

Abstract
Glomerulonephritis is a common cause of end-stage renal disease. Infiltrating leukocytes interacting with renal cells play a critical role during the initiation and progression of glomerulonephritis, but the exact mechanisms are not clearly defined. By using the murine model of nephrotoxic nephritis, we investigated the role of S100A8/A9 [myeloid-related protein (MRP) 8/14, calprotectin] in promoting glomerulonephritis. In nephrotoxic nephritis, wild-type (WT) mice with glomerulonephritis have elevated serum levels of S100A8/A9, whereas mice deficient in MRP14 (S100a9−/−), and hence S100A8/A9, are significantly protected from disease. By using bone marrow transplants, we showed that MRP14 deficiency is required in both the hemopoietic and intrinsic cells for the protective effect. In vitro, both the WT bone marrow–derived macrophages and renal mesangial cells stimulated with S100A8/A9 secrete IL-6, CXCL1, and tumor necrosis factor α; however, Mrp14−/− cells exhibit significantly blunted proinflammatory responses. The interaction of WT bone marrow–derived macrophages with renal microvascular endothelial cells results in increased levels of monocyte chemotactic protein 1, IL-8, and IL-6 cytokines, which is attenuated in Mrp14−/− bone marrow–derived macrophages. Data shows that S100A8/A9 plays a critical role during glomerulonephritis, exerting and amplifying autocrine and paracrine proinflammatory effects on bone marrow–derived macrophages, renal endothelial cells, and mesangial cells. Therefore, complete S100A8/A9 blockade may be a new therapeutic target in glomerulonephritis.
S100A8 and S100A9, members of the S100 family of proteins, are released from phagocytes at sites of inflammation, and are termed damage-associated molecular patterns or alarmins. The two molecules can be expressed as homodimers or form a heterodimer of S100A8/A9 (termed calprotectin), which is expressed in neutrophils, monocytes, and early differentiated macrophages,1 but not resident tissue macrophages.2 S100A8/A9 is an endogenous ligand of Toll-like receptor-4 (TLR4),3 as well as the receptor for advanced glycation end products.4,5 S100A8 is the active component of the heterodimer, whereas S100A9 regulates S100A8 activity.3 Both S100A8 and S100A9 are up-regulated by proinflammatory cytokines tumor necrosis factor α (TNF-α) and IL-1β,6 with lipopolysaccharide (LPS) and interferon-γ7 inducing translocation of S100A8 and S100A9 from the cytoplasm to the cell surface.
We previously demonstrated that patients with active generalized anti-neutrophil cytoplasmic antibody–associated vasculitis had elevated serum S100A8/A9 levels and greater neutrophil and monocyte S100A8/A9 cell surface expression, compared to patients in remission and healthy controls.8 In addition, patients who had persistently elevated S100A8/A9 levels, despite immunosuppressive treatment, were at higher risk of subsequent disease relapse.8 Finally, patients with focal segmental necrotizing or crescentic anti-neutrophil cytoplasmic antibody–associated glomerulonephritis demonstrated glomerular infiltration with predominant S100A8/A9-expressing macrophages, whereas the less inflammatory (sclerotic) class of glomerulonephritis contained S100A8/A9-negative macrophages.8 These data linked S100A8/A9 with active systemic vasculitis and glomerulonephritis.
As well as being a potential disease biomarker, S100A8/A9 exerts several paracrine and autocrine effects, which suggest it may have a more direct pathogenic role in vasculitis and glomerulonephritis. S100A8/A9 stimulation of monocytes/macrophages, acting through TLR4, activates NF-κB and other transcription factors, leading to increased production of metalloproteinases9,10 and proinflammatory cytokines,11 as well as stimulating autoreactive IL-17–producing T cells12 implicated in vasculitic inflammation and glomerulonephritis. In addition, the ability of S100A8/A9 to bind the endothelium via heparin sulfate moieties and carboxylated glycans13,14 results in release of leukocyte chemoattractants, such as CXCL1, and induces endothelial cell (EC) necrosis and apoptosis, leading to disruption of the EC monolayer,15,16 a characteristic feature of vasculitis and crescentic glomerulonephritis.
Experimental glomerulonephritis is characterized by a dynamic interaction between intrinsic renal cells and infiltrating leukocytes. Neutrophils and proinflammatory macrophages play a central role in the initiation and progression of many forms of glomerulonephritis,17,18 whereas other macrophage phenotypes are associated with reparative actions.19,20 Intrinsic renal cell TLR4 stimulation is key in promoting glomerular crescent formation and thrombosis,21 whereas TLR4 deficiency promotes protection from experimental nephrotoxic nephritis (NTN). Similarly, in the anti-myeloperoxidase glomerulonephritis model, administration of highly purified LPS with myeloperoxidase results in augmented glomerular neutrophil influx, an effect mediated through TLR4 expressed on glomerular ECs, and other intrinsic renal cells, including podocytes and mesangial cells.22 Mice deficient in Mrp14−/− (S100a9−/−) also lack MRP8 (S100a8) protein and cannot form the calprotectin (MRP8/14) heterodimer.23
Therefore, we hypothesized that S100A8/A9 (calprotectin) may play a role in the pathogenesis of glomerulonephritis by exerting a proinflammatory effect on infiltrating macrophages and intrinsic renal cells. We tested this hypothesis by performing NTN in wild-type (WT) and Mrp14−/− (calprotectin-lacking) mice.
Materials and Methods
Animals
Mrp14−/− mice were kindly provided by Prof. Fredrik Ivars (University of Lund, Lund, Sweden). Mrp14−/− mice were generated as previously described.24 Homozygous mice, aged 8 to 12 weeks, were used for experiments and were bred at the Central Biological Services Unit, Hammersmith Hospital Campus, Imperial College London (London, UK). Control mice were age and sex matched. C57BL/6 control mice used at the Hammersmith Hospital (London, UK) were obtained from Charles River Laboratories. Control C57BL/6 mice at UCL Centre for Nephrology (London, UK) were bred within the animal house. The mice were bred in a pathogen-free environment with access to food and water. Experiments were performed in accordance with Home Office guidelines.
Serum Levels of Murine S100A8/A9
S100A8/A9 concentration in mouse serum was determined by enzyme-linked immunosorbent assay (ELISA), using an ELISA protocol, as previously described.3
Immunohistochemistry to Detect Expression of S100A8/A9-Positive Cells, Macrophages, CD4+ T Cells, and Granulocytes
Immunoperoxidase staining was performed using the Polink-2 HRP Plus kit (Newmarket Scientific, Newmarket, UK). Sections (5 μm thick) of periodate-lysine-paraformaldehyde fixed tissue were used. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 minutes. Subsequently, after rinsing the sections in distilled water, sections were blocked with skimmed milk (Marvel; Premier International Foods, Spalding, UK) for 30 minutes. The sections were then incubated with the primary antibody diluted in phosphate-buffered saline (PBS): rat anti-mouse CD68 (Serotec, Oxford, UK) and rat anti-mouse CD4 (BD Pharmingen, San Diego, CA), both at a dilution of 1:100, rat anti-mouse S100A8/A9 antibody (a kind gift from Dr. Nancy Hogg, Cancer Research UK London Research Institute, London, UK; dilution 1:120), and the granulocyte marker GR01 (Serotec; dilution 1:50). Negative controls were also included in which the primary antibody was replaced with PBS. The sections were incubated with the primary antibody for 1 hour in a humidified chamber at room temperature. After incubation, sections were washed in PBS and two drops of antibody enhancer were then added to each section for 10 minutes. Slides were washed three times, and two drops of the polymer–horseradish peroxidase were incubated with the sections for a further 10 minutes. Slides were washed three times in PBS, and the detection antibody was made up by adding two drops of the 3,3′-diaminobenzidine chromagen solution to 1 mL of diaminobenzidene substrate. Once brown color development was obtained, slides were rinsed in distilled water and counterstained in filtered Harris hematoxylin (Cell Path PLC, Powys, UK) for 30 seconds. Followed by a rinsing step in distilled water, the slides were briefly placed in acid alcohol, followed by a washing step for 5 minutes until the sections turned blue. Sections were dehydrated by serial immersions for 3 minutes each in 100% ethanol twice and xylene twice, before mounting with DePeX Gurr mounting medium (BDH Prolabo Chemicals–VWR International Ltd, Lutterworth, UK) and coverslip placement.
Immunofluorescence Staining
Direct immunofluorescence was performed on PLP-fixed frozen kidney sections to detect mouse and sheep IgG. Goat serum (20%; Sigma, St. Louis, MO) was applied to block non-specific binding, followed by either goat anti-mouse IgG fluorescein isothiocyanate (Sigma), 1:200, or monoclonal anti-goat/sheep IgG fluorescein isothiocyanate (Sigma), 1:200, for 1 hour in a humidified chamber. Antibodies were diluted in PBS. After washing the sections, they were mounted in Citiflor AF1 solution (VWR, Radnor, PA).
Systemic Immune Response to Sheep IgG Analysis
Serum mouse anti-sheep IgG levels were measured by ELISA. ELISA plates (Nunc Maxisorb; Life Technologies, Paisley, UK) were coated overnight at 4°C with 100 μg/mL of sheep IgG (Sigma) diluted in 100 mmol/L boric acid. After a blocking step, serum was diluted 1:1000 in 1% bovine serum albumin. Alkaline phosphatase–conjugated goat anti-mouse IgG (Southern Biotechnology, Birmingham, AL) at 1:1000 in 1% bovine serum albumin was used as the secondary antibody. All incubations were for 1 hour at 37°C. Plates were developed using p-nitrophenyl phosphate (Sigma-Aldrich) and measured on an ELISA plate reader at 405 nm.
Induction of NTN
To induce accelerated NTN, mice were initially preimmunized with 0.2 mg of sheep IgG (Sigma-Aldrich) made up in 0.9% sterile saline, mixed in a ratio of 1:1 with complete Freund adjuvant (Sigma-Aldrich). Five days after the immunization, the mice were injected with 200 μL sheep nephrotoxic globulin diluted 1:1 in 0.9% NaCl, via the tail vein. Nephrotoxic globulin serum was generated as previously described.25 Mice were placed in metabolic cages for 24 hours 1 day before they were humanely sacrificed 7 to 9 days after nephrotoxic globulin injection. The kidneys were harvested, and urine and blood were collected. For the nonaccelerated model of NTN, mice were not preimmunized with sheep IgG. Instead, mice were injected with 200 μL of nephrotoxic serum diluted 1:1 with sterile saline (0.9%) into the tail vein and sacrificed 2 hours later.
Bone Marrow Transplantation
To obtain donor bone marrow from WT and Mrp14−/− mice, the femurs and tibias were dissected, followed by flushing through the bone with sterile PBS, and bone marrow cells were collected. The cells were resuspended in RPMI 1640 media, and 10 million cells were injected i.v. into the tail vein per mouse.
The female recipients of the bone marrow were irradiated in an IBL 637 irradiator (CIS Bio International, Saclay, France), with a dose of 8 Gy. Within an hour of receiving this dose of radiation, the mice were transplanted with donor bone marrow cells. After transplantation, the mice were kept for 8 weeks before undergoing NTN. Bone marrow reconstitution was confirmed by PCR analysis of circulating leukocytes.
T-Cell Proliferation Assay
After NTN and sacrifice of the animals, the spleens were harvested and single-cell suspensions were isolated, followed by red cell lysis. Splenocytes (1 × 105) were cultured in triplicate in HL-1 serum-free media (Lonza Group Ltd, Basel, Switzerland) alone, with CD28/CD3 beads (Invitrogen–Life Technologies, Carlsbad, CA) or with aggregated sheep IgG (10 and 50 μg/mL) for 72 hours. After the stimulation, 1 μCi of 3H thymidine was added to each well, followed by incubation for a further 24 hours at 37°C. Thymidine uptake was then counted after 24 hours using a 1450 MicroBeta TriLux liquid scintillation and luminescence counter (counts per minute; PerkinElmer, Waltham, MA).
Histological Analysis
Sections were blinded so the animal identity was unknown. To calculate a score for glomerular thrombosis, periodic acid–Schiff kidney sections were used to grade the amount of periodic acid–Schiff–positive material occupying the glomeruli above normal staining. The glomeruli were scored according to the number of quadrants affected, as follows: 0 indicates no thrombosis in any quadrant; 1 indicates one quadrant with thrombosis observed; 2 indicates two quadrants of the glomerulus affected; 3 indicates three of the quadrants affected; 4 indicates that all 4 quadrants were affected (maximum score, 4). Some sections were also stained for fibrin (Abcam, Cambridge, UK) to validate the thrombosis scoring. Glomerular macrophages and T cells identified on immunoperoxidase staining were also counted. Twenty-five glomeruli were counted, and the mean score was then calculated for each mouse. Immunofluorescence sections were scored using Image Pro Plus software (Media Cybernetics, Rockville, MD). To quantitate using Image Pro Plus, images of glomeruli were captured using a QImaging Retiga 2000R Fast 1396 camera (QImaging, Surrey, BC, Canada). For each section, 10 glomeruli were examined and the mean fluorescence intensity recorded. Where Image Pro Plus software was used, results are expressed in arbitrary fluorescence units.
Isolation of BMDMs
After sacrifice of the animals, the femurs and tibias were isolated in a sterile manner. The collected bone marrow was washed with Hanks media and spun at 524 × g for 5 minutes. The cells were resuspended in sterile culture medium of Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 20% fetal calf serum (FCS; Biosera, Boussens, France), 100 U/mL penicillin (Gibco Life Technologies, Grand Island, NY), 100 μg/mL streptomycin, and 25% L929 cell (European Collection of Cell Cultures, Salisbury, UK) conditioned media containing macrophage-specific colony-stimulating factor. The cells were cultured at 37°C, in 5% CO2. After 3 days, the media were replaced with the removal of the nonadherent cells. Fresh media were added. The macrophages were harvested at day 7, and used for stimulation experiments 24 hours later.
Isolation of Mesangial Cells
Kidneys were removed from WT and Mrp14−/− mice and placed in RPMI media (Invitrogen) with penicillin/streptomycin. The kidneys were blended, followed by sequential sieving using size 70-μm, then size 40-μm, sieves. Glomeruli were collected on the smaller sieve and spun down at 233 × g for 5 minutes, followed by a digestion step of collagenase (Sigma-Aldrich) for 30 minutes at 37°C. This was followed by a centrifuge step and resuspension in RPMI media, supplemented with glutamine, 20% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% insulin/selenium/transferrin growth supplement (Sigma-Aldrich), and 20 mmol/L HEPES (Invitrogen). The cells were cultured in tissue culture flasks and incubated at 37°C with 5% CO2. Media were replaced every 2 to 3 days. Mesangial cells were used between passage 6 and 12.
Isolation of WT Kidney ECs
Kidneys from WT mice were harvested and placed in DMEM on ice. The kidneys were blended using a syringe plunger, passed through a 70-μm sieve, and digested using 3 mg/mL collagenase (Sigma-Aldrich) in an agitated water bath at 37°C for 30 minutes. The digested cells were collected and washed twice in DMEM/0.5% FCS and resuspended in media before incubation with rat anti-mouse CD31 and rat anti-mouse CD105 (both from BD Pharmingen) at 4°C for 30 minutes. After two washes, cells were resuspended in 0.5% FCS/DMEM and goat anti-rat microbeads (Miltenyi Biotec, Cologne, Germany), and incubated for 15 minutes at 4°C. After a washing step, the cells were passed through the magnet and retained cells were collected and placed into a 25-cm flask, which had been precoated with 2% gelatin (Sigma-Aldrich). The cells were cultured in GlutaMAX DMEM (Gibco Life Technologies), 20% FCS, endothelial growth supplement (Sigma-Aldrich), 100 U/mL penicillin, and 100 μg/mL streptomycin. Media were changed every 3 days. When confluence was achieved, cells were split into different culture flasks. The cells were used for experiments at passage 8 to 12. The phenotype of the isolated cells was confirmed by positive staining with anti-CD31 immunofluorescence.
Co-Culture of ECs and Macrophages
Cultured ECs were plated into a 6-well plate (Corning; Nunc, Rochester, NY). Cells (320,000 cells per well) were plated out and left to grow in DMEM/10% FCS for 48 hours. The media were then aspirated. Bone marrow–derived macrophages (BMDMs) from WT and from Mrp14−/− mice were grown and were isolated on day 7. BMDMs (1 × 106) were added to the ECs in DMEM. Each group of cells was co-cultured in triplicate. The cells were incubated at 37°C with 5% CO2 for 24 hours. The supernatants were then collected and kept at −20°C before cytokine analysis by ELISA.
Co-Culture and Stimulation of Podocytes
Immortalized mouse podocytes (a gift from Dr. Peter Mundel, Mount Sinai School of Medicine, New York, NY) were cultured on tissue culture plastic coated with 1% Matrigel substrate. For ongoing proliferation, cells were cultured at 33°C in a 5% CO2 incubator in RPMI (Invitrogen) supplemented with 10% FCS (Invitrogen), antibiotics, and 10 U/mL interferon-γ. To induce differentiation, cells were thermoshifted to 37°C and cultured in RPMI in the absence of interferon-γ. Cells were allowed to differentiate under these conditions for 14 days. Cells (50,000 cells per well) were seeded and allowed to attach overnight before incubation for 24 hours with S100A8/A9. Supernatants were collected and used for cytokine analysis.
Mesangial Cell Proliferation Assay
Mesangial cells were used between passage 6 and 12. Proliferation was measured by bromodeoxyuridine cell proliferation assay (Millipore, Billerica, MA) and MTT assay (Life Technologies). Cells were plated at 1 × 104 cells per well of a 96-well flat-bottom plate (Corning: Appleton Woods, Birmingham, UK) in triplicate. After 24 hours of incubation, the cells were washed with warm Dulbecco's PBS (Life Technologies) and incubated in RPMI 1640 medium with l-glutamine, 1% penicillin, and 0.2% fetal bovine serum, with or without S100A8/9. Bromodeoxyuridine reagent was added after 4 hours of serum starvation and cells were incubated for a further 24 hours, when the assay was performed according to the manufacturer's instructions. The MTT assay was performed after 72 hours’ incubation, according to the manufacturer's instructions.
Supernatant ELISAs
Several cytokine ELISAs were used to analyze cytokine production. Supernatants were collected and kept at −20°C until use. The dilutions for each individual cytokine were optimized and varied accordingly. Monocyte chemotactic protein (MCP)-1 and IL-6 were measured using mouse chemokine ligand 2 (MCP-1) ELISA Ready-SET-Go and IL-6 ELISA Ready-SET-Go kit (eBiosciences, Insight Biotechnology Ltd, Wembley, UK), according to the manufacturer's instructions. TNF-α and CXCL1 were analyzed using Duoset ELISA Development System (R&D Systems, Abingdon, UK), according to the manufacturer's instructions.
Statistical Analysis
The animal study results are expressed as median (range). All statistics were performed using GraphPad Prism software version 4.0 (GraphPad Software, San Diego, CA). Nonparametric tests of significance were applied. For comparing two groups, U-test was used, as well as the paired t-test, whereas one-way analysis of variance was performed using nonparametric Kruskal-Wallis test and Dunn's multiple comparison post test for groups of three or more. Two-way analysis of variance was used to analyze differences between groups from more than one experiment. Correlations were assessed using the nonparametric Spearman rank correlation analysis. A significant value was defined as P < 0.05 with 95% confidence.
Results
S100A8/A9 Macrophage Expression and Serum Levels in NTN
We first investigated the infiltration of proinflammatory S100A8/A9-expressing macrophages and measured the serum levels of S100A8/A9 in WT mice after the induction of NTN (n = 9). The autologous model of NTN was used in which mice were preimmunized with sheep IgG, followed by injection of sheep nephrotoxic serum days later. Eight days after disease induction, mice were sacrificed. The mean total number of CD68-positive macrophages per glomerular cross section was counted for each animal and compared with the number of glomerular S100A8/A9-positive cells from the same animals. There was a mean of 3.53 (SD, 1.0) CD68-positive macrophages per glomerular section in WT animals with NTN, whereas there was a mean of 1.22 (SD, 0.6) S100A8/A9 cells per glomerular section (Figure 1). Histologically, the S100A8/9-positive cells were mononuclear and did not have the morphological features of neutrophils, demonstrating that S100A8/A9-positive monocytes/macrophages are recruited into the glomerulus during glomerulonephritis. In addition, serum levels of S100A8/A9 after NTN were significantly elevated, with a median S100A8/A9 level of 2949 ng/mL (range, 309 to 31,428 ng/mL), compared to a level of 3 ng/mL in normal control mice without NTN (range, 0 to 210 ng/mL) (P < 0.0005; Mann-Whitney U -test). Moreover, there were significant positive correlations between S100A8/A9 serum levels and disease outcome measures, such as serum urea (r = 0.74, P < 0.05, Spearman's rank) and glomerular thrombosis (r = 0.9, P < 0.005, Spearman's rank). Overall, this demonstrates there is a subset of S100A8/A9-positive macrophages within inflamed glomeruli, and mice with the most severe disease have the highest serum S100A8/A9 levels, replicating the link between serum S100A8/A9 levels and disease severity found in anti-neutrophil cytoplasmic antibody–associated vasculitis patients.
Figure 1.
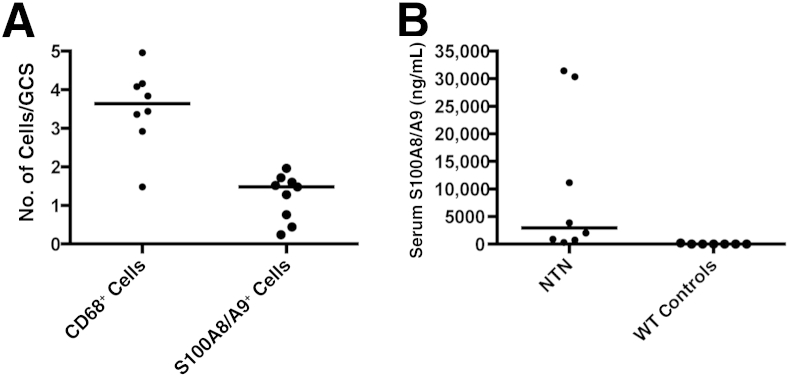
A: Glomerular S100A8/A9 and macrophage CD68 expression in the accelerated nephrotoxic nephritis model in wild-type (WT) mice (n = 9). Number of cells per glomerular cross section was counted (for 25 glomeruli), mean of 3.53 (SD, 1.0) CD68-positive macrophages per glomerular section in WT animals, whereas there was a mean of 1.22 (SD, 0.6) S100A8/A9 cells per glomerular section. B: Graph shows the serum levels of S100A8/A9 in WT mice after nephrotoxic nephritis and in WT controls in which disease has not been induced. The serum level of S100A8/A9 is significantly higher in mice with nephrotoxic nephritis (P < 0.0005). GCS, glomerular cross section; NTN, nephrotoxic nephritis.
Mrp14−/− Mice Demonstrate Significantly Less Disease than WT Mice during NTN
To elucidate whether mice deficient in MRP14 (and therefore deficient in the S100A8/A9 heterodimer) are protected from significant renal inflammation, we induced NTN (autologous model) in WT and Mrp14−/− mice. Three independent experiments were performed and parameters (renal function, glomerular thrombosis, macrophage infiltration, and proteinuria) combined after analysis by two-way analysis of variance. These experiments demonstrated significant differences between the WT (n = 26) and Mrp14−/− (n = 24) mice after NTN induction, with differences in glomerular thrombosis score [WT median, 0.44 (range, 0 to 4); Mrp14−/− median, 0 (range, 0 to 3.28)] (P < 0.005), proteinuria [WT median, 1.532 mg/day (range, 0 to 29.49 mg per day); MRP14−/− median, 0.48 mg/day (range, 0 to 9.48 mg per day)] (P < 0.05), and renal function assessed by serum urea levels [WT median serum urea, 11.55 mmol/L (range, 6.6 to 139 mmol/L); Mrp14−/− median serum urea, 7.55 mmol/L (range, 3.2 to 69 mmol/L)] (P < 0.001) (Figure 2). In addition, fibrin deposition reflected the glomerular thrombosis scores with significantly more fibrin deposition (quadrants affected per glomeruli) in the WT mice compared to Mrp14−/− mice [WT median, 1.6 (range, 0 to 3.2); Mrp14−/−, 0.32 (range, 0 to 0.68)] (P < 0.05).
Figure 2.

A: Thrombosis score of the wild-type (WT) and Mrp14−/− mice in the three experiments, with significantly less thrombosis in the Mrp14−/− mice. B: Combined proteinuria results from the three experiments. C: Serum urea levels in the two animal groups: WT mice have significantly higher urea than Mrp14−/− mice. D: The number of CD68-positive cells per glomerular cross section in WT and Mrp14−/− animals. Medians are shown. E: Photomicrograph shows CD68 macrophage immunohistochemistry in Mrp14−/− mice, with few macrophages infiltrating the glomerulus. F: Photomicrograph shows the CD68 macrophage immunohistochemistry in WT mouse with severe disease. G: Infiltration of S100A8/A9 cells into the glomerulus of WT mice. H: Photomicrographs of glomeruli (periodic acid–Schiff stain) to demonstrate mouse glomerulus without nephrotoxic serum administration. I:Mrp14−/− mouse after nephrotoxic nephritis demonstrating a normal glomerulus. J: WT mouse demonstrating a thrombosed glomerulus and severe disease. ∗P < 0.05, ∗∗∗P < 0.001. Original magnification, ×400 (G). GCS, glomerular cross section.
Assessing immune responsiveness using immunohistochemistry, WT mice demonstrated a significantly greater degree of macrophage infiltration compared with Mrp14−/− animals in all of the experiments, but there were differences in absolute numbers between experiments. Overall, the difference was highly significant, WT median number of macrophages/glomerular cross section was 1.72 (range, 0 to 4.96), compared with Mrp14−/− median, 0.6 (range, 0 to 2.64) (P < 0.001) (Figure 2). By contrast, there was no significant difference in CD4+ T-cell infiltration between WT and Mrp14−/− mice [median number of glomerular CD4+ T cells/glomerular cross section WT, 0.12 (range, 0 to 0.21) versus Mrp14−/−, 0.12 (0 to 0.33)] (not significant). T-cell proliferation was compared between WT and Mrp14−/− mice after NTN. Background splenocyte proliferation (media alone) was subtracted from proliferation in response to anti-CD3/CD28 or sheep IgG. There was no significant difference between the two animal groups in response to anti-CD3/CD28, low-dose sheep IgG (10 μg/mL), and high-dose sheep IgG (50 μg/mL) (data not shown).
Deposited sheep and mouse IgGs were assessed by direct immunofluorescence in all of the experiments, and quantified using image analysis software. Levels were similar in WT and Mrp14−/− mice [median mouse IgG deposition: WT, 96.18 arbitrary units (AUs) (range, 65.35 to 140.2 AU) versus Mrp14−/−, 123.1 AU (range, 66.26 to 165 AU) (not significant); glomerular sheep IgG deposition: WT, 25.96 AU (range, 21.69 to 46.53 AU) versus Mrp14−/−, 38.76 AU (range, 22.14 to 66.44 AU) (not significant)]. Finally, total mouse anti-sheep IgG was measured by ELISA and demonstrated no difference in the IgG response between the two animal groups in the independent NTN experiments (data not shown). This particular nephrotoxic serum resulted in a thrombotic glomerulonephritis rather than a predominantly crescentic phenotype, so glomerular lesions were not scored for crescents. Overall, mice deficient in Mrp14−/− demonstrated significantly less renal injury than WT mice, with preserved renal function, less proteinuria, reduced macrophage infiltration, and attenuated glomerular thrombosis, but with no difference demonstrated in humoral or T-cell responses.
Disease Protection in Mrp14−/− Mice Is Overcome by TLR4 Stimulation Using LPS
Because S100A8/A9 mediates some of its proinflammatory effects through TLR4, we examined if the administration of another TLR4 agonist (LPS) to Mrp14−/−-deficient mice would overcome the deficiency of S100A8/A9 and result in similar renal disease to WT animals. The autologous NTN model was repeated in WT and Mrp14−/− mice, but 3 days after the administration of nephrotoxic serum, each mouse in both groups received 1 μg LPS i.p. Mrp14−/− mice administered LPS developed similar renal damage as WT mice administered LPS, with equivalent glomerular thrombosis scores, proteinuria, renal dysfunction, and glomerular CD68 macrophage infiltration (data not shown). These data demonstrate that MRP14 deficiency and, hence, deficient endogenous TLR stimulation are protective but can be overcome by excess exogenous TLR4 stimulation.
Disease Protection in Mrp14−/− Mice Is Not Due to Defective Neutrophil Recruitment
To assess the early glomerular neutrophil influx in response to nephrotoxic serum and to investigate whether Mrp14−/− failed to recruit glomerular neutrophils, which mediate this early glomerular damage, we performed the heterologous nonaccelerated NTN model in Mrp14−/− and WT mice. The first heterologous phase occurs after injection of the nephrotoxic serum when the sheep antibody is planted on the glomerular basement membrane, resulting in neutrophil infiltration and mild proteinuria, and occurs within the first few hours.26
We examined neutrophil infiltration at 2 hours in WT and Mrp14−/− mice (n = 9 for each), assessed by immunohistochemistry using the granulocyte marker GR-1. No difference in the early neutrophil influx between WT and Mrp14−/− mice was found [median neutrophil number at 2 hours in the WT mice, 0.6 (range, 0.44 to 2) and in Mrp14−/−, 0.71 (range, 0.56 to 1.2); P > 0.05]. This demonstrates that disease protection in Mrp14−/− mice during NTN was not due to diminished glomerular neutrophil chemotaxis.
MRP14 Deficiency in Both Circulating and Intrinsic Cells Is Required for Disease Protection
To understand whether MRP14-deficient circulating cells or resident renal cells are required for disease protection, we performed bone marrow transplants in which Mrp14−/− bone marrow was transplanted into irradiated WT mice, and WT bone marrow into Mrp14−/− mice (n = 9 for each). Controls had WT bone marrow transplanted into WT recipients (n = 8). PCR demonstrated successful reconstitution with donor bone marrow (>95%). Eight weeks after the bone marrow transplantation, NTN was performed. There was no difference between the three groups with regard to disease severity assessed by serum urea [median serum urea in the WT to WT group, 23 mmol/L (range, 4.6 to 53.2 mmol/L); in the Mrp14−/− to WT group, 37.5 mmol/L (range, 6.1 to 210 mmol/L); and in the WT to Mrp14−/− group, 38.9 mmol/L (range, 7.1 to 234 mmol/L)], glomerular thrombosis [median glomerular thrombosis score in the WT to WT group, 1.72 (range, 0 to 3.26); in the Mrp14−/− to WT group, 1.58 (range, 0 to 3.52); and in the WT to Mrp14−/− group, 2.08 (range, 0.3 to 64)], or proteinuria [median 24-hour proteinuria (mg/24 hours) in the WT to WT group, 9.83 mg (range, 2.7 to 16.86 mg); in the Mrp14−/− to WT group, 8.95 mg (range, 0.45 to 32.8 mg); and in the WT to Mrp14−/− group, 2.73 (range, 0.58 to 31.68)] (Figure 3). These data demonstrate that deficiency of MRP14 and, therefore, MRP8/14 (S100A8/9) was necessary in both circulating hemopoietic cells and radioresistant intrinsic cells for disease protection, suggesting that intrinsic cells would be capable of producing and responding to S100A8/9.
Figure 3.

The results of nephrotoxic nephritis in bone marrow chimeras show no difference in glomerular thrombosis (A), proteinuria (mg/24 hours; B), and serum urea levels (mmol/L; C) between the three animal groups [wild type (WT) to WT, Mrp14−/− to WT, and WT to Mrp14−/−].
Mrp14−/− BMDMs and Renal Mesangial Cells Have Diminished Responses to the Proinflammatory Effects of S100A8/A9
We investigated whether S100A8/A9 mediates similar proinflammatory actions in WT and Mrp14−/− BMDMs and intrinsic renal mesangial cells, known to play a critical role during experimental glomerular inflammation, by assessing CXCL1, TNF-α, IL-6, and MCP-1 cytokine secretion. Mesangial cells were characterized by their morphological features and positive staining for myosin and negative staining for pancytokeratin using immunofluorescence (data not shown). On stimulation with S100A8/A9, the Mrp14−/− cells produced significantly less cytokines than WT cells. For BMDMs, the median increase compared to unstimulated cells of CXCL1 in WT cells was 3749 pg/mL (range, 3199 to 4482 pg/mL) and in Mrp14−/−, 659 pg/mL (range, 209 to 792 pg/mL) (P < 0.005); median increase in TNF-α in WT BMDMs, 272 pg/mL (range, 202.6 to 309.9 pg/mL) and in Mrp14−/− BMDMs, 33.4 pg/mL (range, 29.2 to 66.7 pg/mL) (P < 0.05); and median increase in IL-6 in WT BMDMs, 96.5 pg/mL (range, 58 to 106.4 pg/mL) and in Mrp14−/− BMDMs, 12.2 pg/mL (range, 11.5 to 16.2 pg/mL) (P < 0.05) (Figure 4). For mesangial cells, the median increase after S100A8/9 stimulation of CXCL1 in WT cells was 267.8 pg/mL (range, 230.5 to 288.1 pg/mL) and in Mrp14−/− cells, 81.28 pg/mL (range, 67.62 to 87.87 pg/mL); median increase in MCP-1 in WT mesangial cells was 403.7 pg/mL (range, 311.2 to 491.5 pg/mL) and in Mrp14−/−cells, 103 pg/mL (range, 99.4 to 103.9 pg/mL) (all P < 0.005). However, Mrp14−/− mesangial cells produced a greater amount of IL-6 than WT mesangial cells after S100A8/9 stimulation [median increase in IL-6 in WT cells, 0 pg/mL (range, 0 to 2.59 pg/mL) and in Mrp14−/− cells, 6.76 pg/mL (range, 6.7 to 14.29 pg/mL)] (P < 0.05). Figure 5 demonstrates the cytokine production by mesangial cells. Finally, we tested the effect of S100A8/A9 stimulation on mesangial cell proliferation assessed using bromodeoxyuridine incorporation (media alone mean ± SD, 0.538 AU ± 0.01; S100A8/9 mean, 0.545 AU ± 0.006; not significant) and MTT assays (media alone mean, 0.197 AU ± 0.01; S100A8/9 mean, 0.216 AU ± 0.007; not significant), and found no evidence of cell proliferation after exposure to S100A8/9.
Figure 4.

Bone marrow–derived macrophage (BMDM) cytokine production in wild-type (WT) and Mrp14−/− cells, after stimulation with exogenous S100A8/A9: CXCL1 (A), tumor necrosis factor (TNF)-α (B), and IL-6 (C) secretion. There is significantly greater cytokine production in WT BMDMs when stimulated with S100A8/A9 (one-way analysis of variance). Mrp14−/− cells stimulated with S100A8/A9 do not produce a significant increase in cytokine production. Median and range are shown. ∗P < 0.05.
Figure 5.

The increase in wild-type (WT) and Mrp14−/− mesangial cell cytokine production when stimulated with S100A8/A9. Mrp14−/− cells produce significantly less CXCL1 than WT cells (A), less monocyte chemotactic protein (MCP-1; B), but a greater amount of IL-6 than WT cells (C). Median and range are shown. ∗P < 0.05, ∗∗∗P < 0.005.
Therefore, BMDMs and mesangial cells deficient in Mrp14−/− demonstrate decreased inflammatory responses to S100A8/A9. These data suggest that there is a critical requirement for endogenous S100A8/A9 production, to enable cytokine production in response to S100A8/A9 stimulation.
Co-Culture of ECs with BMDM Stimulates Inflammatory Cytokine Production, Which Is Diminished with Mrp14−/− BMDMs Compared to WT Cells
The interaction between the endothelium and leukocytes is critical in the pathogenesis of vasculitis and glomerulonephritis. We investigated the proinflammatory interaction when BMDMs are cultured with renal microvascular ECs and compared the effect of WT and Mrp14−/− BMDMs.
After isolation of renal microvascular ECs from the kidneys of WT mice, the phenotype of the renal ECs was confirmed by positive staining for CD31 by immunofluorescence (data not shown). The following cell conditions were cultured: i) ECs alone, ii) WT BMDMs alone, iii) Mrp14−/− BMDMs alone, iv) ECs co-cultured with WT BMDMs, and v) ECs co-cultured with Mrp14−/− BMDMs. The BMDMs cultured alone produced small quantities of cytokines. Median MCP-1 WT BMDM was 0.843 μg/mL and Mrp14−/− BMDM, 0.942 μg/mL (not significant); median CXCL1 WT BMDM was 41.88 pg/mL and Mrp14−/− BMDM, 63.33 pg/mL (not significant); and median IL-6 WT BMDM was 56.7 pg/mL and Mrp14−/− BMDM, 113.3 pg/mL (P < 0.05). ECs alone produced greater amounts of MCP-1 compared to the two BMDM groups (median MCP-1, 5.34 μg/mL) and greater amounts of CXCL1 (median, 237.2 pg/mL) and less IL-6 (median, 28.7 pg/mL).
However, the greatest amount of cytokine production was after ECs co-culture with WT BMDMs, resulting in significantly greater quantities of MCP-1, CXCL1, and IL-6 compared with ECs co-cultured with Mrp14−/− BMDMs, which were similar to levels found in ECs alone. ECs co-cultured with BMDMs resulted in MCP-1 levels with WT BMDM median, 29.99 μg/mL, and with Mrp14−/− BMDM median, 11.85 μg/mL; CXCL1: WT BMDM median, 1000 pg/mL, and Mrp14−/− BMDM median, 445 pg/mL; and IL-6: WT BMDM median, 870.8 pg/mL, and Mrp14−/− BMDM median, 181.8 pg/mL (all P < 0.01) (Figure 6). Similar data were found after the addition of exogenous S100A8/A9 to ECs, which resulted in augmented secretion of CXCL1 and MCP-1 (data not shown).
Figure 6.

Cytokine production in endothelial cell (EC)–bone marrow–derived macrophage (BMDM) co-culture. CXCL1 (A), monocyte chemotactic protein (MCP-1; B), and IL-6 (C) production during EC co-culture with wild-type (WT) or Mrp14−/− BMDMs. In all cases, cytokine production is significantly greater in EC-WT BMDM co-culture compared with EC-MRP14−/− co-culture. Median and range are shown. ∗∗P < 0.01.
The co-culture experiments illustrate the decreased inflammatory profile of BMDMs lacking S100A8/A9 when interacting with renal microvascular ECs, confirming the critical requirement for S100A8/A9 expression on infiltrating monocytes/macrophages to mediate EC inflammation.
Stimulation of Podocytes with S100A8/A9 Has a Minimal Proinflammatory Effect
Because proteinuria is a feature of experimental glomerulonephritis, we investigated the effect of S100A8/A9 on podocytes. An immortalized podocyte cell line was stimulated with the following: i) LPS (positive control), ii) S100A8/A9, and iii) combined LPS and S100A8/A9. There was no TNF-α detected in any of the stimulation conditions. For MCP-1 secretion, unstimulated cells produced a median of 2002 pg/mL (range, 1988 to 2101 pg/mL), after LPS, 7798 pg/mL (range, 7558 to 7915 pg/mL) (P < 0.05), and after S100A8/A9, 2633 pg/mL (range, 2423 to 2818 pg/mL) (data not significant). There was no synergistic effect of LPS and S100A8/A9, which produced a median of 7150 pg/mL (range, 6710 to 7172 pg/mL). Similar patterns were obtained for IL-6 secretion. Unstimulated podocytes produced no detectable IL-6. LPS stimulation resulted in a median of 44.34 pg/mL (range, 38.72 to 45.34 pg/mL), with S100A8/A9 stimulation resulting in almost no detectable IL-6 [median, 0 (range, 0 to 21.1)]. There was a slight synergistic effect of LPS and S100A8/A9, with a median of 54.61 (range, 47.35 to 54.81). The only statistically significant increase in IL-6 was after the combination of LPS and S100A8/A9 (P < 0.05) (one-way analysis of variance) (Figure 7).
Figure 7.
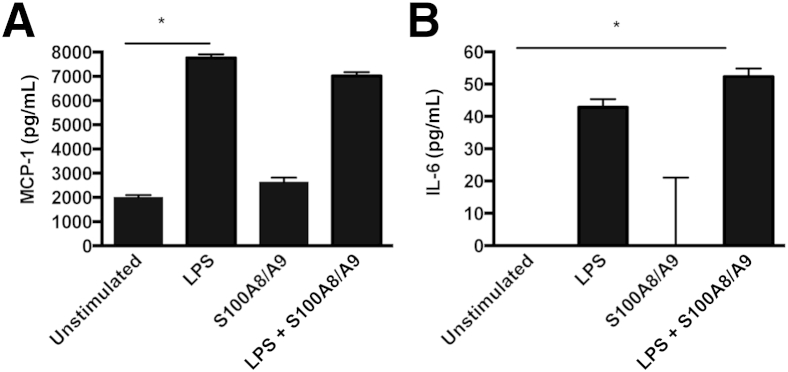
Cytokine production by podocytes. Graph shows monocyte chemotactic protein 1 (MCP-1; A) and IL-6 (B) production after stimulation with lipopolysaccharide (LPS) or S100A8/9. S100A8/A9 alone has little effect on podocyte MCP-1 or IL-6 production, whereas the combination of S100A8/A9 and LPS has a similar effect to LPS alone, increasing MCP-1 and IL-6 production compared to unstimulated cells (one-way analysis of variance). There is a nonsignificant increase in IL-6 after combined treatment, compared to LPS alone. ∗P < 0.05.
Discussion
The results from our experimental model demonstrate that circulating S100A8/A9 serum levels correlate with disease severity, mirroring our previous findings in patients with anti-neutrophil cytoplasmic antibody–associated vasculitis,8 but additionally that S100A8/A9 deficiency in both circulating hemopoietic and intrinsic renal cells protects from development of glomerulonephritis. We investigated the underlying basis for this and found that, although early neutrophil influx and humoral and T-cell immunity were unaffected in Mrp14−/− mice, there was a marked reduction in glomerular macrophage influx. This is consistent with the finding that S100A8/A9 has a role in monocyte adhesion and transmigration, mediated by CD11b expression and activation, as well as an increase in monocyte surface intercellular adhesion molecule-1,27 implying that deficiency in S100A8/A9 could lead to decreased macrophage ability to infiltrate the glomerulus. However, it is perhaps surprising that we found no significant change in neutrophil influx as Mrp14−/−-deficient mice demonstrate both decreased macrophage and neutrophil infiltration, in a mechanical arterial injury model.28
A previous study3 showed no difference in the gene expression pattern of unstimulated WT and Mrp14−/− bone marrow cells. However, crucial differences in several genes after LPS stimulation have been demonstrated with Mrp14−/− phagocytes, demonstrating a reduced inflammatory response.3 Consistent with these data, our results demonstrate a decreased responsiveness to S100A8/A9 in Mrp14−/− BMDMs and renal mesangial cells compared to WT cells. These novel, hitherto unreported, findings suggest that S100A9 synthesis is required for complete cellular responsiveness to the heterodimer S100A8/A9.
After glomerular infiltration, the monocyte/macrophage S100A8/A9 complex appears to exert additional autocrine and paracrine effects to further amplify the inflammatory response, which results in endothelial and mesangial cell activation. These data are consistent with other published data, demonstrating the critical interaction between these S100 proteins and TLR4, which results in activation of a signal transduction cascade and the transcription of several important proinflammatory cytokines, including CXCL1 and TNF-α, an effect abrogated in TLR4−/− cells.10
Interestingly, endogenous TLR4 ligation by S100A8/A9 appears to be required for glomerulonephritis to develop, but this can be overcome by sufficient levels of exogenous TLR4 ligands. Our findings in glomerulonephritis recapitulate the critical role S100A8 and S100A9 have in mediating inflammatory arthritis, which is also related to macrophage-mediated damage.9,29 In addition, a positive feedback loop of cytokine-mediated S100A8 and S100A9 secretion, which, in turn, leads to S100A8-mediated secretion of proinflammatory cytokines, such as IL-6, was demonstrated in chondrocytes. These proinflammatory effects occurred despite low expression of TLR4, and in the presence of an anti-receptor for advanced glycation end products antibody, implicating another unknown receptor(s) in mediating proinflammatory effects of S100A8, at least in the cells tested.30 Although the WT mice develop proteinuria during NTN, the exogenous effect of S100A8/A9 on podocytes, which are known to express TLR4, results in minimal cytokine production. Our co-culture experiments demonstrate a significant proinflammatory role for S100A8/A9-positive macrophages cultured with ECs, consistent with the notion that S100A8/A9 amplifies a proinflammatory response through effects on macrophages and ECs. In addition, stimulation of mesangial cells with S100A8/A9 resulted in the production of proinflammatory cytokines, including MCP-1 and CXCL1, involved in macrophage and neutrophil recruitment to the glomerulus, but this was blunted in the MRP14-deficient cells, with the exception of IL-6 secretion. The bone marrow transplant experiments demonstrate that hemopoietic cell Mrp14−/− deficiency is not sufficient to prevent renal disease, but rather complete deficiency of circulating and intrinsic cells is required for disease protection. These data suggest that the presence of radioresistant bone marrow–derived cells or intrinsic renal cell production of S100A8/A9 is likely to contribute to disease. These observations are reminiscent of the data from bone marrow chimeras used to investigate development of atherosclerosis.31 Bone marrow–specific Mrp14−/− deficiency in an atherosclerosis mouse model was not sufficient to reduce inflammation, insulin resistance, or atherosclerosis, despite finding a reduction in atherosclerosis in mice with total MRP14 deficiency.28 Other nonmyeloid cell types have experimentally been induced to produce S100A8 and/or S100A9, including the murine kidney collecting duct32 and microvascular ECs.33
In conclusion, we have demonstrated a crucial role for S100A8/A9 in glomerular inflammation exerting a proinflammatory effect on macrophages, and intrinsic renal endothelial and mesangial cells, identifying it as a potential target for immunotherapy.
Acknowledgments
We thank Prof. Fredrik Ivars (University of Lund, Lund, Sweden) for kindly providing the Mrp14−/− mice, Dr. Nancy Hogg (Cancer Research UK London Research Institute, London, UK) for providing the rat anti-mouse S100A8/A9 antibody, and Dr. Peter Mundel (Mount Sinai School of Medicine, New York, NY) for providing the immortalized mouse podocytes.
Footnotes
Supported by Medical Research Council grant G0802260/2 for a Clinical Research Training Fellowship (R.J.P.) and Kidney Research UK.
Disclosures: None declared.
References
- 1.Lagasse E., Clerc R.G. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol. 1988;8:2402–2410. doi: 10.1128/mcb.8.6.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Odink K., Cerletti N., Bruggen J., Clerc R.G., Tarcsay L., Zwadlo G., Gerhards G., Schlegel R., Sorg C. Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature. 1987;330:80–82. doi: 10.1038/330080a0. [DOI] [PubMed] [Google Scholar]
- 3.Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M.A., Nacken W., Foell D., van der Poll T., Sorg C., Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 4.Hermani A., De Servi B., Medunjanin S., Tessier P.A., Mayer D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res. 2006;312:184–197. doi: 10.1016/j.yexcr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 5.Bjork P., Bjork A., Vogl T., Stenstrom M., Liberg D., Olsson A., Roth J., Ivars F., Leanderson T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kido J., Hayashi N., Kataoka M., Nagata T. Calprotectin expression in human monocytes: induction by porphyromonas gingivalis lipopolysaccharide, tumor necrosis factor-alpha, and interleukin-1beta. J Periodontol. 2005;76:437–442. doi: 10.1902/jop.2005.76.3.437. [DOI] [PubMed] [Google Scholar]
- 7.Zwadlo G., Schlegel R., Sorg C. A monoclonal antibody to a subset of human monocytes found only in the peripheral blood and inflammatory tissues. J Immunol. 1986;137:512–518. [PubMed] [Google Scholar]
- 8.Pepper R.J., Hamour S., Chavele K.M., Todd S.K., Rasmussen N., Flint S., Lyons P.A., Smith K.G., Pusey C.D., Cook H.T., Salama A.D. Leukocyte and serum S100A8/S100A9 expression reflects disease activity in ANCA-associated vasculitis and glomerulonephritis. Kidney Int. 2013;83:1150–1158. doi: 10.1038/ki.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Lent P.L., Grevers L., Blom A.B., Sloetjes A., Mort J.S., Vogl T., Nacken W., van den Berg W.B., Roth J. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann Rheum Dis. 2008;67:1750–1758. doi: 10.1136/ard.2007.077800. [DOI] [PubMed] [Google Scholar]
- 10.van Lent P.L., Grevers L.C., Schelbergen R., Blom A., Geurts J., Sloetjes A., Vogl T., Roth J., van den Berg W.B. S100A8 causes a shift toward expression of activatory Fcgamma receptors on macrophages via toll-like receptor 4 and regulates Fcgamma receptor expression in synovium during chronic experimental arthritis. Arthritis Rheum. 2010;62:3353–3364. doi: 10.1002/art.27654. [DOI] [PubMed] [Google Scholar]
- 11.Sunahori K., Yamamura M., Yamana J., Takasugi K., Kawashima M., Yamamoto H., Chazin W.J., Nakatani Y., Yui S., Makino H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R69. doi: 10.1186/ar1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loser K., Vogl T., Voskort M., Lueken A., Kupas V., Nacken W., Klenner L., Kuhn A., Foell D., Sorokin L., Luger T.A., Roth J., Beissert S. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med. 2010;16:713–717. doi: 10.1038/nm.2150. [DOI] [PubMed] [Google Scholar]
- 13.Robinson M.J., Tessier P., Poulsom R., Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. 2002;277:3658–3665. doi: 10.1074/jbc.M102950200. [DOI] [PubMed] [Google Scholar]
- 14.Srikrishna G., Panneerselvam K., Westphal V., Abraham V., Varki A., Freeze H.H. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J Immunol. 2001;166:4678–4688. doi: 10.4049/jimmunol.166.7.4678. [DOI] [PubMed] [Google Scholar]
- 15.Viemann D., Strey A., Janning A., Jurk K., Klimmek K., Vogl T., Hirono K., Ichida F., Foell D., Kehrel B., Gerke V., Sorg C., Roth J. Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood. 2005;105:2955–2962. doi: 10.1182/blood-2004-07-2520. [DOI] [PubMed] [Google Scholar]
- 16.Viemann D., Barczyk K., Vogl T., Fischer U., Sunderkotter C., Schulze-Osthoff K., Roth J. MRP8/MRP14 impairs endothelial integrity and induces a caspase-dependent and -independent cell death program. Blood. 2007;109:2453–2460. doi: 10.1182/blood-2006-08-040444. [DOI] [PubMed] [Google Scholar]
- 17.Huang X.R., Tipping P.G., Apostolopoulos J., Oettinger C., D'Souza M., Milton G., Holdsworth S.R. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin Exp Immunol. 1997;109:134–142. doi: 10.1046/j.1365-2249.1997.4091307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffield J.S., Tipping P.G., Kipari T., Cailhier J.F., Clay S., Lang R., Bonventre J.V., Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Wang Y.P., Zheng G., Lee V.W., Ouyang L., Chang D.H., Mahajan D., Coombs J., Wang Y.M., Alexander S.I., Harris D.C. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72:290–299. doi: 10.1038/sj.ki.5002275. [DOI] [PubMed] [Google Scholar]
- 20.Cao Q., Wang Y., Zheng D., Sun Y., Lee V.W., Zheng G., Tan T.K., Ince J., Alexander S.I., Harris D.C. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933–942. doi: 10.1681/ASN.2009060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giorgini A., Brown H.J., Sacks S.H., Robson M.G. Toll-like receptor 4 stimulation triggers crescentic glomerulonephritis by multiple mechanisms including a direct effect on renal cells. Am J Pathol. 2010;177:644–653. doi: 10.2353/ajpath.2010.091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Summers S.A., van der Veen B.S., O'Sullivan K.M., Gan P.Y., Ooi J.D., Heeringa P., Satchell S.C., Mathieson P.W., Saleem M.A., Visvanathan K., Holdsworth S.R., Kitching A.R. Intrinsic renal cell and leukocyte-derived TLR4 aggravate experimental anti-MPO glomerulonephritis. Kidney Int. 2010;78:1263–1274. doi: 10.1038/ki.2010.327. [DOI] [PubMed] [Google Scholar]
- 23.Hobbs J.A., May R., Tanousis K., McNeill E., Mathies M., Gebhardt C., Henderson R., Robinson M.J., Hogg N. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23:2564–2576. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manitz M.P., Horst B., Seeliger S., Strey A., Skryabin B.V., Gunzer M., Frings W., Schonlau F., Roth J., Sorg C., Nacken W. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23:1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robson M.G., Cook H.T., Botto M., Taylor P.R., Busso N., Salvi R., Pusey C.D., Walport M.J., Davies K.A. Accelerated nephrotoxic nephritis is exacerbated in C1q-deficient mice. J Immunol. 2001;166:6820–6828. doi: 10.4049/jimmunol.166.11.6820. [DOI] [PubMed] [Google Scholar]
- 26.Cochrane C.G., Unanue E.R., Dixon F.J. A role of polymorphonuclear leukocytes and complement in nephrotoxic nephritis. J Exp Med. 1965;122:99–116. doi: 10.1084/jem.122.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eue I., Pietz B., Storck J., Klempt M., Sorg C. Transendothelial migration of 27E10+ human monocytes. Int Immunol. 2000;12:1593–1604. doi: 10.1093/intimm/12.11.1593. [DOI] [PubMed] [Google Scholar]
- 28.Croce K., Gao H., Wang Y., Mooroka T., Sakuma M., Shi C., Sukhova G.K., Packard R.R., Hogg N., Libby P., Simon D.I. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–436. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cesaro A., Anceriz N., Plante A., Page N., Tardif M.R., Tessier P.A. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLoS One. 2012;7:e45478. doi: 10.1371/journal.pone.0045478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Lent P.L., Grevers L.C., Blom A.B., Arntz O.J., van de Loo F.A., van der Kraan P., Abdollahi-Roodsaz S., Srikrishna G., Freeze H., Sloetjes A., Nacken W., Vogl T., Roth J., van den Berg W.B. Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum. 2008;58:3776–3787. doi: 10.1002/art.24074. [DOI] [PubMed] [Google Scholar]
- 31.Averill M.M., Barnhart S., Becker L., Li X., Heinecke J.W., Leboeuf R.C., Hamerman J.A., Sorg C., Kerkhoff C., Bornfeldt K.E. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: implications for atherosclerosis and adipose tissue inflammation. Circulation. 2011;123:1216–1226. doi: 10.1161/CIRCULATIONAHA.110.985523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujiu K., Manabe I., Nagai R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J Clin Invest. 2011;121:3425–3441. doi: 10.1172/JCI57582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yen T., Harrison C.A., Devery J.M., Leong S., Iismaa S.E., Yoshimura T., Geczy C.L. Induction of the S100 chemotactic protein, CP-10, in murine microvascular endothelial cells by proinflammatory stimuli. Blood. 1997;90:4812–4821. [PubMed] [Google Scholar]