

Abstract
Background
The birth weight of Black neonates in the United States is consistently smaller than that of their White counterparts. Epigenetic differences between the races may be involved in such disparities. The goal of these analyses was to model the role of IGF1 methylation in mediating the association between race and birth weight. Data was collected on a cohort of 87 live born infants. IGF1 methylation was measured in DNA isolated from the mononuclear fraction of umbilical cord blood collected after delivery. Quantitative, loci-specific methylation was assessed using the Infinium HumanMethylation27 BeadArray (Illumina Inc., San Diego, CA). Locus specific methylation of the IGF1 CpG site was validated on a subset of the original sample (N = 61) using pyrosequencing. Multiple linear regression was used to examine relationships between IGF1 methylation, race, and birth weight. A formal mediation analysis was then used to estimate the relationship of IGF1 methylation to race and birth weight.
Results
Black race was associated with a 7.45% decrease in gestational age-adjusted birth weight (aBW) (P = 0.04) and Black infants had significantly higher IGF1 methylation than non-Black infants (P < 0.05). A one standard deviation increase in IGF1 methylation was associated with a 3.32% decrease in aBW (P = 0.02). Including IGF1 methylation as a covariate, the effect of Black race on aBW was attenuated. A formal mediation analysis showed that the controlled direct effect of Black race on aBW was −6.26% (95% CI = −14.15, 1.06); the total effect of Black race on IGF1 methylation was −8.12% (95% CI = −16.08, −0.55); and the natural indirect effect of Black race on aBW through IGF1 methylation was −1.86% (95% CI = −5.22, 0.18)
Conclusion
The results of the mediation analysis along with the multivariable regression analyses suggest that IGF1 methylation may partially mediate the relationship between Black race and aBW. Such epigenetic differences may be involved in racial disparities observed in perinatal outcomes.
Keywords: Birth weight, IGF1, Methylation, Epigenetics, Race, Disparities, Perinatal
Background
Racial differences in birth outcomes and their health consequences remain among the most persistent and vexing disparities in the United States [1]. For example, Black infants have consistently been shown to have smaller birth weights than their White counterparts [1]. Growing evidence suggests that such perinatal factors can impact health across the life course, including an increased risk of higher blood pressure, insulin resistance and diabetes, abnormal cholesterol profiles, pathologic patterns of fat deposition, and an elevated risk of coronary vascular disease later in life [2]. There is growing evidence that epigenetic changes may mediate the effects of the prenatal environments on short- and long-term health outcomes of the offspring [3].
DNA methylation is an important epigenetic mechanism that helps regulate gene expression and can be influenced by both the environment and the genome. DNA methylation has also been linked to some cancers, complex diseases, and transgenerational effects, and may serve as a potential link between the genome, environment, and disease [4-7]. On a population level, DNA methylation profiles differ across African and European ancestral groups [8]. For example, both at birth and in adulthood, African-Americans have lower genome-wide levels of methylation compared with individuals of European ancestry [9,10]. Such differences may contribute to health disparities observed between the two groups across the life course [11,12]. However, surprisingly few studies have explored racial differences in DNA methylation at birth [9].
Among epigenetically regulated genes, the insulin-like growth factors, including IGF1 and IGF2, have particularly important roles in fetal and placental growth [13-17]. Loss of Igf1 expression is associated with decreased fetal growth in mice [13]. Similarly, IUGR in the rat was associated with altered epigenetic characteristics of the Igf1 gene, altered Igf1 expression, and reduced IGF1 levels in liver and blood [16]. Partial deletion of IGF1 in humans has been associated with impaired pre- and postnatal growth [15], while growth-restricted infants have been shown to have lower umbilical cord blood levels of IGF1 compared to their counterparts with normal growth [17].
The well-established effects of IGF1 on birth weight along with persistently reported racial differences in birth weight and in genome-wide methylation studies have led us to examine the role of IGF1 methylation in the pathway between race and birth weight. We hypothesized that IGF1 methylation may partially explain racial differences in birth weight by mediating the association between race and birth weight. Indeed, our analyses demonstrated that IGF1 methylation partially mediates the relationship between Black race and gestational age-adjusted birth weight (aBW). These results suggest that such epigenetic differences may be involved in racial disparities observed in perinatal outcomes.
Results
Ninety mother-infant pairs were eligible for inclusion in this analysis. The analysis is restricted to 87 mother-infant pairs for whom race is known. Demographic and health characteristics of the 87 infants and their mothers are presented in Table 1. Approximately 24% of the mothers were Black and 76% were non-Black. Black and non-Black mothers were similar for several of the demographic and clinical characteristics considered. A majority of the women had adequate prenatal care and took prenatal vitamins. However, Black mothers were slightly younger than non-Black mothers (mean age 25.6 and 29.2 years, respectively; P = 0.02). While there were not any statistically significant differences in gestational age between the two groups, Black infants had a smaller mean gestational age-adjusted birth weight (P = 0.04).
Table 1.
Maternal and infant characteristics of the cohort by race
| Non-Black | Black | ||||
|---|---|---|---|---|---|
| N | % | N | % | P value | |
| Sample Size | 66 | 75.9 | 21 | 24.1 | |
| Adequate prenatal care | |||||
| No | 13 | 19.7 | 6 | 28.6 | 0.38 |
| Yes | 53 | 80.3 | 15 | 71.4 | |
| Parity | |||||
| Multiparous | 43 | 65.2 | 16 | 76.2 | 0.43 |
| Nulliparous | 23 | 34.9 | 5 | 23.8 | |
| Prenatal vitamin use | |||||
| No | 10 | 15.2 | 6 | 28.6 | 0.20 |
| Yes | 56 | 84.9 | 15 | 71.4 | |
| Smoker | |||||
| No | 58 | 87.9 | 18 | 85.7 | 0.72 |
| Yes | 8 | 12.1 | 3 | 14.3 | |
| Infant gender | |||||
| Female | 28 | 42.4 | 11 | 52.4 | 0.46 |
| Male | 38 | 57.6 | 10 | 47.6 | |
| Mean ± SD | Mean ± SD | ||||
| Maternal age (years) | 29.2 ± 6.6 | 25.6 ± 4.6 | 0.02 | ||
| Gestational age (weeks) | 38.0 ± 2.4 | 38.4 ± 1.7 | 0.49 | ||
| aBW (grams) | 3294.2 ± 455.0 | 3064.5 ± 483.3 | 0.04 | ||
| IGF1 methylation | 0.26 ± 0.05 | 0.29 ± 0.06 | 0.03 | ||
Abbr: aBW = gestational age-adjusted birthweight; SD = standard deviation.
As shown in Table 1, Black infants had significantly higher IGF1 methylation when compared to their non-Black counterparts, (P = 0.03). In Table 2, we analyzed IGF1 methylation and birth weight by maternal and infant characteristics. IGF1 methylation did not differ by adequacy of prenatal care, parity, maternal vitamin use, smoking status, or infant gender. In contrast, birth weight and gestational age-adjusted birth weight were associated with both parity and infant gender as expected.
Table 2.
The relationship of IGF1 methylation and birth weight to select maternal and infant characteristics
| IGF1 methylation | BW (grams) | aBW (grams) | |
|---|---|---|---|
| Mean ± SD | Mean ± SD | Mean ± SD | |
| Adequate prenatal care | |||
| No | 0.27 ± 0.06 | 3060.8 ± 773.3 | 3161.4 ± 500.5 |
| Yes | 0.26 ± 0.05 | 3274.6 ± 623.5 | 3260.3 ± 462.2 |
| Parity | |||
| multiparous | 0.27 ± 0.05 | 3381.8 ± 531.6** | 3331.7 ± 451.3** |
| nulliparous | 0.26 ± 0.06 | 2903.8 ± 787.8 | 3042.8 ± 454.1 |
| Prenatal vitamin use | |||
| No | 0.27 ± 0.05 | 3135.3 ± 568.5 | 3183.1 ± 343.9 |
| Yes | 0.26 ± 0.06 | 3248.8 ± 681.1 | 3251.2 ± 494.7 |
| Smoker | |||
| No | 0.27 ± 0.06 | 3245.7 ± 662.4 | 3253.0 ± 437.9 |
| Yes | 0.26 ± 0.05 | 3105.0 ± 662.3 | 3139.7 ± 668.0 |
| Infant gender | |||
| Female | 0.26 ± 0.07 | 2959.2 ± 721.0** | 3090.2 ± 442.6* |
| Male | 0.27 ± 0.04 | 3446.3 ± 518.4 | 3359.4 ± 460.4 |
| Maternal age | |||
| <30 | 0.27 ± 0.06 | 3123.8 ± 758.5 | 3210.7 ± 490.2 |
| ≥30 | 0.26 ± 0.05 | 3382.6 ± 445.4 | 3280.3 ± 440.9 |
Abbr: BW = birth weight; aBW = gestational age-adjusted birth weight; SD = standard deviation.
*P < 0.05.
**P < 0.01.
We modeled the relationships between race, IGF1 methylation, and gestational age-adjusted birth weight using multivariable linear regression. Figure 1 shows the overall relationship of aBW to IGF1 methylation depicting the individual values by race. Using regression models, IGF1 methylation may be considered as a potential mediator of the relationship between race and aBW if several criteria are satisfied [18]: (a) there is a significant relationship between the independent variable (race) and the mediator (IGF1 methylation); (b) there is a significant relationship between the mediator (IGF1 methylation) and the outcome (aBW); (c) the independent variable (race) significantly affects the outcome (aBW); and (d) the effect of the previously significant relationship between the independent variable (race) and the outcome (aBW) is no longer significant when controlled for the mediator (IGF1 methylation).
Figure 1.
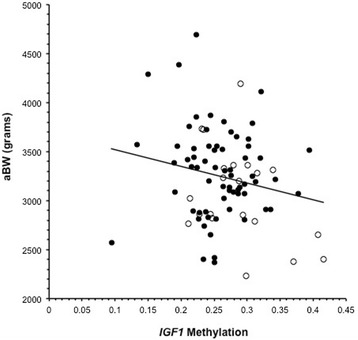
The relationship between gestational age-adjusted birth weight (aBW) and IGF1 methylation. The open circles represent Blacks and the closed circles represent non-Blacks.
As shown in Table 3, Black race is associated with a 7.45% decrease in aBW (P = 0.04) after adjustment for maternal age, parity, and infant gender, and a 0.62 unit standard deviation increase in methylation of IGF1 (P = 0.02) after adjusting for maternal age. A one standard deviation increase in IGF1 methylation was associated with a 3.32% decrease in aBW (P = 0.02). The effect of Black race on aBW was attenuated and no longer significant (β = −5.68%, 95% CI = −12.84, 1.49) after including IGF1 methylation as a covariate to the model. Thus, these results suggest that IGF1 methylation may be a potential mediator of the relationship between race and aBW [18].
Table 3.
The relationships between race, IGF1 methylation, and gestational age-adjusted birth weight 1
| IGF1 Methylation 2 | % change in aBW 3 | |||||
|---|---|---|---|---|---|---|
| β | 95% CI | P value | β | 95% CI | P value | |
| Race | ||||||
| Black race | 0.62 | 0.11, 1.12 | 0.02 | −7.45% | −14.44, −0.45 | 0.04 |
| IGF1 methylation | ||||||
| Methylation z-score | - | - | - | −3.32% | −6.17, −0.47 | 0.02 |
| Joint effects | ||||||
| Black race | - | - | - | −5.68% | −12.84, 1.49 | 0.12 |
| Methylation z-score | - | - | - | −2.70% | −5.63, 0.24 | 0.07 |
1Modeled using standard multivariable linear regression; 2 IGF1 methylation z-score controlled for maternal age; 3gestational age-adjusted birth weight (aBW) controlled for maternal age, parity, and infant gender.
As such, we used mediation analysis (see ‘Methods’) to formally test the hypothesis that IGF1 methylation mediates the relationship between race and aBW as shown in Figure 2 [18]. These analyses were controlled for maternal age, parity, and infant gender. In this model, the total effect (Figure 2A) relating race and aBW was −8.12% (95% CI = −16.08, −0.55). The controlled direct effect relating race to aBW after adjusting for IGF1 methylation status (Figure 2B) was −6.26% (95% CI = −14.15, 1.06). Using these results, the natural indirect effect was −1.86% (95% CI = −5.22, 0.18). The smaller direct effect (−6.26%) compared to the total effect (−8.12%) suggests that IGF1 methylation partially mediates the relationship between Black race and aBW and that the mediation model approaches significance.
Figure 2.
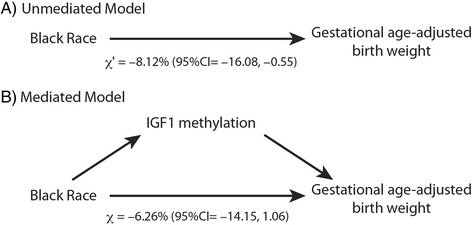
Models for mediation analysis for the relationship of Black race, IGF1 methylation and gestational age-adjusted birth weight. (A) The unmediated model used to calculate the total effect. (B) The mediated model used to calculate the controlled direct effect. The calculated regression coefficients for the total effect, χ’, and the controlled direct effect, χ, are given.
Discussion
The primary aim of these analyses was to model the role of IGF1 methylation as a mediator of the relationship between race and birth weight. It is well established that Black infants have lower mean birth weights than other racial groups in the United States, even after controlling for gestational age. Our data are consistent with these prior observations. It has been thought that epigenetic changes may mediate the effects of adverse birth outcomes on health outcomes of the offspring. Building on these observations, we found that Black infants had higher IGF1 methylation than their non-Black counterparts and that IGF1 methylation was significantly associated with birth weight (Table 3). A formal mediation analysis (Figure 2) suggests that methylation of the IGF1 gene partially mediates the association between Black race and aBW. Moreover, the magnitude of the indirect effect is moderate in size.
The underlying reason for racial differences in DNA methylation levels, specifically at the IGF1 locus, at birth is not known. Such methylation differences may reflect variations in the intrauterine environment, including variation by race in maternal or fetal metabolism, genomic signals for methylation, or environmental exposures. Methylation differences by race may also be related to race-specific variation in the distribution of cell types in the mononuclear fraction of cord blood [19]. Differentiating these factors is beyond the scope of our current study. Irrespective of the mechanism, we hypothesize that DNA methylation differences between races may be related to variation in gene expression by race that influence birth weight and, ultimately, risk of disease across the life course.
Consistent with many prior studies suggesting a relationship between variation in IGF1 expression and birth weight [15,17], our analyses suggest that the higher level of methylation of the IGF1 was significantly associated with a decrease in aBW (Table 3). While we did not measure IGF1 expression per se, the assessed methylation site was near the transcription start site and might be theoretically expected to result in altered expression. It is important to note that IGF1 is regulated by two promoters and multiple transcription start sites [20]. The complex structure of IGF1 makes specific inferences regarding gene expression difficult. Thus additional studies are needed to evaluate how differences in methylation at this and other nearby CpG sites may differentially influence IGF1 expression generally and by race.
Finally, our analyses suggest that the higher level of methylation of the IGF1 locus in Black infants accounts for at least part of the disparity in infant birth weight. Most prior research in this area focuses on relations between the two variables considered separately, and much has been written about the relationship between either race or methylation and their effects on birth weight. Our work is the first to formally model the mediation of the relation between race and birth weight by locus specific methylation.
In our model, we specifically hypothesize that IGF1 methylation is in a causal sequence between race and birth weight. The statistical power to detect mediated effects using these models is typically low [21]. However, while our sample size may not be sufficiently large to achieve statistical significance, analysis of the causal steps in the process using regression models and the formal mediation model suggests that IGF1 methylation partially mediates the relationship between Black race and aBW [22,23]. That is to say, based on established criteria, partial mediation is likely to be present given the following: the total effect of race on aBW is significant; the effect of race on IGF1 methylation is significant; the effect of IGF1 methylation on aBW controlled for race is significant; and, most importantly, the direct effect of race on aBW adjusted for IGF1 methylation is non-significant and smaller than the total effect. The finding of partial mediation implies that other indirect effects are also likely to have a role in mediating racial differences in birth weight.
Conclusions
Racial disparities in birth outcomes remain one of the most vexing public health problems in the United States. Our analyses suggest that IGF1 methylation partially mediates observed racial disparities in birth weight. Our results add to emerging evidence that epigenetic profiles appear to differ across racial groups [10,24,25] and that these differences may contribute to phenotypic differences, such as discordant birth weights. However, birth weight is a complex phenotype influenced by many factors including maternal characteristics, environmental exposures, psychosocial stressors, and infant characteristics. Thus, variation in size at birth results from interaction between maternal genetic factors, fetal genetic factors, the maternal (external) environment, and the intrauterine environment. There may also be effects associated with maternal and paternally derived imprinted fetal genes. While our study did not measure many of these distal factors, our results suggest that they are likely to have a complementary role in mediating racial differences in birth weight. However, differences in DNA methylation and their influence on fetal growth may also represent the end result of the interaction among many of these factors. A more comprehensive examination of the role of epigenetics in the context of these other factors and their influence on racial disparities in perinatal outcomes is warranted.
Methods
Study sample and data collection
Data and biological samples were collected as part of a larger study at Tampa General Hospital in Tampa, Florida. The University of South Florida Institutional Review Board approved this study. All infants born at Tampa General Hospital were initially eligible for inclusion into the study; however, infants born to women whose prenatal tests indicated that they were HIV or Hepatitis B positive were excluded. Samples used for this analysis were restricted to live born, singleton infants without known birth defects. Demographic and clinical variables were abstracted from the medical record using standardized forms as part of the parent study. These variables included the following: gestational age (based on clinical estimate and the date of last menstrual period), infant birth weight, infant gender, pregnancy complications, vitamin use, presence of birth defects, plurality, parity, gravidity, prenatal care usage (adequate or not as recorded in the medical record), maternal age, and race (classified as Black or non-Black). Umbilical cord blood samples were collected after delivery into standard EDTA collection tubes. The mononuclear fraction was isolated via Ficoll-Paque density gradient centrifugation within 24 hours of sample collection. After separation, the mononuclear layer was suspended in fetal bovine serum and 10% DMSO and stored at −80°C until analysis.
DNA isolation and methylation analysis
DNA isolation and methylation assessment were conducted by the Wayne State University Applied Genomics Technology Center. DNA was isolated from the mononuclear fraction of umbilical cord blood using the Qiagen EZ1 DNA tissue kit (Qiagen, Valencia, CA, USA) according to the method of Lum et al. with the exception that PBS was substituted for TE buffer [26]. Bisulfite modified DNA was prepared using the EZ-96 DNA Methylation Kit™ (Zymo Research Corp., Irvine, CA, USA) according to the manufacturer’s instructions. Quantitative, loci-specific methylation was assessed using the Infinium HumanMethylation27 BeadArray (Illumina Inc., San Diego, CA, USA) per the manufacturer’s instructions.
The array interrogates 27,578 loci located in more than 14,000 genes. For each CpG (cytosine-guanine dinucleotide) site, two different probes (one against the methylated site and one against the unmethylated site) were hybridized with the bisulfite-modified DNA. Next, a single-base extension added one of two possible fluorescent probes (one for methylated (C) and one for unmethylated (T) alleles). Methylation status was then represented by a beta value which is calculated from the ratio of fluorescent signals from methylated to the sum of methylated and unmethylated probes and ranges from 0 (unmethylated) to 1 (methylated). Background normalization was done using the GenomeStudio Methylation module according to the guidelines recommended by Illumina. In short, this method subtracts the average signal of the negative control bead types from the probe signals. Normalized beta values were then output for use in subsequent analyses. In this study, our a priori hypotheses focused on the methylation status of IGF1; therefore, we analyzed one CpG locus associated with this gene. The nucleotide position of the IGF1 CpG locus was chr12:101,398,416 according to the NCBI build 36.1.
A subset of samples was run in duplicate in order to assess inter-chip variability. In addition, CpGenome Universal Methylated DNA was used as a positive control (Millipore, Temecula, CA, USA) and was bisulfite treated and run with the methylation assay. The positive control was used to ensure bisulfite conversion and accuracy of methylation measurement. The positive control DNA was almost completely methylated as expected. Inter-chip variability was assessed and was found to be highly reproducible. Pearson correlation coefficients were greater than 0.99 for each set of replicates (P < 0.0001). Internal validity was assessed by examining gender specific methylation of six X-linked housekeeping genes (EFNB1, ELK1, FMR1, G6PD, GPC3, GLA) [27,28]. Overall, methylation of the six aforementioned housekeeping genes was as expected in that females exhibited hemimethylation and males had very little methylation at the loci in these genes (P < 0.0001 for each gene).
Locus-specific methylation of the IGF1 CpG site was validated on a subset of the original sample (N = 61) using pyrosequencing. EpigenDX (Worcester, MA, USA) designed and conducted the pyrosequencing assay according to manufacturer’s instructions. The correlation between the two measures of methylation (pyrosequencing and the Infinium HumanMethylation27 BeadArray) was evaluated using Pearson’s correlation coefficient. The two measures of methylation were significantly correlated (r = 0.36, P = 0.005).
Statistical analyses
SAS version 9.2 (SAS Institute, Cary, NC, USA) was used to perform all analyses. All hypothesis tests were two-tailed with a type 1 error rate fixed at 5%. Demographic and health characteristics were compared using Fisher’s exact test, t-tests, and the Wilcoxon-Mann–Whitney test as appropriate.
Multiple linear regression was used to examine DNA methylation as a mediator of the association between race and birth weight. Thus, the main outcome of interest was birth weight. Since birth weight varies significantly with gestational age, birth weight was regressed onto gestational age. The residual values from each fit were added to the mean birth weight and used to represent the gestational aBW. The gestational age-adjusted birth weight variable did not initially meet all the assumptions of linear regression; therefore, it was log transformed to achieve normality. The log-transformed aBW was ultimately used as the main outcome variable and is interpreted as the percent change (100*coefficient) in aBW for a one-unit increase in the dependent variable. A secondary outcome, methylation of IGF1 was also examined in relation to race. In order to simplify interpretation, the Illumina beta values for IGF1 methylation were converted to z-scores. Potential confounders were included in the adjusted models if the point estimate changed by more than 10% after adjusting for the confounder.
Mediation analysis was conducted using the methods and macro developed by Valeri and VanderWeele [18]. This framework allowed for the decomposition of a total effect into direct and indirect effects, so that we could statistically test mediation of the race-aBW relationship by IGF1 methylation. This method uses a model based on two regression equations representing relationships given in Figure 2B:
| 1 |
| 2 |
where Y is the dependent variable (log-transformed gestational age-adjusted birth weight), x is the independent variable (race), M is the mediator (IGF1 methylation status), y0 and y1 are intercepts, e0 and e1 are the corresponding residuals, and c are covariates. In this model, χ is defined as the controlled direct effect relating the independent variable (race) to the dependent variable (log(aBW)) adjusted for the mediator (IGF1 methylation status) and relevant covariates. The natural indirect effect is the product α × β that is used to test the statistical significance of the mediation model.
An important property of the natural indirect effect and the controlled direct effect is that they can be related to the total effect (Figure 2A), defined as
| 3 |
which relates the independent variable (race) and the dependent variable (log(aBW)), such that
α × β is equivalent to χ ’ − χ (the difference between the total effect and the controlled direct effect).
Acknowledgements
We thank Hamisu Salihu, Kathleen O’Rourke, Erin M. Siegel, Getachew Dagne, Terry Ashmeade, Phil Marty, Jane Carver, Morna Dorsey, Tina Pastoor, Maja Okuka, Celso Silva, Lewis Rubin, Xiaoming Gong, and Doris Wiener for their support and assistance with the collection of the data used in this analysis. This work was supported by a grant from the US Centers for Disease Control to JKS (Grant number 1R36DP001898-01) and a postdoctoral fellowship from Wayne State University to JKS.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JS was responsible for the conception and design of the study, the acquisition of data, methylation analyses, epidemiological modeling, statistical analyses, and interpretation of data. LS carried out the pyrosequencing analyses. MU participated in the interpretation of analyses for methylation data. DPM was involved in epidemiological models, statistical analyses, and interpretation of data. VKM was involved in the interpretation of data. All authors have been involved in drafting the manuscript or revising it critically for important intellectual content. All authors have given final approval of the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Contributor Information
Jennifer K Straughen, Email: jstraug1@hfhs.org.
Levent Sipahi, Email: lsipahi@med.wayne.edu.
Monica Uddin, Email: muddin@illinois.edu.
Dawn P Misra, Email: dmisra@med.wayne.edu.
Vinod K Misra, Email: vmisra@med.wayne.edu.
References
- 1.Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Munson ML. Births: final data for 2003. Natl Vital Stat Rep. 2005;54(2):1–116. [PubMed] [Google Scholar]
- 2.Kuzawa CW, Sweet E. Epigenetics and the embodiment of race: developmental origins of US racial disparities in cardiovascular health. Am J Hum Biol. 2009;21(1):2–15. doi: 10.1002/ajhb.20822. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Chen Q, Tsai HJ, Wang G, Hong X, Zhou Y, et al. Maternal preconception body mass index and offspring cord blood DNA methylation: exploration of early life origins of disease. Environ Mol Mutagen. 2014;55(3):223–30. doi: 10.1002/em.21827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrow TM, Michels KB. Epigenetic epidemiology of cancer. Biochem Biophys Res Commun. 2014;455(1–2):70–83. doi: 10.1016/j.bbrc.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Langevin SM, Kelsey KT. The fate is not always written in the genes: epigenomics in epidemiologic studies. Environ Mol Mutagen. 2013;54(7):533–41. doi: 10.1002/em.21762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rissman EF, Adli M. Minireview: transgenerational epigenetic inheritance: focus on endocrine disrupting compounds. Endocrinology. 2014;155(8):2770–80. doi: 10.1210/en.2014-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakulski KM, Fallin MD. Epigenetic epidemiology: promises for public health research. Environ Mol Mutagen. 2014;55(3):171–83. doi: 10.1002/em.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraser HB, Lam LL, Neumann SM, Kobor MS. Population-specificity of human DNA methylation. Genome Biol. 2012;13(2):R8. doi: 10.1186/gb-2012-13-2-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adkins RM, Krushkal J, Tylavsky FA, Thomas F. Racial differences in gene-specific DNA methylation levels are present at birth. Birth Defects Res A Clin Mol Teratol. 2011;91(8):728–36. doi: 10.1002/bdra.20770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K, et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics. 2011;6(5):623–9. doi: 10.4161/epi.6.5.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olden K, Lin YS, Gruber D, Sonawane B. Epigenome: biosensor of cumulative exposure to chemical and nonchemical stressors related to environmental justice. Am J Public Health. 2014;104(10):1816–21. doi: 10.2105/AJPH.2014.302130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohammed SI, Springfield S, Das R. Role of epigenetics in cancer health disparities. Methods Mol Biol. 2012;863:395–410. doi: 10.1007/978-1-61779-612-8_25. [DOI] [PubMed] [Google Scholar]
- 13.Fowden AL. The insulin-like growth factors and feto-placental growth. Placenta. 2003;24(8–9):803–12. doi: 10.1016/S0143-4004(03)00080-8. [DOI] [PubMed] [Google Scholar]
- 14.Morison IM, Reeve AE. Insulin-like growth factor 2 and overgrowth: molecular biology and clinical implications. Mol Med Today. 1998;4(3):110–5. doi: 10.1016/S1357-4310(97)01197-0. [DOI] [PubMed] [Google Scholar]
- 15.Woods KA, Camacho-Hubner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335(18):1363–7. doi: 10.1056/NEJM199610313351805. [DOI] [PubMed] [Google Scholar]
- 16.Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA. Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. Faseb J. 2009;23(8):2438–49. doi: 10.1096/fj.08-124768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martos-Moreno GA, Barrios V, Saenz De Pipaon M, Pozo J, Dorronsoro I, Martinez-Biarge M, et al. Influence of prematurity and growth restriction on the adipokine profile, IGF1, and ghrelin levels in cord blood: relationship with glucose metabolism. Eur J Endocrinol Eur Federation Endocrine Soc. 2009;161(3):381–9. [DOI] [PubMed]
- 18.Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods. 2013;18(2):137–50. doi: 10.1037/a0031034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir G, et al. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS One. 2012;7(10):1–9. doi: 10.1371/journal.pone.0046705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adamo ML. Regulation of insulin-like growth factor I gene expression: implications for normal and pathological growth. Diabetes Rev. 1995;3(1):2–27. [Google Scholar]
- 21.MacKinnon DP, Fairchild AJ, Fritz MS. Mediation analysis. Annu Rev Psychol. 2007;58:593–614. doi: 10.1146/annurev.psych.58.110405.085542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–55. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 23.Fritz MS, Mackinnon DP. Required sample size to detect the mediated effect. Psychol Sci. 2007;18(3):233–9. doi: 10.1111/j.1467-9280.2007.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Axume J, Smith SS, Pogribny IP, Moriarty DJ, Caudill MA. Global leukocyte DNA methylation is similar in African American and Caucasian women under conditions of controlled folate intake. Epigenetics. 2007;2(1):66–8. doi: 10.4161/epi.2.1.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terry MB, Ferris JS, Pilsner R, Flom JD, Tehranifar P, Santella RM, et al. Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer Epidemiol Biomarkers Prev. 2008;17(9):2306–10. doi: 10.1158/1055-9965.EPI-08-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lum A, Le Marchand L. A simple mouthwash method for obtaining genomic DNA in molecular epidemiological studies. Cancer Epidemiol Biomarkers Prev. 1998;7(8):719–24. [PubMed] [Google Scholar]
- 27.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16(3):383–93. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–4. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]