

Abstract
There is a well-recognized association between obesity, inflammation, and hypertension. Why obesity causes hypertension is poorly understood. We previously demonstrated using a C-reactive protein (CRP) transgenic mouse that CRP induces hypertension that is related to NO deficiency. Our prior work in cultured endothelial cells identified the Fcγ receptor IIB (FcγRIIB) as the receptor for CRP whereby it antagonizes endothelial NO synthase. Recognizing known associations between CRP and obesity and hypertension in humans, in the present study we tested the hypothesis that FcγRIIB plays a role in obesity-induced hypertension in mice. Using radiotelemetry, we first demonstrated that the hypertension observed in transgenic mouse-CRP is mediated by the receptor, indicating that FcγRIIB is capable of modifying blood pressure. We then discovered in a model of diet-induced obesity yielding equal adiposity in all study groups that whereas FcγRIIB+/+ mice developed obesity-induced hypertension, FcγRIIB−/− mice were fully protected. Levels of CRP, the related pentraxin serum amyloid P component which is the CRP-equivalent in mice, and total IgG were unaltered by diet-induced obesity; FcγRIIB expression in endothelium was also unchanged. However, whereas IgG isolated from chow-fed mice had no effect, IgG from high-fat diet–fed mice inhibited endothelial NO synthase in cultured endothelial cells, and this was an FcγRIIB-dependent process. Thus, we have identified a novel role for FcγRIIB in the pathogenesis of obesity-induced hypertension, independent of processes regulating adiposity, and it may entail an IgG-induced attenuation of endothelial NO synthase function. Approaches targeting FcγRIIB may potentially offer new means to treat hypertension in obese individuals.
Keywords: C-reactive protein, hypertension, IgG, inflammation, obesity, serum amyloid P component
There is a well-recognized association between obesity, inflammation, and cardiovascular disease.1 In particular, obesity is a major risk factor for hypertension,2,3 and up to four fifths of the 20% increase in the prevalence of hypertension between 1988 to 1994 and 1994 to 2004 in the National Health and Nutrition Examination Surveys have been attributed to increasing body mass index.2 Regrettably current approaches to combat the underlying obesity, which focus on nutrition and exercise to achieve weight loss and maintenance, have not been effective in the long term.4 In addition, obesity is a risk factor for the development of hypertension that is resistant to single agent therapy.5 As such, new therapeutic approaches are needed for the treatment of hypertension in obese individuals.
Circulating levels of the pentraxin C-reactive protein (CRP), which are often interpreted to indicate the presence of low-grade systemic inflammation, are elevated in obese populations including children, adolescents, and young adults.6,7 Chronic modest elevations in circulating CRP levels are associated with an increased risk of developing hypertension,8 though its direct causal role in human hypertension is unclear.9 We previously demonstrated in studies using a CRP transgenic mouse (TG-CRP), allowing overexpression of the pentraxin, that serum concentrations of CRP comparable with those observed with low-grade systemic inflammation in humans induce hypertension.10 In that work, we further showed that the CRP-induced hypertension is because of the downregulation of endothelial NO synthase (eNOS) activity in vascular endothelial cells,10 and impaired endothelial function is an important contributing factor to the development of hypertension in humans.11 In studies of cultured endothelial cells and endothelial repair and carotid vascular conductance in vivo in mice, we have determined that endothelial actions of CRP are mediated by the IgG receptor Fcγ receptor IIB (FcγRIIB),12–14 which we have demonstrated is expressed in endothelial cells.14,15 Known ligands for FcγRIIB are CRP, the highly homologous pentraxin serum amyloid P component (SAP), which is the acute phase reactant equivalent of CRP in mice,16,17 and IgG, which all bind to identical residues in the extracellular domain of the receptor.18
The present studies using mouse models were designed to better understand the pathogenesis of the hypertension that complicates obesity. Recognizing the associations between CRP and obesity and hypertension, and knowing that FcγRIIB is a biologically relevant CRP receptor, we tested the hypothesis that FcγRIIB mediates obesity-induced hypertension. We first demonstrated that the hypertension caused by elevations in the known FcγRIIB ligand CRP requires the receptor, indicating that FcγRIIB is capable of modifying blood pressure (BP). We then discovered in a model of diet-induced obesity, which yielded equal weight gain and equal increase in body fat with high-fat diet (HFD) feeding in all study groups, that whereas FcγRIIB+/+ mice developed obesity-induced hypertension, FcγRIIB−/− mice were fully protected from hypertension. Thus, independent of any effects on body weight or body composition, FcγRIIB plays a key role in obesity-induced hypertension.
Methods
Detailed methods are available in the online-only Data Supplement.
Animal Models
To study CRP-induced hypertension and FcγRIIB, TG-CRP were crossed with FcγRIIB−/− mice to yield FcγRIIB+/+, FcγRIIB+/+;TG-CRP, FcγRIIB−/−, and FcγRIIB−/−;TG-CRP littermates. All mice were fed standard rodent chow.
To study obesity-induced hypertension, FcγRIIB+/+ and FcγRIIB−/− mice were placed on either control diet (CON) or HFD. Mice were maintained on their respective diet for a minimum of 12 weeks. This resulted in 4 experimental groups: (1) FcγRIIB+/+ CON, (2) FcγRIIB+/+ HFD, (3) FcγRIIB−/− CON, and (4) FcγRIIB−/−HFD. Their care and use were approved by the Institutional Animal Care and Use Committees at Baylor College of Medicine and the University of Texas Southwestern Medical Center.
Body Composition
Body composition was measured by dual-energy x-ray absorptiometry.
BP Measurements
Mice underwent tail-cuff BP measurement for 5 consecutive days, using the mean of the last 10 readings on day 5. For radiotelemetry, mice were anesthetized and instrumented with a radiotelemetry device. Mice were allowed to recover until a return of circadian variation in BP and activity were observed. In the CON chow versus HFD experiments, mice were maintained on their respective study diet throughout the radiotelemetry measurements.
Serum CRP, SAP, and IgG
Serum CRP, SAP, and total IgG were quantified by ELISA.
FcγRIIB Expression
Primary aortic endothelial cells were obtained by collagen digestion, and spleens were harvested and splenic cells dispersed by sieving. Cells then underwent fluorescence-activated cell sorting analysis using monoclonal antibody to FcγRIIB (anti-CD16/32b, 2.4G2) and anti-CD31 (PECAM-1 [platelet-endothelial cell adhesion molecule 1]) or anti-CD45R(B220) antibodies for endothelial cell or B-cell selection, respectively.
NO Synthase Activity in Cultured Endothelial Cells
Total IgG was isolated from wild-type mice after 12 weeks of CON versus HFD feeding. Bovine aortic endothelial cells were incubated with either CON IgG or HFD IgG, and NO synthase activation by vascular endothelial growth factor was then evaluated by measuring 14C-L-arginine conversion to 14C-L-citrulline. Additional experiments were performed in bovine aortic endothelial cell transfected with control small interfering RNA or small interfering RNA targeting FcγRIIB.
Statistical Analysis
Statistical analysis for animal experiments was performed using 2-way ANOVA to interrogate the effects of genotype (FcγRIIB+/+ versus FcγRIIB−/−) and CRP (wild-type versus TG-CRP) or diet (CON versus HFD) and the interaction effect. For endothelial cell culture experiments, 1-way ANOVA was used with Student–Newman–Keuls post-test analysis. Differences were considered significant at P<0.05.
Results
CRP, FcγRIIB, and BP
We previously demonstrated that modest elevations in CRP causes hypertension in mice.10 However, the receptor mediating the hypertension invoked by CRP is unknown, and whether FcγRIIB affects BP is unknown. Radiotelemetry measurements of systolic BP (SBP), diastolic BP (DBP), and mean arterial pressure (MAP) were performed in conscious unrestrained mice to determine whether FcγRIIB is necessary for CRP-induced hypertension. In FcγRIIB+/+;TG-CRP mice, SBP and DBP and MAP were all elevated compared with mice not expressing the CRP transgene. CRP caused the mean SBP to increase from 123 to 133 mm Hg, mean DBP rose from 92 to 100 mm Hg, and mean MAP increased from 108 to 117 mm Hg (Figure 1A–1C). In contrast, SBP, DBP, and MAP were not elevated by CRP in FcγRIIB−/− mice. Heart rate was increased in TG-CRP compared with non–TG-CRP mice (Figure 1D); there was no difference in heart rate related to the presence versus absence of FcγRIIB. These findings reveal that FcγRIIB is required for CRP to cause hypertension in mice.
Figure 1.

Fcγ receptor IIB (FcγRIIB) is required for C-reactive protein (CRP)–induced systolic and diastolic hypertension. Systolic blood pressure (SBP; A), diastolic blood pressure (DBP; B), mean arterial pressure (MAP; C), and heart rate (HR; D) were measured by radiotelemetry in conscious FcγRIIB+/+ control, FcγRIIB+/+;transgenic mouse (TG)-CRP, FcγRIIB−/− control and FcγRIIB−/−;TG-CRP male littermates. Values are mean±SEM (n=8–10). In 2-way ANOVA analysis, SBP, DBP, MAP, and HR were significant for the effect of CRP (P<0.05). Furthermore, SBP and MAP had a significant interaction of the main effects (P<0.05). The effect of genotype was not significant in any comparison (P>0.05). In post-test analysis *P<0.05 vs control, †P<0.05 vs FcγRIIB+/+.
Diet-Induced Obesity Model
Having demonstrated in the TG-CRP mice that FcγRIIB is capable of participation in the pathogenesis of hypertension, we next wished to determine whether the receptor contributes to the disorder in a clinically relevant chronic condition that is commonly complicated by hypertension. Because obesity is a well-recognized risk factor for hypertension in humans2,3 and mice placed on HFD develop hypertension,19,20 we evaluated the possible role of FcγRIIB in hypertension caused by diet-induced obesity in mice. To accomplish this, starting at 5 to 6 weeks of age FcγRIIB+/+ or FcγRIIB−/− mice were placed on either CON or HFD diet. Beginning 2 weeks after the initiation of the diets, predictably the mice on HFD had greater body weight than the mice on CON diet, and the difference was maintained throughout the 12-week period on the diet (Figure 2A). At 2 and 4 weeks on the diets there was a small difference in weight between HFD-fed FcγRIIB+/+ mice and HFD-fed FcγRIIB−/− mice, with the former weighing slightly more than the latter. However, the difference did not persist >4 weeks on diet.
Figure 2.
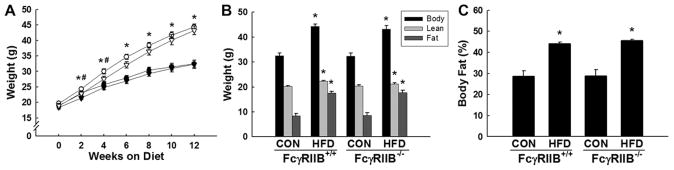
Fcγ receptor IIB (FcγRIIB)+/+ and FcγRIIB−/− mice gain equal weight and fat mass on high-fat diet (HFD). At 5 to 6 weeks of age, FcγRIIB+/+ and FcγRIIB−/− male mice were placed on either a control chow diet (CON) or a HFD. A, Body weight was measured over time in FcγRIIB+/+ CON (●), FcγRIIB+/+ HFD (○), FcγRIIB−/− CON (▼), and FcγRIIB−/− HFD (▽). B and C, After 12 weeks on the respective diets, body weight was measured and lean body mass and fat mass were measured by dual-energy x-ray absorptiometry (DEXA; B), and percentage body fat was calculated (C). Values are mean±SEM (n=8–12), *P<0.05 vs CON diet, #P<0.05 vs FcγRIIB−/− HFD.
Body composition was assessed by dual-energy x-ray absorptiometry after 12 weeks of CON or HFD feeding. HFD-fed FcγRIIB+/+ mice and HFD-fed FcγRIIB−/− mice had body weights that were comparable and increased by 35% above the body weights of mice on CON. The vast majority of the increase in body weight was attributed to an increase in adiposity (Figure 2B), with both FcγRIIB+/+ and FcγRIIB−/− mice displaying a 50% increase in percent body fat on HFD (Figure 2C). Importantly, the adiposity that resulted from HFD feeding was equal in FcγRIIB+/+ and FcγRIIB−/− mice (Figure 2B and 2C), indicating that the presence versus absence of the receptor does not affect body composition.
FcγRIIB and HFD-Induced Hypertension
Tail-cuff BP measurements were first used to assess whether the presence or absence of FcγRIIB affects BP under standard conditions and also the potential role of FcγRIIB in HFD-induced hypertension. Tail-cuff measurements can be used to accurately evaluate SBP in conscious, unanesthetized mice.21 SBP was similar in control chow (CON)–fed FcγRIIB+/+ and FcγRIIB−/− mice (Figure 3A). In FcγRIIB+/+ mice, SBP was 10 mm Hg greater in HFD-fed versus CON-fed mice. The increase in SBP was similar in degree to that previously reported in HFD-fed versus CON-fed mice.19,20 In contrast, in FcγRIIB−/− mice, SBP was similar in HFD-fed and CON-fed animals.
Figure 3.

Fcγ receptor IIB (FcγRIIB) is required for obesity-induced systolic and diastolic hypertension. Tail-cuff systolic blood pressure (SBP) was measured in conscious, restrained FcγRIIB+/+ and FcγRIIB−/− mice after control chow (CON) or high-fat diet (HFD) feeding for 16 weeks. The mice were acclimated to the BP monitor for 4 consecutive days before data were collected on the fifth day. Values are mean±SEM (n=12–13). B to F, A separate group of mice underwent carotid artery cannulation with a radiotelemetry device for measurement of SBP (B), diastolic blood pressure (DBP; C), mean arterial pressure (MAP; D), and heart rate (HR; E) after being on their respective diets for 12 weeks. In B to F, values are mean±SEM (n=4–7). In 2-way ANOVA analysis, cuff SBP and heart rate were significant for the effect of diet (P<0.05). More importantly, DBP and MAP had a significant interaction of diet and genotype (P<0.05). The effect of genotype was not significant in any comparison (P>0.05). In post-test analysis, *P<0.05 vs CON diet and †P<0.05 vs FcγRIIB+/+ HFD mice.
To confirm the findings for SBP obtained by tail cuff, and to also evaluate DBP, which is not accurately evaluated by cuff devices,22 further experiments were performed using radiotelemetry. SBP by radiotelemetry was increased by 13 mm Hg in HFD-fed compared with CON-fed FcγRIIB+/+ mice, but statistical significance was not achieved (P=0.06 for diet in 2-way analysis; Figure 3B). In contrast, diet-induced obesity did not alter SBP in FcγRIIB−/− mice. Diastolic BP was elevated 9 mm Hg by HFD in FcγRIIB+/+ mice but not in FcγRIIB−/− mice (P<0.05 for interaction in 2-way ANOVA; Figure 3C). MAP in the 4 experimental groups displayed the same pattern, increased 11 mm Hg by HFD in wild-type mice but not in receptor null mice (P<0.05 for interaction; Figure 3D). Heart rate was significantly increased by HFD (HFD versus CON; P<0.05; 2-way ANOVA), but there was no effect of genotype (P>0.05; Figure 3E).
Additional analyses were performed to determine whether the impact of diet-induced obesity on BP, or the impact of FcγRIIB genotype on the BP response to obesity, occurs primarily during daytime or during nighttime, which represent periods of low versus high activity, respectively. Figure 4 shows the measurements during light hours (open bars) and dark hours (filled bars). In all comparisons, BP was increased at night compared with day as expected. SBP was increased during the day in FcγRIIB+/+ HFD-fed mice (Figure 4A), and in contrast there was no impact of HFD on SBP in FcγRIIB−/− mice, either during daytime or during nighttime. Diastolic BP and MAP (Figure 4B and 4C) were raised by HFD in FcγRIIB+/+ mice both during daytime and during nighttime, whereas diet had no effect on either of those parameters during either 12-hour period in FcγRIIB−/− mice. Therefore, increased BP caused by HFD is not related to a change in the diurnal BP pattern, and the presence versus absence of FcγRIIB influences obesity-induced hypertension equally throughout the 24-hour day. Activity monitoring confirmed day–night variation in activity in the 4 experimental groups (Figure 4D). There was a trend for mice on HFD to be less active at night compared with CON-fed mice (P=0.06). Collectively these finding reveal that FcγRIIB participates in the pathogenesis of the hypertension that accompanies diet-induced obesity.
Figure 4.

Modulation of blood pressure by high-fat diet (HFD) and Fcγ receptor IIB (FcγRIIB) occur during both light and dark cycles. A to C, Radiotelemetric systolic (A), diastolic (B), and mean arterial pressure (C) measurements in FcγRIIB+/+ and FcγRIIB−/− mice after control chow (CON) or HFD feeding were grouped and averaged by 12-hour light (open bars) and 12-hour dark cycles (filled bars). D, Relative activity levels were also quantified. Values are mean±SEM (n=4–7), *P<0.05 vs CON diet and †P<0.05 vs FcγRIIB+/+ HFD mice.
Diet-Induced Obesity and FcγRIIB Ligands
The known ligands for FcγRIIB are the pentraxins CRP and SAP and the Fc region of IgG, which have shared binding sites on Fc receptors.18 Seeking to identify the FcγRIIB ligand that is responsible for HFD-induced hypertension and recognizing that obesity is a chronic inflammatory state, we hypothesized that ≥1 of these mediators of inflammatory response is elevated in the setting of HFD feeding and resulting obesity in mice. Analyzing serum from FcγRIIB+/+ mice fed CON versus HFD for 12 weeks, we found that there was no change in serum CRP in response to HFD feeding and the obesity that it causes (Figure 5A). CRP was surprisingly lower in HFD-fed versus CON-fed FcγRIIB−/− mice. Serum SAP also did not change in response to HFD in FcγRIIB+/+ mice (Figure 5B). However, serum SAP rose modestly with HFD feeding in FcγRIIB−/− mice. Total IgG additionally was similar in CON-fed versus HFD-fed FcγRIIB+/+ mice (Figure 5C). Likely related to the lack of the inhibitory Fc receptor in B cells in the FcγRIIB−/− mice, the null mice had marked elevations in total IgG compared with wild-type mice; however, the elevations in total IgG in FcγRIIB−/− were similar in CON-fed versus HFD-fed mice. Thus, there is not a demonstrable change in the circulating level of a known Fcγ receptor ligand in the setting of HFD feeding and resulting obesity in mice.
Figure 5.
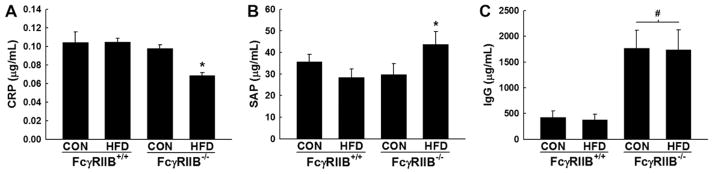
Impact of high-fat diet (HFD)–induced obesity on serum C-reactive protein (CRP), serum amyloid P component (SAP), and total IgG. Concentrations of CRP (A), SAP (B), and total IgG (C) were measured by ELISA in serum obtained from Fcγ receptor IIB (FcγRIIB)+/+ and FcγRIIB−/− mice after control chow (CON) or HFD feeding for 12 weeks. Values are mean±SEM (n=8–12), *P<0.05 vs CON diet, #P<0.05 vs FcγRIIB+/+.
FcγRIIB Expression
Possible effects of HFD on receptor expression were evaluated by fluorescence-activated cell sorting analysis in endothelial cells, in which we previously demonstrated their activation antagonizes eNOS,14 and in B cells in which FcγRIIB attenuates B-cell receptor–mediated processes.23 In both endothelial cells and B cells, FcγRIIB expression was similar in cells obtained from CON versus HFD-fed mice (Figure S1 in the online-only Data Supplement). Thus, diet-induced obesity does not affect FcγRIIB expression, particularly in endothelium in which the receptor is known to alter vascular function.
Endothelial Actions of IgG
Observing that circulating levels of CRP, SAP, and IgG, and also FcγRIIB expression were not altered by HFD feeding, possible changes in IgG function were evaluated by studying IgG effects on eNOS in cultured endothelial cells. Treatment with IgG from CON-fed mice did not alter vascular endothelial growth factor activation of eNOS; in contrast, HFD IgG caused marked attenuation of eNOS activation (Figure 6A). To determine whether the actions of HFD IgG are mediated by FcγRIIB, the receptor was silenced by small interfering RNA (Figure 6B). Whereas HFD IgG blunted eNOS activation in control small interfering RNA–transfected cells, it had no effect in FcγRIIB-depleted endothelial cells (Figure 6C). Thus, diet-induced obesity yields IgG that antagonizes eNOS via FcγRIIB.
Figure 6.
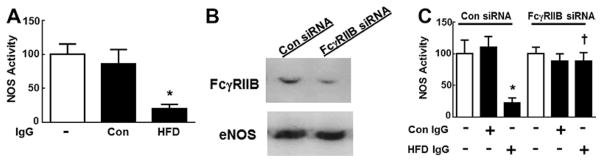
IgG from high-fat diet (HFD)–fed mice antagonizes vascular endothelial growth factor (VEGF) activation of endothelial NO synthase (eNOS) via Fcγ receptor IIB (FcγRIIB). A, Bovine aortic endothelial cells (BAECs) were incubated with IgG isolated from control chow (CON) versus HFD-fed mice (10 μg/mL), and VEGF (100 ng/mL) activation of eNOS was evaluated by quantifying 14C-L-arginine conversion to 14C-L-citrulline during 15-minute incubations (n=6–12). B, BAECs were transfected with either sham small interfering RNA (siRNA) or siRNA targeting FcγRIIB, and receptor abundance was evaluated by immunoblot analysis. C, The effects of CON vs HFD IgG on VEGF activation of eNOS were compared in cells transfected as in B (n=5). In A and C, values are mean±SEM, *P<0.05 vs CON IgG, †P<0.05 vs Con siRNA.
Discussion
Seeking to better understand the basis for obesity-induced hypertension, we designed experiments in mice to determine the role of FcγRIIB in the pathogenesis of the disorder. First testing whether a known ligand for FcγRIIB alters BP via the receptor, we demonstrated that CRP-induced hypertension in the mouse is fully dependent on FcγRIIB. We then discovered in a model of diet-induced obesity with equal increases in body weight and body fat in wild-type and null mice that whereas obese FcγRIIB+/+ mice developed hypertension, obese FcγRIIB−/− mice were fully protected from hypertension. These observations indicate that independent of any effect on body weight or body composition, FcγRIIB plays a key role in obesity-induced hypertension.
The basis for the CRP-induced hypertension that we now show is mediated by FcγRIIB was elucidated in our previous studies.10 TG-CRP mice displayed exaggerated BP elevation in response to angiotensin II and a reduction in vascular angiotensin II receptor subtype 2 expression. In contrast, the decline in BP with angiotensin receptor subtype 1 antagonism and vascular angiotensin receptor subtype 1 abundance were unaltered in the TG-CRP mice, indicating a selective effect of CRP on vascular angiotensin II receptor subtype 2. Additional experiments showed that the CRP-induced decrease in vascular angiotensin II receptor subtype 2 is caused by NO deficiency, and earlier studies indicated that NO deficiency results from potent antagonism of eNOS by the CRP-FcγRIIB tandem.14 The eNOS antagonism by CRP entails the coupling of FcγRI to FcγRIIB by Src kinase, and the activation of SH2 domain–containing inositol 5′-phosphatase 1, which attenuates signaling downstream of PI3 kinase, thereby preventing Akt activating phosphorylation at Ser473.12,13 Thus, we now know that FcγRIIB activation by CRP initiates a series of alterations in intracellular signaling in endothelial cells, and that the actions of the pentraxin-receptor pair ultimately cause hypertension in mice.
Along with an important role for endothelial dysfunction,24 altered sympathetic nervous system activity may participate in obesity-related hypertension.25 Interestingly, we observed that heart rate is increased in TG-CRP mice and in HFD-fed obese mice, and that whereas FcγRIIB deletion normalizes BP in the setting of either CRP elevation or diet-induced obesity, it has modest, if any, effect on the heart rate elevation. The finding that FcγRIIB participates in the hypertension but not in the tachycardia observed with raised CRP or obesity, with the heart rate changes likely mediated by sympathetic nervous system activation,25 suggests a peripheral (non-CNS, non–sympathetic nervous system) site of action of FcγRIIB. As such, the distinct impact of the receptor on BP versus heart rate is consistent with a role in hypertension for FcγRIIB in endothelium, which is the cell type implicated in our prior studies of the vascular mechanisms of action of CRP.12–14
Observing the parallel requirement for FcγRIIB in CRP- and obesity-induced hypertension, we were then prompted to identify the FcγRIIB ligand that is operative in the obesity-induced disorder. In wild-type mice we found that there is no change in serum CRP in response to HFD feeding and the obesity that it causes. This is consistent with prior reports that CRP is not elevated in mice in response to a HFD.26,27 Recognizing that the acute phase reactant in mice is the highly homologous pentraxin SAP,16 we also assessed whether SAP is altered by HFD in wild-type mice, and found that it is unaffected. Surprisingly, SAP was modestly increased in FcγRIIB−/− mice on HFD. However, SAP levels in healthy mice are between 25 and 60 μg/mL and during an acute inflammatory reaction they reach 250 μg/mL,16,28 and the highest SAP concentration observed in HFD-fed FcγRIIB−/− was only 55 μg/mL. Because neither CRP nor SAP levels were elevated by HFD in association with the hypertension that HFD invokes in wild-type mice, we determined serum concentrations of total IgG as it is also a ligand for FcγRIIB. HFD did not affect total IgG in wild-type mice, and predictably total IgG was increased in FcγRIIB−/− mice because the receptor is expressed in B cells in which it negatively regulates IgG production.29 In mice others have observed either no effect of HFD on IgG or an increase in total IgG.30,31 In humans, total IgG may rise with obesity,32 and it falls after weight loss after gastric banding.33 We additionally evaluated possible effects of HFD on FcγRIIB abundance, and found that expression in both endothelial cells, where the receptor alters vascular function, and B cells was not modified by HFD feeding. Alternatively, we discovered that the IgG obtained from HFD-fed mice potently antagonizes eNOS activation, and furthermore that the eNOS antagonism is entirely FcγRIIB dependent. These findings mirror those we previously made on the role of the receptor in eNOS antagonism by CRP,12 and they are consistent with the prior observation that aortic endothelial cells isolated from HFD-fed mice display decreased activating phosphorylation of eNOS at Ser1176 compared with endothelial cells from control diet–fed mice.24 The particular nature of the IgG generated during diet-induced obesity that antagonizes eNOS can now be interrogated in future studies. One possibility is that the culprit is IgG autoantibodies, which increase with obesity and are thought to link obesity with autoimmune disorders.31
Obesity-related hypertension is a perplexing clinical problem, and efforts to combat the underlying obesity regrettably can often be unsuccessful. We have discovered FcγRIIB as a new mediator of obesity-induced hypertension that participates in the disorder in mechanisms that are unrelated to the control of body weight or body composition. Novel therapeutic approaches targeting FcγRIIB-related processes may offer valuable new means to combat obesity-related hypertension independent of strategies to promote weight loss.
Perspectives
The mechanisms that link obesity and hypertension are incompletely understood. The mainstay of clinical management for obesity-related conditions is to encourage weight loss. However, such efforts are often unsuccessful and therapies that complement weight loss regimens would be useful. Having discovered that FcγRIIB is critically involved in obesity-induced hypertension, approaches targeting FcγRIIB may offer potentially new means to treat hypertension in obese individuals.
Supplementary Material
FACS was performed on primary aortic endothelial cells (A) and spleen-derived B cells (B) from control chow (CON) or high fat diet (HFD) fed mice. Antibodies employed were 2.4G2 to detect FcγRIIB, and anti-CD31 (PECAM-1) or anti-CD45R(B220) antibodies to identify endothelial cells or B cells, respectively. Values are mean±SEM, n=4–8.
Novelty and Significance.
What Is New?
We demonstrate a novel role for the inflammatory receptor, Fcγ receptor IIB (FcγRIIB), in obesity-induced hypertension.
What Is Relevant?
The mainstay of clinical management for obesity-related conditions is to encourage weight loss. However, such efforts are often unsuccessful and therapies that complement weight loss regimens would be useful. Having discovered that FcγRIIB plays an important role in obesity-induced hypertension, approaches targeting FcγRIIB may offer potentially new means to treat hypertension in obese individuals.
Summary
We have discovered FcγRIIB participates in the development of obesity-induced hypertension by mechanisms that are unrelated to adiposity. Therefore, novel therapeutic approaches targeting FcγRIIB-related processes may offer valuable new means to combat obesity-related hypertension independent of strategies to promote weight loss.
Acknowledgments
Sources of Funding
The work was supported by National Institutes of Health grant HL115122 (P.W. Shaul), by the Children’s Medical Center Foundation (N.C. Sundgren), by the Physician-Scientist Training Program at UT Southwestern (N.C. Sundgren), and by the Bad Pants Open Fund (N.C. Sundgren).
Footnotes
The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.114.04670/-/DC1.
Disclosures
None.
References
- 1.Lowe GD. The relationship between infection, inflammation, and cardiovascular disease: an overview. Ann Periodontol. 2001;6:1–8. doi: 10.1902/annals.2001.6.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Cutler JA, Sorlie PD, Wolz M, Thom T, Fields LE, Roccella EJ. Trends in hypertension prevalence, awareness, treatment, and control rates in United States adults between 1988–1994 and 1999–2004. Hypertension. 2008;52:818–827. doi: 10.1161/HYPERTENSIONAHA.108.113357. [DOI] [PubMed] [Google Scholar]
- 3.Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH American Heart Association; Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898–918. doi: 10.1161/CIRCULATIONAHA.106.171016. [DOI] [PubMed] [Google Scholar]
- 4.Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults--the evidence report. National Institutes of Health. Obes Res. 1998;6(suppl 2):51S–209S. [PubMed] [Google Scholar]
- 5.Gupta AK, Nasothimiou EG, Chang CL, Sever PS, Dahlöf B, Poulter NR ASCOT Investigators. Baseline predictors of resistant hypertension in the Anglo-Scandinavian Cardiac Outcome Trial (ASCOT): a risk score to identify those at high-risk. J Hypertens. 2011;29:2004–2013. doi: 10.1097/HJH.0b013e32834a8a42. [DOI] [PubMed] [Google Scholar]
- 6.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Low-grade systemic inflammation in overweight children. Pediatrics. 2001;107:E13. doi: 10.1542/peds.107.1.e13. [DOI] [PubMed] [Google Scholar]
- 7.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 8.Niskanen L, Laaksonen DE, Nyyssönen K, Punnonen K, Valkonen VP, Fuentes R, Tuomainen TP, Salonen R, Salonen JT. Inflammation, abdominal obesity, and smoking as predictors of hypertension. Hypertension. 2004;44:859–865. doi: 10.1161/01.HYP.0000146691.51307.84. [DOI] [PubMed] [Google Scholar]
- 9.Virdis A, Ghiadoni L, Plantinga Y, Taddei S, Salvetti A. C-reactive protein and hypertension: is there a causal relationship? Curr Pharm Des. 2007;13:1693–1698. doi: 10.2174/138161207780831365. [DOI] [PubMed] [Google Scholar]
- 10.Vongpatanasin W, Thomas GD, Schwartz R, Cassis LA, Osborne-Lawrence S, Hahner L, Gibson LL, Black S, Samols D, Shaul PW. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation. 2007;115:1020–1028. doi: 10.1161/CIRCULATIONAHA.106.664854. [DOI] [PubMed] [Google Scholar]
- 11.Chrostowska M, Szyndler A, Hoffmann M, Narkiewicz K. Impact of obesity on cardiovascular health. Best Pract Res Clin Endocrinol Metab. 2013;27:147–156. doi: 10.1016/j.beem.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Tanigaki K, Mineo C, Yuhanna IS, Chambliss KL, Quon MJ, Bonvini E, Shaul PW. C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of FcgammaRIIB and SHIP-1. Circ Res. 2009;104:1275–1282. doi: 10.1161/CIRCRESAHA.108.192906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sundgren NC, Zhu W, Yuhanna IS, Chambliss KL, Ahmed M, Tanigaki K, Umetani M, Mineo C, Shaul PW. Coupling of Fcγ receptor I to Fcγ receptor IIb by SRC kinase mediates C-reactive protein impairment of endothelial function. Circ Res. 2011;109:1132–1140. doi: 10.1161/CIRCRESAHA.111.254573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mineo C, Gormley AK, Yuhanna IS, Osborne-Lawrence S, Gibson LL, Hahner L, Shohet RV, Black S, Salmon JE, Samols D, Karp DR, Thomas GD, Shaul PW. FcgammaRIIB mediates C-reactive protein inhibition of endothelial NO synthase. Circ Res. 2005;97:1124–1131. doi: 10.1161/01.RES.0000194323.77203.fe. [DOI] [PubMed] [Google Scholar]
- 15.Tanigaki K, Vongpatanasin W, Barrera JA, Atochin DN, Huang PL, Bonvini E, Shaul PW, Mineo C. C-reactive protein causes insulin resistance in mice through Fcγ receptor IIB-mediated inhibition of skeletal muscle glucose delivery. Diabetes. 2013;62:721–731. doi: 10.2337/db12-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pepys MB, Baltz M, Gomer K, Davies AJ, Doenhoff M. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature. 1979;278:259–261. doi: 10.1038/278259a0. [DOI] [PubMed] [Google Scholar]
- 17.Mold C, Baca R, Du Clos TW. Serum amyloid P component and C-reactive protein opsonize apoptotic cells for phagocytosis through Fcgamma receptors. J Autoimmun. 2002;19:147–154. doi: 10.1006/jaut.2002.0615. [DOI] [PubMed] [Google Scholar]
- 18.Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature. 2008;456:989–992. doi: 10.1038/nature07468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, Cassis LA. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension. 2012;60:1524–1530. doi: 10.1161/HYPERTENSIONAHA.112.192690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L, Zhang Y, Cohen SB, DiPetrillo K. TrkB agonist antibody dose-dependently raises blood pressure in mice with diet-induced obesity. Am J Hypertens. 2010;23:732–736. doi: 10.1038/ajh.2010.49. [DOI] [PubMed] [Google Scholar]
- 21.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25:1111–1115. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- 22.Geddes LA. The Direct and Indirect Measurement of Blood Pressure. Chicago: New York Medical Publishers Inc; 1970. pp. 135–170. [Google Scholar]
- 23.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 24.Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. doi: 10.1016/j.cmet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 25.Lohmeier TE, Iliescu R, Liu B, Henegar JR, Maric-Bilkan C, Irwin ED. Systemic and renal-specific sympathoinhibition in obesity hypertension. Hypertension. 2012;59:331–338. doi: 10.1161/HYPERTENSIONAHA.111.185074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br J Nutr. 2009;102:514–519. doi: 10.1017/S000711450820749X. [DOI] [PubMed] [Google Scholar]
- 27.Teupser D, Weber O, Rao TN, Sass K, Thiery J, Fehling HJ. No reduction of atherosclerosis in C-reactive protein (CRP)-deficient mice. J Biol Chem. 2011;286:6272–6279. doi: 10.1074/jbc.M110.161414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pepys MB, Baltz ML, Musallam R, Doenhoff MJ. Serum protein concentrations during Schistosoma mansoni infection in intact and T-cell deprived mice. I. The acute phase proteins, C3 and serum amyloid P-component (SAP) Immunology. 1980;39:249–254. [PMC free article] [PubMed] [Google Scholar]
- 29.Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. 2000;290:84–89. doi: 10.1126/science.290.5489.84. [DOI] [PubMed] [Google Scholar]
- 30.Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arai S, Maehara N, Iwamura Y, et al. Obesity-associated autoantibody production requires AIM to retain the immunoglobulin M immune complex on follicular dendritic cells. Cell Rep. 2013;3:1187–1198. doi: 10.1016/j.celrep.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, Fernandez-Merino C, Vidal C. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol. 2008;151:42–50. doi: 10.1111/j.1365-2249.2007.03545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakçak I, Avşar MF, Hamamci EO, Bostanoğlu S, Sonişik M, Bostanoğlu A, Erdem NZ, Coşgun E. Comparison of early and late changes in immunoglobulins and acute phase reactants after laparoscopic adjustable gastric banding in patients with morbid obesity. Obes Surg. 2010;20:610–615. doi: 10.1007/s11695-009-0061-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FACS was performed on primary aortic endothelial cells (A) and spleen-derived B cells (B) from control chow (CON) or high fat diet (HFD) fed mice. Antibodies employed were 2.4G2 to detect FcγRIIB, and anti-CD31 (PECAM-1) or anti-CD45R(B220) antibodies to identify endothelial cells or B cells, respectively. Values are mean±SEM, n=4–8.