

Abstract
Objective
To investigate the role of Toll-like receptor 2 (TLR2), TLR4, TLR9 and myeloid differentiation factor 88 (MyD88) on alveolar macrophages in ventilator-induced lung injury (VILI).
Methods
Male, adult pathogen-free Sprague-Dawley rats weighing 300-350 g were used in this study. Animals were tracheotomized and allowed to breathe spontaneously for 4 h or mechanically ventilated for 4 h with low or high tidal volume (7 or 40 mL/kg). TLR2, TLR4, and TLR9, MyD-88 and NF-κΒ of alveolar macrophages’ expression under the different ventilation conditions were detected. Pulmonary permeability, lung inflammatory, IL-6 and IL-1β were assessed as well.
Results
Rats subjected to high tidal volume showed significantly greater pulmonary permeability and lung inflammatory than the control rats. Alveolar macrophages from rats subjected to high tidal volume also showed significantly higher protein expression of TLR2 (0.59±0.049 vs. 0.35±0.036 and 0.36±0.031, both P<0.001), TLR4 (0.845±0.0395 vs. 0.401±0.026 and 0.403±0.020, both P<0.001), TLR9 (0.727±0.074 vs. 0.383±0.039 and 0.367±0.043, both P<0.001), MyD-88 (1.01±0.060 vs. 0.485±0.045 and 0.507±0.046, both P<0.001) and NF-κΒ (0.776±0.067 vs. 0.448±0.043 and 0.481±0.047, both P<0.001), as well as significantly higher concentrations of IL-6 (7.32±0.24 vs. 2.42±0.13 and 2.44±0.32, both P<0.001) and IL-1β (139.95±9.37 vs. 53.63±5.26 and 53.55±6.63, both P<0.001) than the control and low tidal volume group.
Conclusions
The overexpression of TLR2, TLR4, and TLR9 on alveolar macrophages and release of pro-inflammatory cytokines play a role in VILI.
Keywords: Toll like receptor (TLR), myeloid differentiation factor 88 (MyD88), alveolar macrophages, ventilator-induced lung injury (VILI)
Background
Mechanical ventilation facilitates surgical interventions during general anesthesia and is a life-saving therapy in patients with acute lung injury or with the most severe form of this injury, acute respiratory distress syndrome. At the same time, mechanical ventilation can exacerbate lung injury; the resulting damage has been termed ventilator-induced lung injury (VILI) (1). This type of acute lung injury is characterized by inflammation associated with robust release of proinflammatory mediators and activation of inflammatory signaling pathways (2).
Various inflammatory cytokines are released into the pulmonary distal airspaces during acute lung injury; among these, IL-1β initiates and amplifies inflammation (2,3). This cytokine has also recently been implicated in the pathogenesis of VILI, in which it is a major contributor to alveolar barrier dysfunction (4). In fact, clinical studies have identified IL-1β as one of the best markers of ventilator-induced lung inflammation (5).
Alveolar macrophages account for 5% of peripheral lung cells and reside in the alveolar space, where they account for 90% of all leukocytes, with the remainder being mainly dendritic cells and T cells (6). Alveolar macrophages play a crucial role in the maintenance of immunological homeostasis and host defense, and they are the primary producers of proinflammatory cytokines in lungs following exposure to noxious stimuli (2,7,8). These macrophages are rapidly activated by mechanical ventilation, suggesting that they may play a critical role in the pathogenesis of VILI. Indeed, removing alveolar macrophages from rats exposed to mechanical ventilation at high tidal volume attenuates the resulting alveolar barrier dysfunction and inflammatory lung injury (4,8,9).
Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns (PAMPs), which refer to biomolecules produced by invading microbes or released from damaged tissue. These biomolecules, which include lipopolysaccharides, peptidoglycans, lipoteichoic acid and CpG-containing oligonucleotides (CpG ODNs), serve as danger signals that initiate an inflammatory immune response. TLRs trigger inflammation mediated by complement, macrophages, and neutrophils. These cells produce chemokines or cytokines that mediate systemic immune responses and recruit leukocytes to the sites of inflammation. This injury-induced inflammation can lead to systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS) (10-12).
TLR2, TLR4 and TLR9 are crucial for PAMP signaling. These receptors work through the adaptor protein myeloid differentiation factor 88 (MyD88), which activates nuclear factor-κB (NF-κB) and ultimately stimulates the production of proinflammatory cytokines (13,14). Rat alveolar macrophages are known to be sensitive to mechanical and CpG ODN (15) stimulation, but we are unaware of studies examining whether they are sensitive to ventilation-induced injury. If so, this would implicate TLR- and MyD88-mediated signaling in alveolar macrophages in the pathogenesis of VILI.
To address this question, we examined the effects of mechanical ventilation on the expression of TLR2, TLR4 and TLR9 genes in rat alveolar macrophages, as well as on activation of MyD88 and NF-κB.
Materials and methods
Animal procedures
Healthy, pathogen-free male Sprague-Dawley rats (300-350 g) were obtained from the Animal Center of Guangxi Medical University (Nanning, China), and kept in an environmentally controlled room (23±2 °C, 55%±10% humidity) with a 12-h light/dark cycle and allowed free access to food and water. Rats were anesthetized initially by intraperitoneal injection of pentobarbital (50 mg narcoren/kg body weight; Merial, Halbergmoos, Germany) and fentanyl (0.05 mg/kg body weight; Janssen-Cilag, Neuss, Germany), and then placed in a supine position on an adjustable warming pad to maintain a temperature of 37±1 °C. Anesthesia was maintained by supplementing with one third of the initial dose of anesthetic agents approximately every 45 min during the experimental period. Temperature was monitored continuously using a rectal probe. Anesthetized animals were randomly allocated into three groups (with 10 rats for each) using table of random number: non-ventilated, ventilated with low tidal volume (7 mL/kg, low VT), and ventilated with high tidal volume (40 mL/kg, high VT). The study was performed in accordance with the guiding principles for the care and use of laboratory animals approved by Institutional Animal Care and Use Committee of Tumor Hospital of Guangxi Medical University.
All animals were tracheotomized and then allowed to breathe spontaneously for 4 h or ventilated mechanically for 4 h using a small animal ventilator (TOPO, Kent Scientific, Torrington, USA) set at a positive end-expiratory pressure of 0. The ventilation rate was 80 breaths/min at low tidal volume and 60 breaths/min at high tidal volume. All rats were supplemented with oxygen at approximately 40-50% (16) of the inspired oxygen fraction, as determined by oxygen monitoring; the amount of oxygen was always less than 55% of the inspired oxygen fraction. Fluid-filled polyurethane catheters (inside diameter, 50.58 mm; outside diameter, 50.96 mm; SIMS Portex Ltd., Hythe, UK) were inserted into the right femoral vein for infusion of saline (0.01 mL/g/h body weight) and glycopyrrolate (0.1 μg/g body weight) to maintain intravascular volume, and into the right femoral artery for measurement of blood pressure and blood gas analysis.
After 4 h, all animals were sacrificed by lethal dose of anesthetic agent, and tissue samples were harvested. All animal procedures were performed with special care to minimize activation of an inflammatory response.
Lung histopathology and determination of wet/dry ratio
The middle lobe of the right lung was fixed by injection of 10% formaldehyde solution through the right middle bronchus at a pressure of 20 cmH2O. After fixation, the tissue was embedded in paraffin and 4-μm sections were prepared and stained with hematoxylin and eosin, then examined by light microscopy. To score lung injury we used a modified VILI histopathology scoring system as previously described (17). VILI was scored according to the following four main items: alveolar congestion; haemorrhage; infiltration or aggregation of neutrophils in airspace or vessel wall; and thickness of the alveolar wall/hyaline membrane formation. A score of 0 represented normal lungs; 1 represented mild, less than 25% lung involvement; 2 represented moderate, 25% to 50% lung involvement; 3 represented severe, 50% to 75%lung involvement; and 4 represented very severe, more than 75% lung involvement. An overall score of VILI was obtained based on the summation of all the scores from normal or ventilated lungs (n=10 per group).
Pulmonary wet/dry ratios were measured as an index of pulmonary congestion. Immediately after rats were killed, the right lower lobe was weighed and then dried to a constant weight at 60 °C for 24 h.
Collection and analysis of bronchoalveolar lavage fluid (BALF) and alveolar macrophages
Alveolar macrophages were isolated as described previously (18). In brief, the trachea of killed animals was cannulated and the lungs were flushed once with 5 mL of cold phosphate-buffered saline (Dulbecco’s PBS; Gibco BRL, Grand Island, USA) to collect BALF for mediator analysis (BALF 1 fraction). The lungs were subsequently flushed another eight times with 10 mL PBS to obtain alveolar macrophages (BALF 2 fraction). The two BALF fractions were centrifuged, and the BALF 1 supernatant was stored at −80 °C for enzyme-linked immunosorbent assay (ELISA) and determination of total protein using the bicinchoninic acid (BCA) assay according to the manufacturer’s instructions (Pierce, Rockford, USA).
BALF 1 and 2 pellets were combined, resuspended in Dulbecco’s modified Eagle medium (DMEM, Gibco, USA), counted, and transferred to 24-well culture plates (BD, Franklin Lakes, NJ, USA). After incubation for 60 min at 37 °C in a 5% CO2 atmosphere, cultures were washed with DMEM to remove nonadherent cells. The adherent cells were counted using a hemocytometer, viability was determined using a 0.2% trypan blue exclusion assay, and cell differentiation and aggregation were examined by counting 500 cells on a Wright-Giemsa-stained slide. These cultures also served as the source of RNA and protein for analyzing mRNA and protein expression in alveolar macrophages.
Cytokine analysis in BALF and plasma
Concentrations of IL-1β, IL-6 and IL-10 were determined in BALF and plasma using a commercial ELISA according to the manufacturer’s recommendations (R&D Systems, Minneapolis, USA). The manufacturer-specified detection limits with this kit were IL-1β, 15.6 pg/mL; IL-6, 0.078 pg/mL; and IL-10, 0.78 pg/mL.
RNA isolation and real-time reverse transcriptase-polymerase chain reaction
Total RNA was extracted from alveolar macrophages using TRIzol (Invitrogen, Carlsberg, USA). RNA quality and quantity were determined by spectrophotometry. RNA samples were reverse-transcribed into cDNA using a reverse transcription kit (Toyobo, Osaka, Japan). The following primers were used to determine mRNA expression levels: TLR2, 5’-GGGATACAGGCCGTCAAGAC-3’ (forward) and 5’-CAGGAGCAGATGAAATGGTTGT-3’ (reverse); TLR4, 5’-CGCTCTGGCATCATCTTCAT-3’ (forward) and 5’-CTCCTCAGGTCAAAGTTGTTGC-3’ (reverse); TLR9, 5’-CCTGGCACACAATGACATTCA-3’ (forward) and 5’-TAAAGGTCCTCCTCGTCCCA-3’ (reverse); MyD88, 5’-GAGATCCGCGAGTTTGAGAC-3’ (forward) and 5’-TTGTCTGTGGGACACTGCTC-3’ (reverse); NF-κΒ, 5’-GAGGACTTGCTGAGGTTGG-3’ (forward) and 5’-TGGGGTGGTTGATAAGGAGTG-3’ (reverse); and glyceraldehyde phosphate dehydrogenase (GAPDH), 5’-GGCACAGTCAAGGCTGAGAATG-3’ (forward) and 5’-ATGGTGGTGAAGACGCCAGTA-3’ (reverse). Real-time reverse transcriptase PCR was performed as previously described (19). The level of each target gene was normalized relative to that of GAPDH in each sample using the ΔCt method. Relative differences in gene expression among groups of the alveolar macrophages were determined using the comparative Ct (ΔΔCt) method and fold expression was calculated by the formula 2−ΔΔCt, where ΔΔCt represents ΔCt values normalized relative to the mean ΔCt of healthy control samples. All fragments were checked for the specificity by direct sequencing of both strands with an ABI PRISM 310 Genetic Analyzer using Big Dye Terminator kit v 3.1 (Applied Biosystems, CA, USA).
Protein extraction and Western blotting
Proteins were extracted from alveolar macrophages using RIPA buffer and quantitated using the BCA protein assay kit. For Western blot detection of TLR2, TLR4, TLR9, MyD-88 and NF-κΒ, samples (50 μg) were electrophoresed on a 10% SDS-polyacrylamide gel, electroblotted onto nitrocellulose membranes, and blocked at 4 °C overnight with 5% bovine serum albumin (BSA) dissolved in Tris-buffered saline (TBS) containing 0.1% Tween-20. Membranes were incubated for 2 h with rabbit anti-rat monoclonal antibodies (Cell Signaling Technology, Danvers, USA) against TLR2 (1:1,000), TLR4 (1:2,000), TLR9 (1:1,000), MyD-88 (1:1,000) and NF-κΒ (1:1,000). Blots were washed several times, and then incubated with horseradish peroxidase-conjugated mouse anti-rabbit antibody (1:5,000 in PBS containing 5% nonfat dry milk; Pierce, USA). Blots were again washed several times and visualized by enhanced chemiluminescence (Pierce, USA). Blots were also incubated with rabbit anti-rat GAPDH antibody (1:10,000, Santa Cruz Biotechnology, CA, USA) as a control for protein loading.
Sample size and statistical analysis
We estimated that a total of 30 rats would be needed to detect a difference between groups, with a 2-tailed α of 0.05 and a 1-β of 0.80, for a comparison of two independent proportions if there was an absolute decrease of 15% in the composite outcome measure. The Data and Safety Monitoring Board, blinded to treatment group, reviewed two formal interim analyses and regular reports of our primary composite outcome as well as serious adverse events. All data were analyzed using SPSS software (version 16.0; IBM, Chicago, USA). All quantitative data were expressed as mean ± SD. Multiple comparisons were analyzed using one-way ANOVA, followed by the Friedman test and the Wilcoxon signed-rank test. P values less than 0.05 were considered significant.
Results
Outcomes and overall pathophysiology assessment
All animals survived the 4-h period of spontaneous breathing or mechanical ventilation at low or high tidal volume. Lungs from animals ventilated with high tidal volume showed acute inflammatory infiltration and perivascular edema, whereas no major histological differences were observed between animals ventilated with low tidal volume or spontaneously breathing control animals. The lung histopathology score was higher in high VT rats as compared with the controls and low VT. However, no differences were noted between the low VT and the controls (Figure 1 and Table 1).
Figure 1.
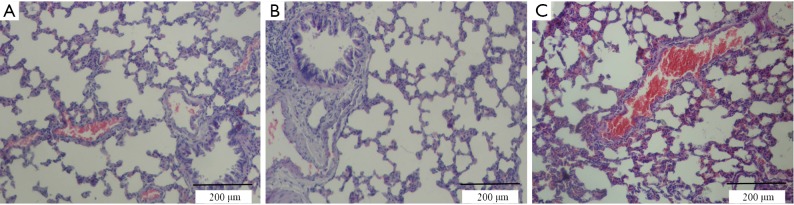
Lung tissue histopathology in spontaneously breathing control rats (A) and rats mechanically ventilated with low (B) or high (C) tidal volume. Sections were stained with hematoxylin-and-eosin. Tissue ventilated with high tidal volume showed much more inflammatory cell infiltration, alveolar 2 septal thickening, and pulmonary edema than did tissue in the other two groups.
Table 1. Levels of total protein, IL-6 and IL-1β in BALF after ventilation-induced lung injury and lung histopathology score.
| Parameter | Non-ventilated control | Ventilated groups |
|
|---|---|---|---|
| Low tidal volume | High tidal volume | ||
| Wet/dry ratio | 4.88±0.18 | 4.93±0.26 | 5.74±0.21* |
| Protein content (μg/μL) | 567±46 | 593±57 | 988±39* |
| BALF (pg/mL) | |||
| IL-6 | 2.42±0.13 | 2.44±0.32 | 7.32±0.24* |
| IL-1β | 53.63±5.26 | 53.55±6.63 | 139.95±9.37* |
| Plasma | |||
| IL-6 (pg/mL) | 1.70±0.23 | 1.67±0.21 | 2.08±0.11* |
| VILI-score | 0.30±0.10 | 0.50±0.15 | 3.5±0.5* |
Data are presented as mean ± SD (n=10 in all groups). *, P<0.001 vs. non-ventilated animals and animals ventilated with low tidal volume. BALF, bronchoalveolar lavage fluid; IL-1β, interleukin-1β; IL-6, interleukin-6; VILI, ventilator-induced lung injury.
Counts of isolated AMs
In the high VT group, significantly more AMs were isolated than in the low VT group and non-ventilated animals (Figure 2). And the macrophage percentages were more than 90% counted by the hemocytometer.
Figure 2.
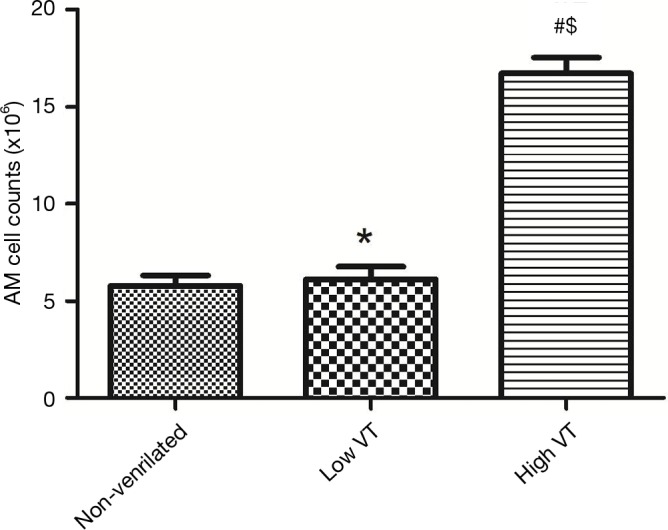
Alveolar macrophage cell counts. Male rats were subjected to non-ventilate or low VT or high VT. After 4 h of the procedure, AM cells were isolated from the lung. Numbers of viable cells were determined by trypan blue staining and counted by a hemocytometer. Data are mean ± SD, n=10. #, P<0.001 vs. non-ventilated animals; $, P<0.001 vs. low VT; *, P>0.05 vs. non-ventilated animals.
Wet/dry ratios and BALF analysis
Wet/dry ratios, BALF total protein levels were significantly higher in the group of high VT than in the control and low VT (Table 1). These three parameters were similar for rats ventilated with low tidal volume and for spontaneously breathing rats.
Pro-inflammatory cytokine concentrations in BALF and plasma
Cytokine profiles were similar in animals ventilated with low tidal volume and control animals (Table 1). IL-10 were undetectable (IL-10 detection threshold was 0.78 pg/mL) in either BALF or plasma in the three groups, and plasma IL-1β levels were below the level of detection (IL-1β detection threshold was 15.6 pg/mL) in control and ventilation groups. These animals showed significantly higher levels of IL-6 levels in BALF and plasma in the high VT group than did the other two groups of animals (Table 1).
Mechanical ventilation induced MyD-88 and NF-κB expression and up-regulated expression of TLR2, TLR4 and TLR9
Expression of mRNA and protein for TLR2, TLR4, TLR9, MyD-88 and NF-κB were similar for animals ventilated with low tidal volume and for spontaneously breathing controls (Figures 3,4). The mRNA and protein levels of all these genes, however, were significantly higher for animals ventilated with high tidal volume than the control and low tidal volume group. mRNA levels as follow, TLR2 (4.78±0.98 vs. 1±0.22 and 1.15±0.30, both P<0.001), TLR4 (5.15±0.78 vs. 1±0.19 and 1.23±0.35, both P<0.001), TLR9(10.75±1.80 vs. 1±0.27 and 0.91±0.28, both P<0.001), MyD-88 (3.97±0.75 vs. 1±0.20 and 0.96±0.24, both P<0.001), NF-κΒ (5.79±0.98 vs. 1±0.23 and 1.08±0.25, both P<0.001). Protein levels as follow, TLR2 (0.59±0.049 vs. 0.35±0.036 and 0.36±0.031, both P<0.001), TLR4 (0.845±0.0395 vs. 0.401±0.026 and 0.403±0.020, both P< 0.001), TLR9 (0.727±0.074 vs. 0.383±0.039 and 0.367±0.043, both P<0.001), MyD-88 (1.01±0.060 vs. 0.485±0.045 and 0.507±0.046, both P<0.001) and NF-κΒ (0.776±0.067 vs. 0.448±0.043 and 0.481±0.047, both P<0.001).
Figure 3.
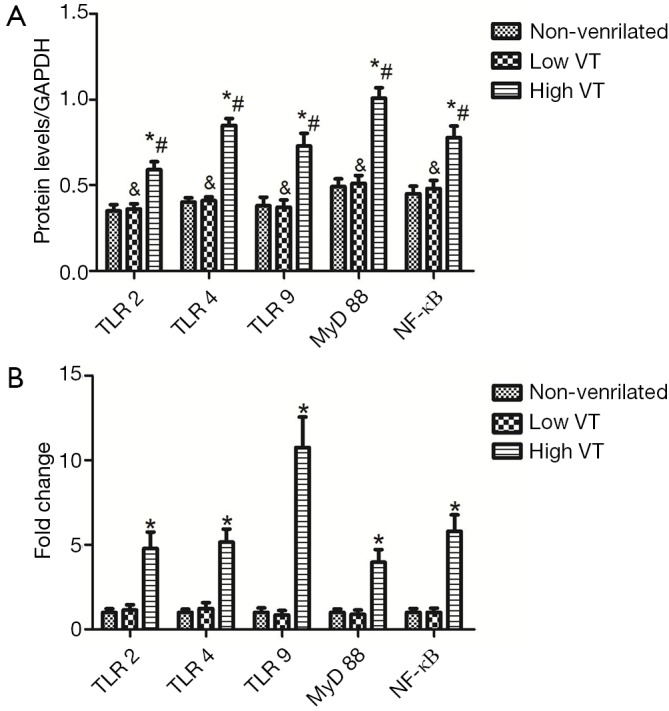
Fold changes in TLR2, TLR4, TLR9, MyD88 and NF-κB mRNA levels in alveolar macrophages of healthy rats after 4 h of spontaneous breathing (non-ventilated) or 4 h of mechanical ventilation with tidal volumes of 7 mL/kg (low VT) or 40 mL/kg (high VT) are shown in (B). Data are mean ± SD, n=10. Results were normalized to the values in GAPDH control amplifications, and expressed as fold changes relative to the non-ventilated control animals. *, P<0.001 vs. non-ventilated animals. TLR, Toll-like receptor; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor-κB.
Figure 4.
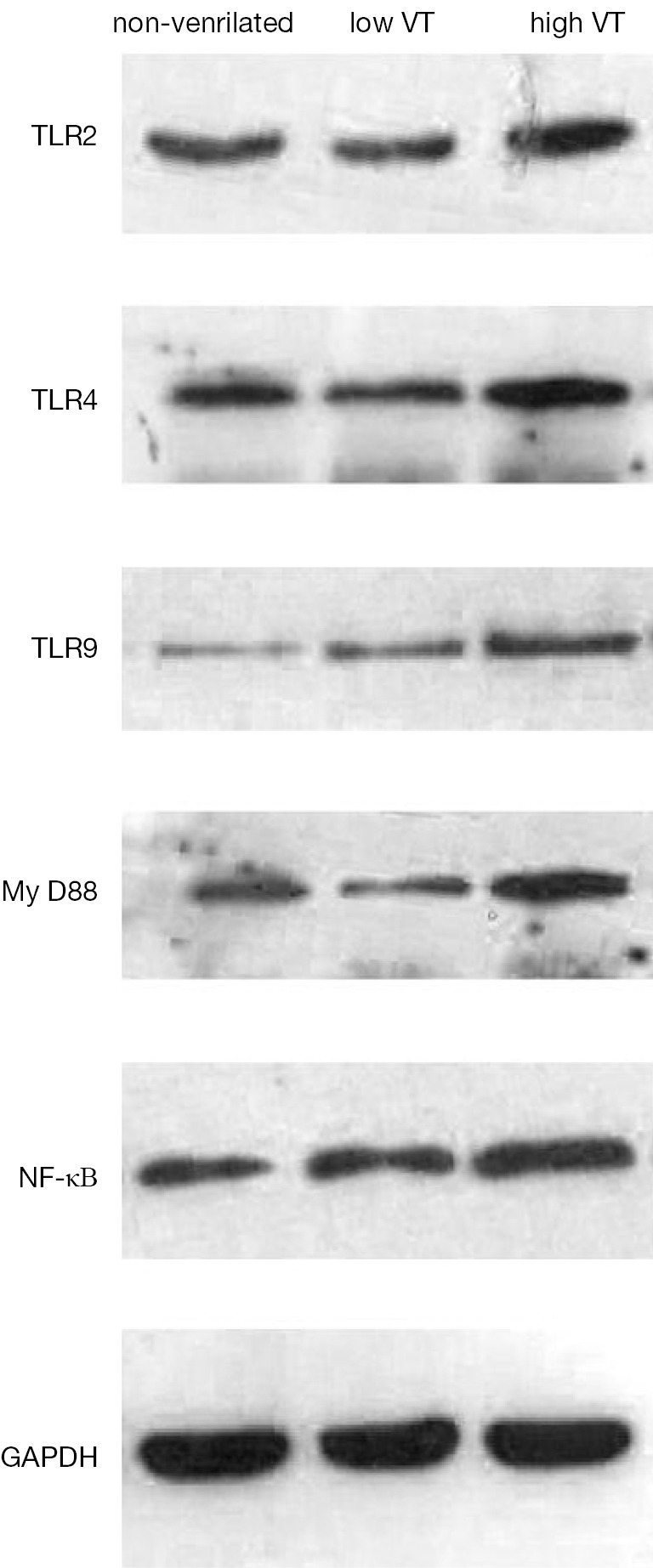
Changes in TLR2, TLR4, TLR9, MyD88 and NF-κB protein levels on alveolar macrophages of healthy rats after 4 h of spontaneous breathing (non-ventilated) or 4 h of mechanical ventilation with tidal volumes of 7 mL/kg (low VT) or 40 mL/kg (high VT). Representative Western blot results are shown above, and quantitation of TLR2, TLR4, TLR9, MyD88 and NF-κB protein levels independent experiments is shown in (Figure 3A). Data are mean ± SD, n=10. Results were normalized to levels of GADPH protein detected on the same blots. *, P<0.001 vs. non-ventilated animals; #, P<0.001 vs. animals ventilated with low tidal volume; &, P>0.05 vs. non-ventilated animals. TLR, Toll-like receptor; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor-κB.
Discussion
Here we investigated the hypothesis that mechanical ventilation activates alveolar macrophage TLR receptors that are normally involved in initiating innate immune responses to PAMPs. TLR activation should up-regulate expression of the adaptor protein MyD-88, stimulating production of NF-κB and ultimately the secretion of proinflammatory cytokines. As predicted, we found that 4-h mechanical ventilation of tracheotomized rats using a high tidal volume increased the expression of the major PAMP sentinels TLR2, TLR4 and TLR9, expression of MyD-88 and NF-κΒ, and concentrations of IL-1β and IL-6 in the BALF. Our findings implicate TLR-mediated signaling in ventilation-induced lung injury and suggest that the pathogenesis occurs through a mechanism similar to that induced by infectious stimuli.
Our rat model of mechanical ventilation with high tidal volume reproduced the pathologic changes consistent with VILI, including increased alveolar permeability (Figure 1) and increased alveolar macrophage cell counts (Figure 2) and protein levels in BALF (Table 1) in the absence of infection. In this way, mechanical ventilation appears to stimulate the immune system akin to severe bacterial infections (12). These findings are consistent with various experimental models of VILI, in which the authors have proposed that alveolar overdistension triggers an innate immune inflammatory reaction similar to that caused by pathogens (20-22). This reaction involves changes in the growth, differentiation, migration, remodeling, and gene expression of various pulmonary cell types, including alveolar macrophages, alveolar epithelial cells, endothelial cells and fibroblasts. One consequence of these changes is an increase in pulmonary and systemic pro-inflammatory cytokine levels, as we observed in our measurements of cytokine levels in BALF and serum after ventilation with high tidal volume.
Our results are consistent with previous studies suggesting that mechanical ventilation, alone or in combination with infection, induces inflammation by the synthesis of NF-κΒ and proinflammatory cytokines (12,23-25). For example, a study with isolated, perfused mouse lung showed that combining 150-min lung overinflation with a tidal volume of 32 mL/kg and lipopolysaccharide treatment activated NF-κΒ and induced the release of cytokines known to be dependent on NF-κB (24). Indeed we focused on alveolar macrophages in our study because they are involved in initiating innate immune responses and may activate NF-κΒ in acute lung injury and inflammation (26). Removing alveolar macrophages from rats exposed to mechanical ventilation at high tidal volume attenuates the resulting alveolar barrier dysfunction and inflammatory lung injury (4,8,9).
Our animals ventilated with low tidal volume were indistinguishable from control animals breathing spontaneously. This is consistent with the widespread use of ventilation with low tidal volume to protect against VILI (16,27,28). Our finding contrasts with studies in animals suggesting that even ventilation performed with low tidal volume can induce lung inflammation and increase the risk of ventilator-induced injury. For example, computed tomography studies suggest that low tidal volume can still cause tidal hyperinflation in patients with acute respiratory distress syndrome because of the anatomical heterogeneity of the damaged lung (16). These differences are likely due to differences between laboratory animals and humans, and should be explored further to optimize tidal volume for vulnerable patients.
Our study indicates that mechanical ventilation with high tidal volume upregulates the expression of TLR2, TLR4 and TLR9. TLRs have long been recognized to play a predominant role in the innate immune response to infection and tissue injury (16,29,30). TLR2 is widely expressed in first-line host defense cells, including monocytes, macrophages, dendritic cells, and polymorphonuclear leukocytes. TLR2 is thought to mediate inflammatory responses to components of Gram-positive and -negative bacteria and mycobacteria such as peptidoglycan, lipoteichoic acid, bacterial lipoproteins, lipopeptides, and lipoarabinomannan (31). TLR4 activates MyD88-dependent and Toll/IL-1 receptor domain containing adapter inducing interferon (TRIF)-dependent signaling pathways. It is activated in response to mechanical ventilation with low tidal volume (32), and it plays an important role in acute lung injury induced by ventilation with high tidal volume (33), lipopolysaccharide (34), acid aspiration (35), hemorrhage (36), and ischemia and reperfusion injury (37). TLR9 mediates cellular responses to CpG ODN and to endogenous ligands released during noninfectious tissue injury, including high-mobility group box 1 proteins released from necrotic cells, oxidized phospholipids created by locally generated reactive oxygen species, low molecular weight hyaluran and fibrinogen released from degraded extracellular matrix, heat shock proteins released from necrotic cells, and surfactant protein-A (16,38,39). In this way, our study extends the proinflammatory role of TLRs to the specific case of VILI.
Upon binding to endogenous activators, TLR2, TLR4 and TLR9 recruit the downstream adaptor molecule MyD88, which activates NF-κΒ and thereby induces transcription of pro-inflammatory genes, ultimately leading to cytokine production (40). Consistent with this sequence of events, we found that mechanical ventilation with high tidal volume upregulated TLR expression, NF-κΒ and MyD88 activation in the lung, and levels of IL-1β and IL-6 in BALF and serum. Our findings suggest that signaling involving TLR2, TLR4, TLR9 and MyD88 may contributes to the pro-inflammatory response during ventilation induced lung injury.
One limitation of our approach is that stresses due to the experimental manipulations may have triggered the same TLR-mediated signaling that we wished to study. To minimize this risk, we continuously monitored oxygen saturation and hemodynamic stability in the anesthetized rats. We also administered oxygen to the rats under anesthesia and maintained the inspired oxygen fraction below 55% to avoid both hypoxia and hyperoxia (16,41,42). The fact that all outcomes that we measured were similar between the group ventilated with low tidal volume and the control group suggests that our precautions were effective.
Although our data may imply roles for the TLR2, TLR4 and TLR9/MyD88 signaling in regulating multiple pro-inflammatory cytokines during MV, we acknowledge some limitations to this study. First, we did not explore whether repression of MyD88 expression during high VT MV could be reversed by returning to low VT. However in the patients with ALI, pulmonary and systemic inflammatory responses induced by temporary application of high VT mechanical ventilation can be reversed by reinstitution of lung protective MV (12), at least over the time frame of a few hours. Second, we do not know whether inhibition of TLR2, TLR4 and TLR9 with blocking antibodies affect the MyD88 response. We can not say that our data fully demonstrate that TLR2, TLR4 and TLR9 pathway is conclusively involved in increased inflammation associated with the use of high-VT ventilation because the experiments did not examine the effects of disrupting these pathways. Third, there is a possibility that the repression of MyD88 expression could be unrelated to the activation of TLR2, TLR4 and TLR9 signaling and could be governed by other molecules capable of regulation of inflammation.
Conclusions
Mechanical ventilation with tidal volumes of 40 mL/kg increased lung permeability and induced inflammation. The mechanism of action of mechanical ventilation may involve increasing in TLR2, TLR4, and TLR9 and MyD88 expressions. And our data supports an interaction between TLR2, TLR4, and TLR9 and MyD88 signaling pathway for the overexpression and release of pro-inflammatory cytokines during VILI. Further investigation to identify the exact functions of TLR families will provide crucial insight into designing new interventions to limit lung injury induced by mechanical ventilation.
Acknowledgements
Ethics: All animal studies were approved by the Institutional Animal Care and Use Committee of Tumor Hospital of Guangxi Medical University.
Authors’ contributions: Linghui Pan designed the overall study. Huijun Dai carried out experiments, collected and analyzed data, and co-wrote the paper. Fei Lin carried out experiments and collected and analyzed data with Huijun Dai. Wanyun Ge, Wei Li and Sheng He carried out experiments, and analyzed data with Huijun Dai and Fei Lin. All authors read and approved the final manuscript.
Funding: This study was supported by the National Natural Science Foundation of China [81060008].
Disclosure: The authors declare no conflict of interest.
References
- 1.Vaneker M, Joosten LA, Heunks LM, et al. Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 2008;109:465-72. [DOI] [PubMed] [Google Scholar]
- 2.Wu J, Yan Z, Schwartz DE, et al. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol 2013;190:3590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganter MT, Roux J, Miyazawa B, et al. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res 2008;102:804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank JA, Pittet JF, Wray C, et al. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 2008;63:147-53. [DOI] [PubMed] [Google Scholar]
- 5.Conway Morris A, Kefala K, Wilkinson TS, et al. Diagnostic importance of pulmonary interleukin-1beta and interleukin-8 in ventilator-associated pneumonia. Thorax 2010;65:201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holt PG, Strickland DH, Wikström ME, et al. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol 2008;8:142-52. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Nikrad MP, Travanty EA, et al. Innate immune response of human alveolar macrophages during influenza A infection. PLoS One 2012;7:e29879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eyal FG, Hamm CR, Parker JC. Reduction in alveolar macrophages attenuates acute ventilator induced lung injury in rats. Intensive Care Med 2007;33:1212-8. [DOI] [PubMed] [Google Scholar]
- 9.Frank JA, Wray CM, McAuley DF, et al. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 2006;291:L1191-8. [DOI] [PubMed] [Google Scholar]
- 10.Zhai Y, Shen XD, O'Connell R, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol 2004;173:7115-9. [DOI] [PubMed] [Google Scholar]
- 11.Oppeltz RF, Rani M, Zhang Q, et al. Burn-induced alterations in toll-like receptor-mediated responses by bronchoalveolar lavage cells. Cytokine 2011;55:396-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villar J, Cabrera NE, Casula M, et al. Mechanical ventilation modulates TLR4 and IRAK-3 in a non-infectious, ventilator-induced lung injury model. Respir Res 2010;11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao W.Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol 2009;296:H1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beutler BA. TLRs and innate immunity. Blood 2009;113:1399-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm CK, Paludan SR, Fitzgerald KA. DNA recognition in immunity and disease. Curr Opin Immunol 2013;25:13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Su X, Yan X, et al. Toll-like receptor 4-myeloid differentiation factor 88 signaling contributes to ventilator-induced lung injury in mice. Anesthesiology 2010;113:619-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolthuis EK, Vlaar AP, Choi G, et al. Mechanical ventilation using non-injurious ventilation settings causes lung injury in the absence of pre-existing lung injury in healthy mice. Crit Care 2009;13:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seitz DH, Palmer A, Niesler U, et al. Altered expression of Fas receptor on alveolar macrophages and inflammatory effects of soluble Fas ligand following blunt chest trauma. Shock 2011;35:610-7. [DOI] [PubMed] [Google Scholar]
- 19.Herold S, Tabar TS, Janssen H, et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am J Respir Crit Care Med 2011;183:1380-90. [DOI] [PubMed] [Google Scholar]
- 20.Vogel V, Sheetz M.Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol 2006;7:265-75. [DOI] [PubMed] [Google Scholar]
- 21.Tremblay LN, Miatto D, Hamid Q, et al. Injurious ventilation induces widespread pulmonary epithelial expression of tumor necrosis factor-alpha and interleukin-6 messenger RNA. Crit Care Med 2002;30:1693-700. [DOI] [PubMed] [Google Scholar]
- 22.Pelosi P, Rocco PR. Effects of mechanical ventilation on the extracellular matrix. Intensive Care Med 2008;34:631-9. [DOI] [PubMed] [Google Scholar]
- 23.Martin TR. Interactions between mechanical and biological processes in acute lung injury. Proc Am Thorac Soc 2008;5:291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Held HD, Boettcher S, Hamann L, et al. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med 2001;163:711-6. [DOI] [PubMed] [Google Scholar]
- 25.Wurfel MM. Microarray-based analysis of ventilator-induced lung injury. Proc Am Thorac Soc 2007;4:77-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haeberle HA, Takizawa R, Casola A, et al. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis 2002;186:1199-206. [DOI] [PubMed] [Google Scholar]
- 27.Matthay MA. Treatment of acute lung injury: clinical and experimental studies. Proc Am Thorac Soc 2008;5:297-9. [DOI] [PubMed] [Google Scholar]
- 28.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol 2005;33:319-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar H, Kawai T, Akira S.Toll-like receptors and innate immunity. Biochem Biophys Res Commun 2009;388:621-5. [DOI] [PubMed] [Google Scholar]
- 30.Belperio JA, Keane MP, Lynch JP, 3rd, et al. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med 2006;27:350-64. [DOI] [PubMed] [Google Scholar]
- 31.Chua BY, Johnson D, Tan A, et al. Hepatitis C VLPs delivered to dendritic cells by a TLR2 targeting lipopeptide results in enhanced antibody and cell-mediated responses. PLoS One 2012;7:e47492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gharib SA, Liles WC, Klaff LS, et al. Noninjurious mechanical ventilation activates a proinflammatory transcriptional program in the lung. Physiol Genomics 2009;37:239-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu G, Malik AB, Minshall RD. Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 2010;38:194-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanimura N, Saitoh S, Matsumoto F, et al. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun 2008;368:94-9. [DOI] [PubMed] [Google Scholar]
- 35.Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008;133:235-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lv T, Shen X, Shi Y, et al. TLR4 is essential in acute lung injury induced by unresuscitated hemorrhagic shock. J Trauma 2009;66:124-31. [DOI] [PubMed] [Google Scholar]
- 37.Zanotti G, Casiraghi M, Abano JB, et al. Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol Physiol 2009;297:L52-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006;26:174-9. [DOI] [PubMed] [Google Scholar]
- 39.Hoque R, Sohail M, Malik A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011;141:358-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An H, Yu Y, Zhang M, et al. Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology 2002;106:38-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bellani G, Messa C, Guerra L, et al. Lungs of patients with acute respiratory distress syndrome show diffuse inflammation in normally aerated regions: a [18F]-fluoro-2-deoxy-D-glucose PET/CT study. Crit Care Med 2009;37:2216-22. [DOI] [PubMed] [Google Scholar]
- 42.Vaneker M, Heunks LM, Joosten LA, et al. Mechanical ventilation induces a Toll/interleukin-1 receptor domain-containing adapter-inducing interferon beta-dependent inflammatory response in healthy mice. Anesthesiology 2009;111:836-43. [DOI] [PubMed] [Google Scholar]