ensure that more current would go through the cell–cell junction.30,31 TEER was calculated by multiplying the resistance by the surface area of the electrode.
In Vivo Alveolar Barrier Permeability Measurement
The permeability assay was modified from previously described methods.32 WT and p120EKO mice at 7 weeks post-Dox treatment were anesthetized in a bell jar with 2.5% isoflurane in room air supplied at 2 L per minute. After induction, anesthesia was continued by means of a nose cone, and a tracheotomy was performed. The trachea was rapidly cannulated for ventilation of the lung with the anesthetic gas mixture (peak inspiratory pressure 12 cm H2O, positive end-expiratory pressure 2 cm H2O, frequency 120 min−1). Heparin (50 U) was injected into the jugular vein for anticoagulation. The pulmonary artery and the left atrium were cannulated with custom-made glass cannulae (Radnoti, Monrovia, CA). Lungs were perfused in situ through the pulmonary artery at constant flow (1.6 to 1.8 mL per minute), temperature (37°C), and venous pressure (0 cm H2O) with ventilation continuing at a peak inspiratory pressure of 14 cm H2O. The perfusing liquid consisted of phenol red–free RPMI 1640 medium (Sigma-Aldrich) supplemented with 1 g/100 mL bovine serum albumin (Fraction V, 99% pure and endotoxin-free; Sigma-Aldrich). The perfusate flow rate was measured at the beginning of each experiment by collecting and weighing the venous effluent.
The tracheal cannula had a concentric design permitting ventilation through an 18-Ga stainless-steel tube and intratracheal instillation via an indwelling PE10 tube, whose position was adjustable. After an equilibration perfusion of 30 minutes, the ventilator was turned off, which caused the lung to collapse. The PE10 tube was advanced to the bifurcation of the main bronchus, and secured to the distal trachea with a ligature to prevent instillate back flow. 125I-lableled human serum albumin (0.25 to 0.5 μCi; Anazao Health Corp., Tampa, FL) in 0.4 mL of RPMI, supplemented with 0.3 mg/mL Evans Blue dye as a distribution marker, was instilled into the lung at a rate of 0.2 mL per minute with the aid of a syringe drive. Because the instilled volume of 0.4 mL is comparable to the normal tidal volume in an adult mouse, the instillate in most cases uniformly re-expanded the lung; an additional 0.1 mL of air was injected via the inner tube to clear the dead space. Only lung preparations exhibiting a uniform light blue color (indicative of even tracer distribution) were used.
Immediately after completing the instillation protocol, pulmonary venous effluent samples (volume 1.5 to 2.0 mL) were collected into stoppered tubes at 10-minute intervals for 60 minutes. The radioactivity in the venous effluent samples, instillate, and lung tissue was determined with the aid of a gamma counter (Model 2470; Perkin-Elmer, Shelton, CT). Tracer albumin permeability–surface area product (PS) was calculated for each time point by where Cas and Cperf are the concentrations of albumin tracer [counts per minute (cpm)] in airspace fluid and perfusing liquid, respectively, and solute flux was calculated from the slope of the tracer efflux curve (cpm) since the preceding sample. Cas (cpm/mL) was calculated from the ratio of tracer counts remaining in the lung at each time point and the instilled volume (mL). This volume was estimated as total instilled tracer counts at zero time divided by the measured tracer concentration (cpm/mL) in an instillate sample. PS values were stable after 30 minutes, and the PS product for each lung preparation was calculated from the slope of the tracer efflux curve between 40 and 60 minutes. PS is reported in units of nL per minute.
Mean Linear Intercept Calculation
The mean linear intercept was calculated as previously described using sections stained with hematoxylin and eosin.33,34 Briefly, two lines of the same known length were crossed in a microscopic field of a lung section. Ten fields were measured. Every time the transverse crossed an alveolar wall, a single intercept was counted. A cut in or out of a blood vessel was counted as half of an intercept. The number of intercepts was counted in both vertical and horizontal lines. The mean linear intercept was calculated by the formula in which L is the length of the transverses, m is the sum of all intercepts, and N is the number of times the transverses are placed on the lung.
Pseudomonas aeruginosa–Induced Pneumonia in Mice
Pseudomonas aeruginosa (strain 103) was prepared as described.26 Mice were challenged with P. aeruginosa through intratracheal instillation. Each mouse was injected with 2 × 104 live P. aeruginosa mixed with 2 × 106 dead P. aeruginosa. Dead P. aeruginosa was generated by storage at −20°C for 1 week.
Statistics
Each data point is presented as the means ± SEM. Differences between groups were tested for statistical significance using a t-test. P < 0.05 was considered statistically significant.
Results
Alveolar Type II Cell–Specific Deletion of p120 in Mice
We used the SPC-rtTA/TetO-Cre/p120EKO mouse line to specifically disrupt the p120 gene in type II cells. Expression of Cre recombinase was activated in alveolar type II cells in adult mice by the Dox-inducible rtTA/TetO system coupled with a type II cell–specific promoter, SPC.24–26 Cre recombinase mediates the disruption of p120 through the loxP sites that flank introns 2 to 8 of the p120 gene (Figure 1A).25 The Dox-inducible system enabled the disruption of p120 specifically in type II cells of adult mouse lungs. Previous studies indicated that approximately 70% of type II cells expressed Cre recombinase in this system.26 We found that p120 protein expression in mutant type II cells freshly isolated from p120EKO mouse lungs was not changed at 3 weeks after Dox treatment (Figure 1B) but was significantly decreased at 7 weeks post-Dox treatment (Figure 1C and Supplemental Figure S1). The delayed disruption of p120 may reflect the slow turnover rate of p120 protein and/or type II cells themselves.14,35 Immunohistochemistry showed that, although the disruption of p120 expression occurred in type II cells, expression in other cell types, such as type I cells, was unchanged (Figure 1, D–K). Because p120 protein levels were decreased after 7 weeks post-Dox treatment, all of the following experimental analyses were performed at this time point.
Figure 1.

Type II cell–specific disruption of p120 in mice. A: Schematic diagram showing the type II cell–specific deletion system for disrupting p120. The rtTA protein is produced from a type II cell–specific surfactant protein (Sp)-C promoter. After interacting with doxycycline (Dox), rtTA induces the production of Cre recombinase from the tetO site in type II cells. Cre then disrupts p120 through the two loxP sites flanking introns 2 to 8 of the p120 gene. B and C: Western blot analysis using type II cells isolated from wild-type (WT; black bars) or p120EKO (white bars) lungs at 3 weeks (B) and 7 weeks (C) post-Dox treatment. Relative expression levels of p120 were quantified by measuring signal intensities and normalizing to β-actin level. D–K: Lung sections from WT and p120EKO mice at 7 weeks post-Dox treatment were stained with antibodies to Sp-C, T1α, and p120. E, G, I, and K are enlarged images of the boxed areas in D, F, H, and J, respectively. D–G: Lung sections were stained with Sp-C (red) and p120 (green). p120 expression is present in Sp-C–expressing cells in WT lungs (arrows in D and E), but is absent in Sp-C+ cells in p120EKO lungs (arrows in F and G). H–K: p120 is not deleted in cells that expressed T1α (pink, arrowheads) in both WT (H and I) and p120EKO (J and K) lung sections. Data points are presented as means ± SEM as a percentage of control (WT) (B and C). Data are representative of two independent observations (D–K). n = 3 mice per group (B); n = 6 mice (C). ∗P < 0.05 versus WT. Scale bar = 10 μm.
Severe Lung Inflammatory Phenotype of p120EKO Mice
We first analyzed BAL isolated from control and p120EKO mice at 7 weeks post-Dox treatment. BAL from mutant mice showed an increased total cell count (Figure 2A) and a higher ratio of neutrophils to the total cell number (Figure 2B). To confirm the observed inflammatory phenotype, we measured myeloperoxidase activity36 and found that the lung lysates from p120EKO mice had significantly increased myeloperoxidase levels compared with WT mice (Figure 2C). Assays for NF-κB activity were performed on whole lung lysates by comparing the ratio of phospho-p65 to total p65. This assay showed that p120EKO lungs had increased NF-κB activity compared with WT lungs, whereas the amount of total p65 did not show a change (Figure 2, D and E). Transcript levels of several proinflammatory targets downstream of NF-κB were also increased in p120EKO lungs, including macrophage inflammatory protein-2 (MIP-2 or CXCL2) (Figure 2F), intercellular adhesion molecule 1 (ICAM1) (Figure 2G), and TLR4 (Figure 2H). Because ICAM1 is an important mediator of neutrophil migration,37 we also showed by Western blot analysis that ICAM1 protein expression was augmented in p120EKO whole-lung lysates compared with lysates from WT mice (Figure 2, I and J).
Figure 2.

Inflammatory phenotype of lungs from p120EKO mice. A and B: Mice were subjected to bronchoalveolar lavage (BAL). p120EKO lungs display increased total numbers of cells (A) and a greater ratio of neutrophils to total cells (B) in BAL. C:p120EKO lung lysates show augmented myeloperoxidase (MPO) activity compared with wild-type (WT) lung lysates. D: Lungs were processed for Western blotting for phospho-NF-κBp65 and total NF-κBp65. E: The mutant lungs showed an enhanced ratio of phospho-p65 to total p65, as quantified by measuring band intensities by Western blot analysis. F–H: Quantitative RT-PCR using WT and mutant whole-lung RNA shows higher macrophage inflammatory protein-2 (MIP-2; F), intercellular adhesion molecule 1 (ICAM1; G), and Toll-like receptor 4 (TLR4; H) mRNA levels in mutant lungs compared with WT lungs. I and J: Western blot analysis using whole-lung lysates shows that ICAM1 protein levels are increased in mutant lungs compared with WT lungs. Data points indicate the means ± SEM. n = 3 to 8 mice per group (A–C, E–H, and J). ∗P < 0.05, ∗∗P < 0.01 versus WT.
Because p120 was only disrupted in type II cells, we isolated type II cells from WT and p120EKO lungs 7 weeks post-Dox and examined NF-κB signaling by Western blot analysis and real-time PCR analysis. In contrast to the results obtained using whole-lung lysates, p120EKO mutant type II cells showed similar levels of NF-κB activity as WT controls (Supplemental Figure S2A). Mutant type II cells also expressed levels of TLR4, MIP-2, and ICAM1 transcripts similar to WT cells (Supplemental Figure S2, B–D). The expression level of ICAM1 protein in mutant type II cells was slightly higher than WT, but the value was not statically significant (Supplemental Figure S2E). Thus, the increase in NF-κB activity and MIP-2, ICAM1, and TLR4 expression (Figure 2, D–H) occurred nonautonomously in other lung cells that retained normal p120 expression.
Deletion of Type II Cell p120 Disrupts Alveolar Epithelial Barrier
Because p120 has been shown to stabilize E-cadherin at the AJ,12 we examined the protein levels of E-cadherin in type II cells freshly isolated from WT and p120EKO mice by Western blot analysis. We found that there was a significant reduction of E-cadherin expression in mutant type II cells compared with control (Figure 3A). Because E-cadherin is an important component of AJs, we next examined whether p120 disruption induced alveolar epithelial barrier defect. We found that p120EKO lungs had a higher protein concentration in the BAL (Figure 3B), suggesting severe leakage of capillary–alveolar epithelial barriers. To assess alveolar epithelial permeability, we measured transepithelial albumin flux. Mouse lungs were ventilated and perfused from the pulmonary artery to the pulmonary vein, and 125I-labeled albumin was instilled through the trachea (Figure 3C). The amounts of 125I-bovine serum albumin that crossed the epithelial barrier at different time points were measured by recording radioactivity in venous effluent samples. Permeability was determined by calculating the tracer albumin PS. We observed greater PS in mutant lungs compared with WT lungs (P < 0.05) (Figure 3, D and E). Because the PS value can be affected by the surface area, we estimated the alveolar surface area of WT and mutant lungs by measuring the mean linear intercept value from sections stained with hematoxylin and eosin that were prepared from lungs of both genotypes.33,34 We found that there was no significant difference in either morphology or mean linear intercept values between the WT and the mutant (Supplemental Figure S3). Therefore, the epithelial permeability of the mutant lung was significantly increased compared with WT.
Figure 3.

Leakiness of alveolar epithelial barrier in p120EKO mice. A: Type II cells were freshly isolated from 7 weeks post-doxycycline (Dox)-treated wild-type (WT) and p120EKO mice, and expression levels of E-cadherin in these cells were assessed by Western blot analysis. β-actin level was used as loading control. E-cadherin levels were not quantified because of the extremely faint signal for mutant cells. B: Post-Dox treatment mice subjected to bronchoalveolar lavage (BAL) analysis show increased BAL protein concentrations in the mutant. C–E:In vivo trans-alveolar epithelial 125I-albumin permeability assay. C: A schematic diagram showing the experimental model. Mouse lungs were isolated under anesthesia and ventilated; next, 125I-albumin (125I-bovine serum albumin) was injected intratracheally. Lungs were also perfused with RPMI from the pulmonary artery (PA), and effluent was collected in the pulmonary vein (PV). The trans-alveolar epithelial flux of 125I-albumin was recorded by measuring the radioactivity of venous effluent at different time points. D: Representative tracer washout curves in lungs of WT and p120EKO mice. E:125I-albumin permeability–surface area product (PS) is a measurement of lung alveolar epithelial protein permeability and was calculated from the slope of the washout curve at 40 to 60 minutes. PS was found to be approximately 90 nL per minute in control mice and approximately 220 nL per minute in the mutant mice. F: Albumin levels increased in lavage fluid of p120EKO mice. Data points indicate the means ± SEM (B, E, and F). Data are representative of three independent experiments (A). n = 3 to 8 mice per group (D and E); n = 5 to 6 mice per group (F). ∗P < 0.05, ∗∗P < 0.01 versus WT.
To assess whether deletion of p120 from type II cells could also lead to increased permeability in both endothelial and epithelial barriers, we measured albumin concentration in the BAL. Albumin BAL levels were significantly increased in lavage fluid of p120EKO lung compared to WT (Figure 3F), suggesting increased lung epithelial and vascular endothelial permeability.
Next, we assessed whether p120 deletion in type II cells interfered with their ability to form a restricted monolayer leading to exaggerated protein leak observed in In Vivo Alveolar Barrier Permeability Measurement. Type II cells were isolated from WT or p120EKO mice at 7 weeks post-Dox treatment. Equal numbers of type II cells were cultured on gold-plated electrodes, and real-time changes in TEER were recorded by a method developed by Tiruppathi et al.29 This method used an AC current of 4000 Hz such that more current goes through the cell–cell junctions (paracellular) rather than directly through the cell body (transcellular).29–31 We grew cells for ≥110 hours until they formed a confluent monolayer (Figure 4A). We found from the time course of recorded TEER change that in WT type II cells, TEER rapidly increased until 110 hours after culture initiation, after which it reached a plateau. However, TEER values did not increase in p120-deficient type II cells even after 120 hours (Figure 4B). We also found that the final TEER of mutant type II cells was significantly lower than that of type II monolayers derived from WT lungs (Figure 4C). These findings demonstrate that loss of p120 impaired the formation of restricted epithelial monolayer, ie, increased epithelial permeability was retained in cultured cells obtained from the mutant mice.
Figure 4.

Reduced transepithelial resistance (TEER) and defective adherens junctions in p120-deficient type II cells. A: Alveolar type II cells were freshly isolated from wild-type (WT) and p120EKO mice and cultured until the cells formed a confluent monolayer. B and C: Type II cells from WT and p120EKO mice were cultured on gelatin-coated gold electrodes for TEER measurement. B: TEER was measured at different time points between 80 and 140 hours after culture initiations. Note that TEER values reached a plateau at 110 hours after starting culture. C: TEER measured at 110 hours after culture initiation. D and E: Alveolar type II cells isolated from WT and p120EKO mice were cultured and stained for p120, E-cadherin, and zona occludens-1 (ZO-1). D: Cells in which p120 was depleted do not show E-cadherin staining (arrows). E: Cells in which p120 was disrupted show normal ZO-1 staining (arrows). Data are presented as means ± SEM. n = 3 to 4 mice per group (B); n = 4 to 6 mice per group (C). Data are representative of two independent observations (D and E). ∗P < 0.05 versus WT. Scale bars: 10 μm (A); 5 μm (D and E).
To examine cell–cell adhesion in mutant alveoli epithelial cells, we analyzed these cells by immunofluorescence. We stained for E-cadherin and ZO-1, major components of AJs and TJs, respectively, in cultured type II cells isolated from WT or p120EKO mice. The p120 mutant type II cells showed decreased E-cadherin staining in cell junctions (Figure 4D), consistent with Figure 3A and indicating a role for p120 in stabilizing E-cadherin at AJs.12 By contrast, the expression or localization of ZO-1 remained unaltered in p120-deficient cells (Figure 4E and Supplemental Figure S4), indicating that p120 catenin deletion in alveoli type II cells specifically altered AJs as opposed to TJs.
Augmented Production of Surfactant Proteins Sp-B, Sp-C, and Sp-D in p120EKO Lungs
Because alveolar type II cell–specific p120 depletion could induce lung inflammation by modulating the production of surfactant proteins, we investigated whether the generation of these proteins was altered in p120EKO lungs. We observed by real-time PCR and Western blot analysis that whole lung extracts showed no difference in Sp-A expression between p120EKO and WT mice (Figure 5, A and E), whereas Sp-B, Sp-C, and Sp-D expression were increased in p120EKO lungs compared with WT lungs (Figure 5, B–D, and F–H). By culturing isolated WT and mutant type II cells, and measuring Sp-D levels in the supernatant, we observed that significantly greater amounts of Sp-D was produced by mutant type II cells compared with control cells (Supplemental Figure S5).
Figure 5.

Augmented expression of surfactant proteins (Sp)-B, -C, and -D in p120EKO lungs. Lungs from control (black bars) and p120EKO (white bars) mice were processed for quantitative RT-PCR (A–D) and Western blot (E–H) analysis. A–D: The amount of Sp-A (A), Sp-B (B), Sp-C (C), and Sp-D (D) transcripts were examined by quantitative RT-PCR. E–H: Protein levels of Sp-A (E), Sp-B (F), Sp-C (G), and Sp-D (H) were assessed by Western blot analysis. Expressions of Sp-A, Sp-B, Sp-C, and Sp-D were normalized to β-actin expression and plotted as a percentage of controls [wild type (WT)]. Data points indicate the means ± SEM. n = 3 to 7 mice per group. ∗P < 0.05 versus WT.
Severe Inflammation in Response to P. aeruginosa Pneumonia in p120EKO Mice
Because p120EKO lungs showed inflammatory phenotypes in the unchallenged basal state, we further investigated whether these mutants elicited severe inflammatory responses toward lung injury induced by pneumonia. We used a P. aeruginosa pneumonia model that we described earlier.26 At 48 hours after P. aeruginosa administration, p120EKO lungs exhibited increased myeloperoxidase activity compared with WT (Figure 6A). BAL collected from the mutant lungs at this time contained significantly higher number of cells than control (Figure 6B), and for both control and mutant, >95% of the cells in BAL were neutrophils (Figure 6, C and D).
Figure 6.

Severe inflammation in p120EKO lungs in mice with Pseudomonas aeruginosa–induced pneumonia. Wild-type (WT; black bars) and p120EKO (white bars) mice at 7 weeks post-doxycycline (Dox) were injected with P. aeruginosa through intratracheal instillation, and inflammatory responses were analyzed at 48 hours after the P. aeruginosa challenge. A: Myeloperoxidase (MPO) activities were measured using lung lysates. The data are plotted as the relative values compared to the mean values of WTs in each independent experiment. B: Bronchoalveolar lavage (BAL) was collected at 48 hours after P. aeruginosa, and numbers of cells in BAL were counted. C and D: Hema 3 staining shows that most cells from BAL of both WT (C) and mutant (D) were neutrophils. Data are plotted as means ± SEM (A and B). n = 3 mice from two independent experiments (A and B). ∗P < 0.05, ∗∗P < 0.01 versus WT. Scale bar = 20 μm (C and D).
Deletion of p120 Spares Facultative Progenitor Function of Type II Cells
Type II cells have the potential to serve as facultative progenitor cells that proliferate and transdifferentiate into type I cells14,35 To examine whether p120 plays a role in type II cell progenitor activities, we isolated type II cells from control and p120EKO lungs, and examined the transcription levels of cyclin B1 and CDC25C, two factors that are known to participate in type II cell proliferation.26 The expression levels of these genes were not changed in the mutant type II cells compared with WT (Figure 7, A and B). The expression level of proliferating nuclear antigen in the mutant and WT type II cells was also similar (Figure 7C). When control and mutant type II cells were cultured, both differentiated into flat type I–like cells (Figure 7, D–G) and expressed similar transcript levels of Aqp5, the type I cell marker (Figure 7H).26
Figure 7.

p120-disrupted type II cells show normal proliferation and type II-to-I cell differentiation. A–C: Type II cells were isolated from wild type (WT; black bars) and p120EKO (white bars) mice at 7 weeks post-doxycycline (Dox) and analyzed by quantitative RT-PCR (A and B) or Western blot analysis (C). Type II cells from p120EKO mice do not show differences in the levels of mRNA for cyclin B1 (A) or CDC25C (B). C: Proliferating nuclear antigen (PCNA) protein levels were compared between WT and p120-deficient type II cells. D–H: Type II cells were isolated from mice at 7 weeks post-Dox and cultured. D–G: Type II cells from both WT (D and F) and p120EKO lungs (E and G) were able to differentiate into flat type I-like cells at a similar time frame. F and G are higher magnification of images D and E. H: After culture, p120-deficient type II cells had similar levels of Aqp5 mRNA compared to WT cells, as accessed by quantitative RT-PCR. Data are presented as means ± SEM. Images are representative of more than three experiments (D–G). n = 4 to 6 mice per group (A–C and H). Scale bar = 10 μm (D–G).
Discussion
Here, we disrupted p120 specifically in alveolar epithelial type II cells using the Dox-inducible Cre/loxP system. Lungs from p120EKO mice manifested a defective epithelial barrier, as indicated by increased trans-alveolar epithelial albumin permeability and decreased TEER values compared to controls. The mutant lungs also showed marked inflammation, as was evident by augmented neutrophil influx, phosphorylation of NF-κB p65, and increased expression of MIP-2, ICAM1, and TLR4. In addition, mutant type II cells produced far greater amounts of surfactant proteins, especially Sp-D. These results together showed that p120 expressed in type II alveolar cells has a fundamental role in the regulation of innate immune functions in the lung.
The inflammatory phenotype of p120EKO mice required a period of 7 weeks post-Dox treatment to become apparent. This timing correlated with decreased levels of p120 protein in type II cells that appeared within this time frame. The severity of the phenotype was also correlated with the reduction of p120 levels. The delayed loss of p120 and appearance of inflammatory phenotype likely reflects the slow turnover rate of p120 protein and/or type II cells14,35 The consequences of disrupting p120-catenin in type II cells appeared to be specific to certain type II cell functions such as alveolar epithelial barrier, lung's host defense, and surfactant production, but p120 deletion had no effect on type II cell proliferation and potential of type II cells to differentiate into type I cells.
Although type II cells occupy only approximately 5% of the alveolar surface area, it is surprising that type II cell–specific disruption of p120 caused such pervasive defects in lung immunity and alveolar epithelial integrity. The level of phospho-NF-κB and expressions of MIP-2, TLR4, and ICAM1 mRNA were markedly increased in lungs from p120EKO mice. The severity of the barrier leakiness might result from both loss of AJs in type II cells and injury to the epithelial barrier induced by the inflammation. Interestingly, the TJs and ZO-1 expression appeared normal in mutant type II cells, consistent with the effect of p120-catenin deletion in the intestinal epithelium.38
The loss of alveolar epithelial barrier integrity might be caused by defective stabilization of E-cadherin. p120 could inhibit E-cadherin internalization because it binds to the same C-terminal site on E-cadherin that binds to the clathrin protein AP-1, thereby masking this endocytic signal.39 The loss of E-cadherin in p120-deficient type II cells is in accord with this concept that E-cadherin was internalized and degraded in the absence of p120. Thus, a likely scenario for the severe lung inflammation seen in p120EKO mice is that p120 ablation-induced E-cadherin instability caused a breach in the alveolar epithelial barrier, resulting in the influx of antigens into alveoli, and the activation of lung inflammation. The lung inflammation could indirectly increase the permeability of endothelial barrier.40 In this sense, p120 may be a crucial inflammation dampening mechanism of the alveolar barrier. This is also evident from our finding showing the mutant lungs had severely exaggerated inflammation response to P. aeruginosa pneumonia as compared to WT.
In our study, to compare the transepithelial permeability of WT and p120 mutant cells, we used the ECIS plates to measure the TEER of type II cell monolayers. Our results showed consistently that the TEER values of WT type II cell monolayer were significantly greater than that of p120-depleted cells, indicating reduced barrier function of p120 mutant type II cells. The values obtained in our study were lower than some previous measurements using Ussing chamber systems,41–43 reflecting differences in methods used. The discrepancy may be related to most of the current passing through cell–cell junctions (paracellular) rather than passing the cell bodies (transcellular) in our ECIS system,29–31 whereas the Ussing chamber approach cannot distinguish these two kinds of currents.41–43 Second, in most experiments using a Ussing chamber, the cells were cultured on polycarbonate filter supports,41–43 and the contacts between cells and polycarbonate filter generate significantly increased resistance on top of the resistance imposed by TJs.44 In our system, cells were cultured on gold-plated microelectrodes, and the value we obtained contains little cell-substrate component.44
We also found that p120 ablation in alveoli type II cells resulted in increased production of the surfactant proteins Sp-B, Sp-C, and Sp-D, suggesting that p120 modulates the production of these proteins important for innate immunity. Sp-D plays a protective role against lung infection, allergy, asthma, and inflammation by modulating host defense and inflammation.23,45,46 Sp-D can bind to bacteria, viruses, and dead cells to enhance uptake by macrophages.47 It can also modulate the production of inflammatory mediators by binding to cell surface receptors, including SP-A receptor 210, TLR2, TLR4, and signal-inhibitory protein.23 Sp-D mutant mice exhibit delayed microbial clearance and deregulated expression of proinflammatory cytokines.47 The mechanisms that underlie the overproduction of surfactant proteins by mutant type II cells are unclear; however, because p120 is coupled to the transcription factor Kaiso,48 it is possible that unchecked Kaiso activity in p120-deficient type II cells transcriptionally increases the generation of these surfactant proteins. Augmented Sp-D expression may itself have a role in the pathogenesis of the lung inflammation observed in p120EKO mice. Sp-D binds and activates TLR449; thus, the observed increases of both Sp-D and TLR4 could synergistically contribute to the severe inflammation observed in the lungs of p120EKO mice.
Our findings add support to the role of p120 as an indispensable immune modulator perhaps because it connects the AJs to innate immunity.50–52 Other studies showed that knockdown of p120 by siRNA in lung endothelial cells enhances the lung inflammatory response induced by lipopolysaccharide.52 p120 down-regulated lipopolysaccharide-mediated NF-κB activation through TLR4 signaling in endothelial cells.52 Other studies using mouse models of p120-specific depletion in the epidermis show that these mutant mice display chronic inflammation which is associated with the activation of NF-κB in the absence of infection.50,51 In mice with conditional p120 deletion in the intestine, epithelial barrier defects occur and generate a microenvironment conducive to chronic inflammation and tumor formation.38,53 Our findings not only reinforce these observations but also importantly show that p120 in type II alveolar epithelial cells has a central role in regulating the host defense functions of lungs.
Acknowledgments
We thank Dr. Guo-Chang Hu for discussions and insight; Drs. Marcelo Bonini, Reddy Sekhar, and Chinnaswamy Tiruppathi for providing reagents; Dr. Sukriti Baveja for assistance with TEER measurements; and Dr. Varsha Suresh Kumar for P. aeruginosa injections. Anti-p120 antibody was provided by Dr. Albert B. Reynolds (Vanderbilt University Medical Center, Nashville, TN).
Footnotes
Supported by CAPES fellowship (Coodernação de Aperfeiçoamento de Pessoal de Nível Superior) from Brazil (A.Z.C.); NIH grants HL105947-01 (Y.L.), HL07829-16 (A.B.M.), HL090152 (A.B.M.); and the Department of Pharmacology, University of Illinois College of Medicine, Chicago.
Disclosures: None declared.
Supplemental Data
Supplemental Figure S1.
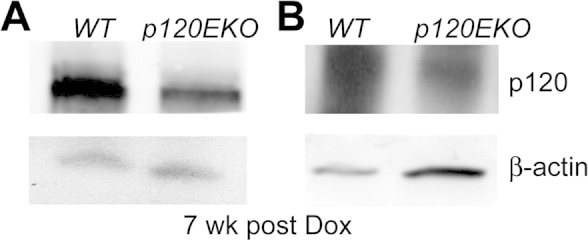
Type II cells isolated from p120EKO lungs show decreased levels of p120 protein. Freshly isolated type II cells from wild-type (WT) and p120EKO lungs at 7 wks post-doxycycline (Dox) treatment were subjected to Western analysis to access the protein amount of p120. β-actin was used as loading control. A and B are two examples of the Western analysis.
Supplemental Figure S2.
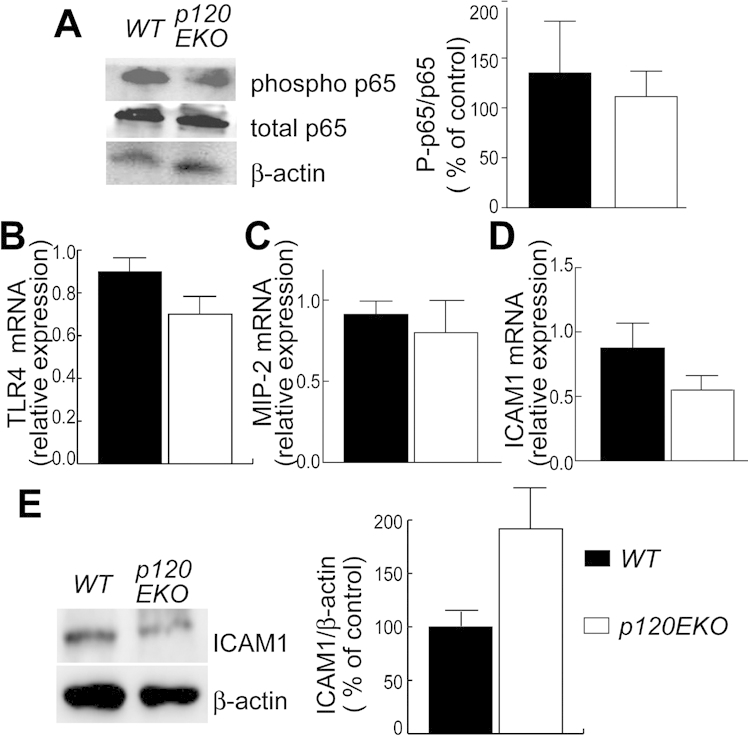
p120-deficient type II cells show similar levels of inflammatory factors. Alveolar type II cells were isolated from 7 wks post-doxycycline lungs and processed for Western blot (A and E) or real-time RT-PCR analysis (B–D). A: Activity of NF-κB was assessed by Western blot analysis using phospho-p65 over total p65 ratio (P-p65/p65). NF-κB activity was estimated by comparing signal intensity of phospho to total p65 and presented as percentage of control (WT). β-Actin is shown as a loading control. p120EKO lung-derived type II cells show similar levels of NF-κB activity. B–D: Transcripts levels of TLR4 (B), MIP-2 (C), and ICAM1 (D) in freshly isolated p120 deficient type II cells were analyzed by quantitative RT-PCR and compared to WT controls. E: ICAM1 protein expression was assessed in fresh isolated alveolar type II cells and was normalized to β-actin expression. Data are presented as means ± SEM. n = 4 to 8 mice per group.
Supplemental Figure S3.
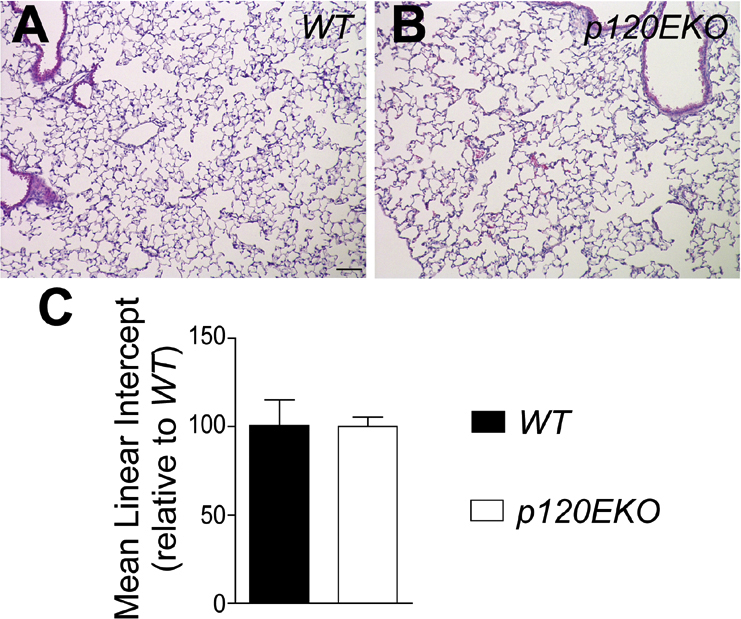
Mean linear intercept does not change in p120EKO lungs. A and B: Wild-type (WT) and p120EKO mouse lung sections were stained with H&E. C: The mean linear intercept was measured using the H&E-stained pictures to estimate alveolar surface area. Data are presented as means ± SEM (C). n = 3 mice per group (C).
Supplemental Figure S4.
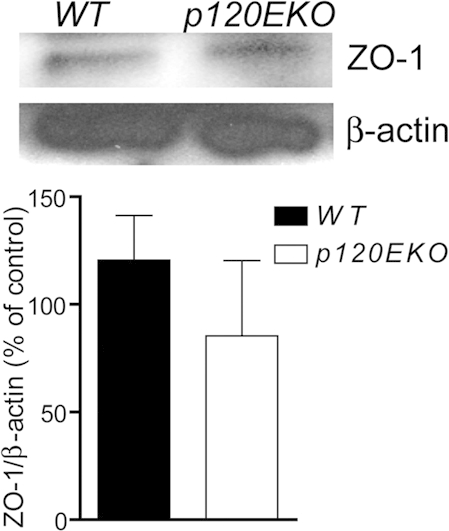
ZO-1 expression of cultured type II cells. WT and p120−/− type II cells were cultured and processed for Western analysis. The signal intensity of the bands was measured using ImageJ and normalized with the β-actin levels. Data are presented as means ± SEM. n = 3 mice per group.
Supplemental Figure S5.

Cultured mutant type II cells produce greater amount of Sp-D than wild type (WT). Type II cells were isolated from WT and p120EKO lungs and cultured; supernatant from the culture was used for Western analysis for the presence of Sp-D protein. Ponceau staining was used as loading control. Data are representative of two independent results.
References
- 1.dos Santos G., Kutuzov M.A., Ridge K.M. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol. 2012;303:L627–L633. doi: 10.1152/ajplung.00225.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizgerd J.P. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am J Respir Crit Care Med. 2012;186:824–829. doi: 10.1164/rccm.201206-1063PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthay M.A., Ware L.B., Zimmerman G.A. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noble P.W., Barkauskas C.E., Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herzog E.L., Brody A.R., Colby T.V., Mason R., Williams M.C. Knowns and unknowns of the alveolus. Proc Am Thorac Soc. 2008;5:778–782. doi: 10.1513/pats.200803-028HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weibel E.R. What makes a good lung? Swiss Med Wkly. 2009;139:375–386. doi: 10.4414/smw.2009.12270. [DOI] [PubMed] [Google Scholar]
- 7.Evans S.M., Blyth D.I., Wong T., Sanjar S., West M.R. Decreased distribution of lung epithelial junction proteins after intratracheal antigen or lipopolysaccharide challenge: correlation with neutrophil influx and levels of BALF sE-cadherin. Am J Respir Cell Mol Biol. 2002;27:446–454. doi: 10.1165/rcmb.4776. [DOI] [PubMed] [Google Scholar]
- 8.Ohta H., Chiba S., Ebina M., Furuse M., Nukiwa T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L193–L205. doi: 10.1152/ajplung.00349.2010. [DOI] [PubMed] [Google Scholar]
- 9.Wheelock M.J., Johnson K.R. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–235. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 10.Borghi N., Sorokina M., Shcherbakova O.G., Weis W.I., Pruitt B.L., Nelson W.J., Dunn A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc Natl Acad Sci U S A. 2012;109:12568–12573. doi: 10.1073/pnas.1204390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flozak A.S., Lam A.P., Russell S., Jain M., Peled O.N., Sheppard K.A., Beri R., Mutlu G.M., Budinger G.S., Gottardi C.J. {beta}-catenin/TCF signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem. 2010;285:3157–3167. doi: 10.1074/jbc.M109.070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reynolds A.B. p120-catenin: past and present. Biochim Biophys Acta. 2007;1773:2–7. doi: 10.1016/j.bbamcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu G. p120-Catenin: a novel regulator of innate immunity and inflammation. Crit Rev Immunol. 2012;32:127–138. doi: 10.1615/critrevimmunol.v32.i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason R.J. Biology of alveolar type II cells. Respirology. 2006;11:S12–S15. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 15.Droemann D., Goldmann T., Branscheid D., Clark R., Dalhoff K., Zabel P., Vollmer E. Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem Cell Biol. 2003;119:103–108. doi: 10.1007/s00418-003-0497-4. [DOI] [PubMed] [Google Scholar]
- 16.Haddad J.J. Recombinant TNF-alpha mediated regulation of the I kappa B-alpha/NF-kappa B signaling pathway: evidence for the enhancement of pro- and anti-inflammatory cytokines in alveolar epithelial cells. Cytokine. 2002;17:301–310. doi: 10.1006/cyto.2002.1017. [DOI] [PubMed] [Google Scholar]
- 17.Chuquimia O.D., Petursdottir D.H., Rahman M.J., Hartl K., Singh M., Fernández C. The role of alveolar epithelial cells in initiating and shaping pulmonary immune responses: communication between innate and adaptive immune systems. PLoS One. 2012;7:e3212. doi: 10.1371/journal.pone.0032125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farnand A.W., Eastman A.J., Herrero R., Hanson J.F., Mongovin S., Altemeier W.A., Matute-Bello G. Fas activation in alveolar epithelial cells induces KC (CXCL1) release by a MyD88-dependent mechanism. Am J Respir Cell Mol Biol. 2011;45:650–658. doi: 10.1165/rcmb.2010-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L.F., Greene W.C. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 20.Hayden M.S., Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Christman J.W., Sadikot R.T., Blackwell T.S. The role of nuclear factor-kappa B in pulmonary diseases. Chest. 2000;117:1482–1487. doi: 10.1378/chest.117.5.1482. [DOI] [PubMed] [Google Scholar]
- 22.Andreeva A.V., Kutuzov M.A., Voyno-Yasenetskaya T.A. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L259–L271. doi: 10.1152/ajplung.00112.2007. [DOI] [PubMed] [Google Scholar]
- 23.Wright J.R. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 24.Perl A.T., Wert S., Nagy A., Lobe C.G., Whitsett J. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A. 2002;99:10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis M.A., Reynolds A.B. Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev Cell. 2006;10:21–31. doi: 10.1016/j.devcel.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y., Sadikot R.T., Adami G.R., Kalinichenko V.V., Pendyala S., Natarajan V., Zhao Y.Y., Malik A.B. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J Exp Med. 2011;208:1473–1484. doi: 10.1084/jem.20102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y.Y., Liu Y., Stan R.V., Fan L., Gu Y., Dalton N., Chu P.H., Peterson K., Ross J.J., Chien K.R. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobbs L.G. Isolation and culture of alveolar type II cells. Am J Physiol. 1990;258:L134–L147. doi: 10.1152/ajplung.1990.258.4.L134. [DOI] [PubMed] [Google Scholar]
- 29.Tiruppathi C., Malik A.B., Del Vecchio P.J., Keese C.R., Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci U S A. 1992;89:7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giaever I., Keese C.R. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci U S A. 1991;88:7896–7900. doi: 10.1073/pnas.88.17.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandoval R., Malik A.B., Minshall R.D., Kouklis P., Ellis C.A., Tiruppathi C. Ca(2+) signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol. 2001;533:433–445. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.John T.A., Vogel S.M., Minshall R.D., Ridge K., Tiruppathi C., Malik A.B. Evidence for the role of alveolar epithelial gp60 in active transalveolar albumin transport in the rat lung. J Physiol. 2001;533(Pt 2):547–559. doi: 10.1111/j.1469-7793.2001.0547a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunnill M.S. Quantitative methods in the study of pulmonary pathology. Thorax. 1962;17:320–328. doi: 10.1136/thx.17.4.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooney T.P., Thurlbeck W.M. The radial alveolar count method of Emery and Mithal: a reappraisal 1–postnatal lung growth. Thorax. 1982;37:572–579. doi: 10.1136/thx.37.8.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barkauskas C.E., Michael J., Cronce M.J., Rackley C.R., Bowie E.J., Keene D.R., Stripp B.R., Randell S.H., Noble P.W., Hogan B.L.M. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mullane K.M., Kraemer R., Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods. 1985;14:157–167. doi: 10.1016/0160-5402(85)90029-4. [DOI] [PubMed] [Google Scholar]
- 37.Hu G., Vogel S.M., Schwartz D.E., Malik A.B., Minshall R.D. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ Res. 2008;102:e120–e131. doi: 10.1161/CIRCRESAHA.107.167486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smalley-Freed W.G., Efimov A., Burnett P.E., Short S.P., Davis M.A., Gumucio D.L., Washington M.K., Coffey R.J., Reynolds A.B. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J Clin Invest. 2010;120:1824–1835. doi: 10.1172/JCI41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nanes B.A., Chiasson-MacKenzie C., Lowery A.M., Ishiyama N., Faundez V., Ikura M., Vincent P.A., Kowalczyk A.P. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komarova Y., Malik A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 41.Chen L., Bosworth C.A., Pico T., Collawn J.F., Varga K., Gao Z., Clancy J.P., Fortenberry J.A., Lancaster J.R., Jr., Matalon S. DETANO and nitrated lipids increase chloride secretion across lung airway cells. Am J Respir Cell Mol Biol. 2008;39:150–162. doi: 10.1165/rcmb.2008-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazrak A., Chen L., Jurkuvenaite A., Doran S.F., Liu G., Li Q., Lancaster J.R., Jr., Matalon S. Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am J Respir Cell Mol Biol. 2012;46:342–354. doi: 10.1165/rcmb.2011-0309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen L., Song W., Davis I.C., Shrestha K., Schwiebert E., Sullender W.M., Matalon S. Inhibition of Na+ transport in lung epithelial cells by respiratory syncytial virus infection. Am J Respir Cell Mol Biol. 2009;40:588–600. doi: 10.1165/rcmb.2008-0034OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo C.M., Keese C.R., Giaever I. Cell-substrate contact: another factor may influence transepithelial electrical resistance of cell layers cultured on permeable filters. Exp Cell Res. 1999;250:576–580. doi: 10.1006/excr.1999.4538. [DOI] [PubMed] [Google Scholar]
- 45.Gaunsbaek M.Q., Rasmussen K.J., Beers M.F., Atochina-Vasserman E.N., Hansen S. Lung surfactant protein D (SP-D) response and regulation during acute and chronic lung injury. Lung. 2013;191:295–303. doi: 10.1007/s00408-013-9452-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qaseem A.S., Sonar S., Mahajan L., Madan T., Sorensen G.L., Shamji M.H., Kishore U. Linking surfactant protein SP-D and IL-13: implications in asthma and allergy. Mol Immunol. 2012;54:98–107. doi: 10.1016/j.molimm.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 47.Wang J.Y., Reid K.B. The immunoregulatory roles of lung surfactant collectins SP-A, and SP-D, in allergen-induced airway inflammation. Immunobiology. 2007;212:417–425. doi: 10.1016/j.imbio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Daniel J.M. Dancing in and out of the nucleus: p120(ctn) and the transcription factor Kaiso. Biochim Biophys Acta. 2007;1773:59–68. doi: 10.1016/j.bbamcr.2006.08.052. [DOI] [PubMed] [Google Scholar]
- 49.Ohya M., Nishitani C., Sano H., Yamada C., Mitsuzawa H., Shimizu T., Saito T., Smith K., Crouch E., Kuroki Y. Human pulmonary surfactant protein D binds the extracellular domains of Toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry. 2006;45:8657–8664. doi: 10.1021/bi060176z. [DOI] [PubMed] [Google Scholar]
- 50.Perez-Moreno M., Davis M.A., Wong E., Pasolli H.A., Reynolds A.B., Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124:631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Moreno M., Song W., Pasolli H.A., Williams S.E., Fuchs E. Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci U S A. 2008;105:15399–15404. doi: 10.1073/pnas.0807301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y.L., Malik A.B., Sun Y., Hu S., Reynolds A.B., Minshall R.D., Hu G. Innate immune function of the adherens junction protein p120-catenin in endothelial response to endotoxin. J Immunol. 2011;186:3180–3187. doi: 10.4049/jimmunol.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smalley-Freed W.G., Efimov A., Short S.P., Jia P., Zhao Z., Washington M.K., Robine S., Coffey R.J., Reynolds A.B. Adenoma formation following limited ablation of p120-catenin in the mouse intestine. PLoS One. 2011;6:e19880. doi: 10.1371/journal.pone.0019880. [DOI] [PMC free article] [PubMed] [Google Scholar]