

Abstract
Structure-based design, synthesis, biological evaluation and X-ray structural studies of fluorine containing HIV-1 protease inhibitors are described. The synthesis of both enantiomers of the gem-difluoro-bis-THF ligands was carried out in a stereoselective manner using a Reformatskii-Claisen reaction as the key step. Optically active ligands HIV-1LAI were converted to protease inhibitors. Two of these inhibitors (3 and 4) exhibited HIV-1 protease inhibitory Ki’s in picomolar range. Both inhibitors showed very potent antiviral activity with EC50 values of 0.8 nM and 3.1 nM respectively against the laboratory strain HIV-1LAI. Both inhibitors exhibited improved lipophilicity profiles compared to darunavir. Also, both inhibitors showed much improved blood-brain-barrier permeability in an in vitro model. A high resolution X-ray structure of inhibitor 4-bound HIV-1 protease was determined. The X-ray structure revealed that fluoro ligand makes extensive interactions with the HIV-1 protease S2 subsite, including hydrogen-bonding interactions with the protease backbone atoms. Also, both fluorine atoms on the bis-THF ligand formed strong interactions with the flap Gly48 carbonyl oxygen.
Keywords: HIV-1 protease inhibitor, multidrug-resistant HIV-1 strains, blood-brain-barrier, fluoro-bis-THF, ligands, X-ray structure
Introduction
The introduction of fluorine in bioactive molecules is an important strategy in medicinal chemistry.[1,2] Fluorine can improve metabolic stability, membrane permeation and protein-ligand interactions. [3,4] There are numerous approved drugs that contain fluorine atoms. In our continuing interest in the design and synthesis of nonpeptide HIV-1 protease inhibitors with clinical potential, we reported a number of exceptionally potent HIV-1 protease inhibitors that incorporated a variety of novel ligands and scaffolds targeting the active site HIV-1 protease backbones. [5,6] This molecular design strategy resulted in inhibitors that maintain robust activity against HIV-1 variants resistant to the currently approved HIV-1 protease inhibitors. One of these inhibitors is darunavir (1, DRV, Figure 1), which has been used clinically world-wide as a first-line therapy for the treatment of HIV/AIDS.[7–9] In darunavir, we incorporated a structure-based designed and stereochemically defined P2-ligand, 3(R), 3a(S), 6a(R)-bis-tetrahydrofuranylurethane (bis-THF) which forms a number of strong hydrogen bonds, through ligand oxygens, with the backbone atoms of the active site of HIV-1 protease. Furthermore, the bicyclic ligand effectively fills the hydrophobic pocket in the S2 subsite.[10–11]
Figure 1.

Structure of darunavir (1) and PIs 2–4.
Based upon the X-ray structural insight of the molecular interactions of bis-THF, we subsequently planned to investigate the effect of fluorine atoms at the C4 position of bis-THF ring carbon.[12] In particular, we speculated that fluorine would increase lipophilicity which in turn may improve drug penetration in the central nervous system (CNS). Since the CNS is a major sanctuary for HIV-1 infection, improving drug concentration in the CNS may be an important strategy to reduce HIV-1 associated dementia and other CNS-related disorders.[13,14] With prolonged patient survival, HIV-1 associated neurocognitive disorders (HAND) are increasing, possibly due to poor CNS penetration of current anti-HIV therapies.[15] Furthermore, subtherapeutic drug concentration in CNS may also play a role in the development of viral resistance.[16] While combined antiretroviral therapy (cART) has been effective in reducing morbidity and mortality of HIV/AIDS patients, at present, there appears to be no cure or eradication of HIV readily possible, in part due to viral reservoirs in tissues and CNS.
In our structure-based design strategies, we plan to preserve key backbone hydrogen-bonding interactions through bis-THF ring oxygens. We also presume that our structure-based fluorine substitution may improve molecular binding properties in the HIV-1 protease active site through noncovalent interactions involving fluorine.[17,18] This may result in potent antiviral activity against multi-drug resistant HIV-1 variants. Herein, we report an enantioselective synthesis of gem-difluoro-bis-THF ligands and their conversion to gem-difluoro-bis-THF containing HIV-1 protease inhibitors resembling darunavir. We have also carried out X-ray structural studies of inhibitor-bound HIV-1 protease and the biological evaluation of inhibitors.
Synthesis of ligands and inhibitors
The synthesis of stereochemically defined gem-difluoro derivatives of bis-THF is shown in Scheme 1. As shown, DIBAL-H reduction of optically active methyl ester 5 provided dibenzyl-L-glyceraldehyde.[19] Horner-Emmons reaction of the resulting aldehyde with NaH and triethyl phosphonoacetate afforded α,β-unsaturated ester 6 in 88% yield over two-steps. It is important to note that the crude dibenzyl-L-glyceraldehyde be employed immediately to Horner-Emmons reaction without purification to avoid racemization of the aldehyde. Reduction of α,β-unsaturated ester 6 with DIBAL-H in CH2Cl2 at −78 °C provided the corresponding allylic alcohol.[20] Treatment of the resulting alcohol with chlorodifluoroacetic acid in CHCl3 at reflux afforded difluoro acetate derivative 7 in 90% yield over 2 steps. Optical rotation of ester 6 ([α]D23 −15.4 (c 1.08, CHCl3)) was comparable to the reported value for the corresponding enantiomeric ester (lit. [20] [α]-D23 +13.4 (c 1.75, CHCl3)). Ester 7 was then exposed to Reformatskii-Claisen reaction[21,22] by treatment with TMSCl and activated Zn-dust in CH3CN at reflux for 24 h. Subsequent esterification of the crude acid product with a catalytic amount of concentrated H2SO4 in ethanol at 50 °C furnished a 2:1 mixture (by 1H-NMR analysis) of diastereomers 8 in 80% yield over 2-steps. This mixture could not be separated by silica gel column chromatography. However, the mixture can be separated after its conversion to the Weinreb amides. Thus, reaction of the mixture 8 with HN(Me)OMe.HCl and n-BuLi in THF at −78 °C for 3 h provided the corresponding mixture of Weinreb amides. These amide diastereomers were separated by silica gel chromatography to provide syn-diastereomer 9a as the major product and anti-diastereomer 9b as the minor product in a 2:1 ratio and 80% combined yield. The assignment of stereochemistry was carried out based upon reported 1H-NMR data for the corresponding enantiomer.[20]
Scheme 1.

Reformatskii-Claisen route to gem-difluoro derivatives 9a,b.
Both syn-diastereomer 9a and anti-diastereomer 9b were converted to respective gem-difluoro derivatives of bis-THF as shown in Scheme 2. Reduction of Weinreb amide 9a was carried out with LAH in THF at −78 °C and the resulting crude aldehyde was reduced with the addition of NaBH4 in a one-pot operation to provide difluoro alcohol 10 in near quantitative yield. Ozonolysis of the olefin in 10 followed by reductive cleavage with triphenylphosphine provided crude cyclic acetal 11, upon cyclization. The crude cyclic acetal was purified in a short silica gel column using CH2Cl2 as the eluent. The resulting cyclic acetal was then subjected to catalytic hydrogenation over Pearlman’s catalyst under a hydrogen-filled balloon in ethyl acetate to furnish the corresponding triol. Treatment of the resulting triol with catalytic amount of camphorsulfonic acid (CSA) in CH2Cl2 at 23 °C for 12 h afforded optically active difluoro-bis-THF 12 in 68% yield over 3 steps. Anti-diastereomer 9b was converted to its corresponding enantiomeric ligand. Weinreb amide 9b was reduced to alcohol 13 which was converted to the corresponding difluoro-bis-THF derivative 14 following the same reaction sequence as for synthesis of 12. Bis-THF derivative 14 was obtained in 73% yield over 3-steps from alcohol 13. This was converted to the enantiomer of 12 (ent-12) by Dess-Martin oxidation followed by reduction with L-selectride in THF at −78 °C providing ent-12 in 82% yield over 2-steps.
Scheme 2.

Synthesis of gem-difluoro bis-THF derivatives 12 and ent-12.
The synthesis of inhibitors containing difluoro-bis-THF ligands is shown in Scheme 3. Both bis-THF enantiomers and alcohol 14 were reacted with p-nitrophenyl chloroformate in CH2Cl2 in the presence of pyridine at 0 °C to 23 °C for 12 h to provide the corresponding activated carbonate 15, ent-15 and 16 respectively. [23] These activated carbonates were reacted with known (R)-hydroxyethylsulfonamide isostere 17[23] in the presence of triethylamine to furnish inhibitors 3, 19 and 20 in good yields. Treatment of known amine 18[24] and activated carbonates 15 and ent-15 provided inhibitors 4 and 21 in good yield.
Scheme 3.

Synthesis of inhibitors 3, 4,19, 20and 21 with gem-difluorides.
Results and Discussion
Our examination of the X-ray crystal structure of darunavir-bound HIV-1 protease[12] and subsequent modeling suggested that substitution of fluorine at the C4 position of bis-THF ligand may lead to strong non-bonded interactions with the Gly48 carbonyl oxygen. Also, fluorine may effectively fill the hydrophobic pocket in the S2 site as well. For synthesis and stability purposes, we designed gem-difluoride at the C4 position. As can be seen in Table 1, two fluoro-bis-THF derived inhibitors showed exceptional enzyme inhibitory protency, using the assay protocol reported by Toth and Marshall. [25] Inhibitor 3 with a p-methoxysulfonamide as the P2′-ligand showed a Ki of 22 pM. The corresponding p-amino derivative 4 displayed a Ki value of 5.8 pM. The enantiomeric fluoro bis-THF ligand as a P2 ligand and a p-methoxysulfonamide as the P2′-ligand provided inhibitor 19. This inhibitor showed a significant reduction in enzyme inhibitory potency. Furthermore, C-3 epimeric ligand at P2 and p-amino sulfonamide as the P2′ ligand resulted in inhibitor 20 with a dramatic increase in enzyme Ki. Antiviral activity of these gem-difluoro derivatives was determined in MT-2 human T-lymphoid cells exposed to HIVLAI.[26] As shown, both inhibitors 3 and 4 displayed EC50 values of 0.0008 μM and 0.003 μM, respectively. In comparison, both Darunavir and saquinavir showed antiviral EC50 values of 0.005 μM and 0.021 μM, respectively. Inhibitor 19 showed significant reduction of antiviral activity over inhibitor 3. Inhibitor 21 was not active in our antiviral assay. Inhibitors 3 and 4 showed cytotoxicity only at high concentration with CC50 values of 17.5 μM and 37 μM, respectively. The selectivity index (CC50/EC50) for compounds 3 and 4 was 21,875 and 12,333, respectively compared to darunavir which showed a selectively index of >20,000.
Table 1.
Enzymatic inhibitory and antiviral activity of difluoro-bis-THF-derived inhibitors
| Entry | Inhibitor | Ki(nM) | IC50(μM)ab |
|---|---|---|---|
| 1. |
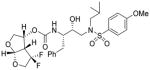 3 |
0.022 | 0.0008 |
| 2. |
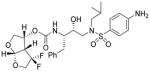 4 |
0.0058 | 0.0031 |
| 3. |
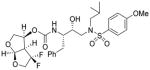 19 |
0.21 | 0.02 |
| 4. |
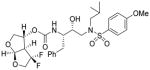 20 |
3.9 | 0.64 |
| 5. |
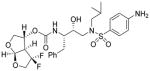 21 |
5.14 | >1.0 |
Ki values represents at least 5 data points. Standard error in all cases less than 7%. Darunavir exhibited Ki = 16 pM.
Values are means of at least three experiments.
Human T-lymphoid (MT-2) cells were exposed to 100 TCID50 values of HIV-1LAI and cultured in the presence of each PI, and IC50 values were determined using the MTT assay. Standard error in all cases less than 5%. Darunavir exhibited IC50 = 1.6 nM.
We then examined inhibitors 3 and 4 against clinical wild-type X4-HIV-1 isolate (HIV-1ERS104pre) along with a panel of highly multi-PI-resistant primary HIV-1 strains, HIV-1MDR/B (multidrug resistant, MDR), HIV-1MDR/C, HIV-1MDR/G, HIV-1MDR/TM, HIV-1MDR/ISI and HIV-1MDR/UM as shown in Table 2. These were isolated from HIV/AIDS patients who had failed a number of anti-HIV therapeutic regimens after receiving 9 to 11 anti-HIV drugs over 32 to 83 months.[27] These primary strains possessed 9 to 14 amino acid substitutions in the protease-encoding region of the HIV-1 genome. These substitutions have been associated with HIV-1 resistance against various approved protease inhibitors. These are noted in the footnote of Table 2. As can be seen, the EC50 values of both inhibitors 3 and 4 are significantly more potent than amprenavir (APV) and comparable to the potency of darunavir (DRV) against wild-type HIV-1ERS104pre using PHA-PBMC as target cells and p24 production as the end point. Interestingly, both PIs 3 and 4 displayed EC50 values ranging from 0.021 μM to 0.002 μM against the panel of six multidrug resistant clinical HIV-1 variants. In comparison, APV was less active with EC50 values and fold-differences ranging between 0.21 μM to 0.63 μM and 7 to 22 respectively. The EC50 values of inhibitors 3 and 4 are comparable or better than the activity of DRV against these panel of variants. Both inhibitors 3 and 4 are also effecitve against PI selected laboratory HIV-1-variants. A detailed study has been reported recently.[28] We have also evaluated apparent blood-brain-barrier permeability coefficients of inhibitors 3 and 4 and compared with that for darunavir. The assay protocol involved a triple cell co-culture system with rat astrocytes, pericytes and monkey endothelial cells. The model kit represents an in vitro BBB model for drug transport assay, as described by Nakagawa and co orkers.[29] In this assay, test inhibitor was added to the luminal interface (termed the blood side) of the microtiter culture wells under the optimal conditions for transendothelial electrical resistance (TEER) determination. The concentration of each inhibitor that permeated into the abluminal interface (termed brain side) was determined using a spectrophotometer 30 min after the addition of each inhibitor to the wells. As shown in Table 3, both fluorinated inhibitors 3 and 4 showed significantly higher drug concentration in the abluminal interface of the microtiter culture wells compared to darunavir (0.62 μM). The apparent permeability co-efficient (Papp) is referred as a brain uptake index. It is a way to measure the penetration efficiency of a drug across the BBB model quantitatively and qualitatively.[30] As can be seen, both fluoro derivatives 3 and 4 have shown Papp values significantly better than Papp values of DRV.
Table 2.
Antiviral activity of Pis 3 and 4 against multi-drug resistant clinical isolates in PHA-PBMs (EC50, μM)
| Virus (phenotype) | APV | DRV | 3 (GRL-04810) | 4 (GRL-05010) |
|---|---|---|---|---|
| HIV-1ERS104pre (wild-type X4) | 0.0295 ± 0.0004 | 0.004 ± 0.001 | 0.0023 ± 0.0001 | 0.0027 ± 0.0003 |
| HIV-1MDR/B(X4) | 0.49 ± 0.05 (15) | 0.021 ± 0.001 (5) | 0.014 ± 0.001 (7) | 0.011 ± 0.001 (3) |
| HIV-1MDR/C(X4) | 0.21 ± 0.02 (7) | 0.005 ± 0.001 (1) | 0.002 ± 0.001 (1) | 0.002 ± 0.001 (1) |
| HIV-1MDR/G(X4) | 0.31 ± 0.08 (11) | 0.014 ± 0.009 (4) | 0.004 ± 0.001 (2) | 0.004 ± 0.001 (1) |
| HIV-1MDR/TM(X4) | 0.328 ± 0.001 (12) | 0.03 ± 0.01 (9) | 0.004 ± 0.001 (2) | 0.004 ± 0.001 (2) |
| HIV-1MDR/MM(R5) | 0.630 ± 0.009 (22) | 0.025 ± 0.002 (5) | 0.021 ± 0.004 (10) | 0.020 ± 0.0002 (7) |
| HIV-1MDR/JSL(R5) | 0.27 ± 0.01 (9) | 0.010 ± 0.001 (3) | 0.002 ± 0.001 (1) | 0.003 ± 0.001 (1) |
The amino acid substitutions identified in the protease-encoding region of HIV-1ERS104pre, HIV-1B, HIV-1C, HIV-1G, HIV-1TM, HIV-1MM, HIV-1JSL compared to the consensus type B sequence cited from the Los Alamos database include L63P; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90M, I93L; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, L89M; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, L90M; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M, I93L; L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, Q92K; and L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, V82A, respectively. HIV-1ERS104pre served as a source of wild-type HIV-1. The EC50 values were determined by using PHA-PBMs as target cells and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of EC50 values for each isolate compared to the IC50 values for wild-type HIV-1ERS104pre. All assays were conducted in duplicate, and the data shown represent mean values (± 1 standard deviations) derived from the results of two or three independent experiments.
Table 3.
Apparent permeability blood brain barrier coefficient for inhibitors 3 and 4 in in vitro model.
| Compound | Initial luminal tracer concentration (μM) | Final abluminal tracer concentration (μM) | Papp (10−6 cm/s) |
|---|---|---|---|
| GRL-04810 | 100 | 3.33 ±0.70 | 50.38 ± 10.61 |
| GRL-05010 | 100 | 4.01 ± 0.27 | 60.84 ± 3.97 |
| DRV | 100 | 0.62 ± 0.15 | 9.32 ± 2.25 |
In the in vitro model using a triple co-culture of rat astrocytes, pericytes and monkey endothelial cells, GRL-04810, GRL-05010, DRV (all 100 μM) were added to the luminal interface (termed blood side) of duplicate wells. The mathematical formula used for the calculation of Papp is described in Materials and Methods. Results show average values ±1 S.D. of duplicated determinations.
To obtain molecular insight into the interactions of fluorinated inhibitors, we co-crystallized inhibitor 4 (GRL-05010) with HIV-1 protease and the structure was refined at the high resolution of 1.3 Å (PDB ID: 4U8W). The structure contains the HIV-1 protease dimer and the inhibitor with orientations related by 180° rotation with 55/45% relative occupancies. The protease dimer structure closely resembles our previously reported structure of protease-darunavir complex with RMSD of 0.19 Å for all Cα atoms.[12] Since this inhibitor is a difluoro derivative of darunavir, the majority of inhibitor interactions in the active site are similar to those for darunavir. As shown in Figure 2, the inhibitor is bound in the active site cavity via a network of strong hydrogen bonding interactions with backbone atoms, as well as with the catalytic aspartates of HIV-1 protease. Both oxygens of the bis-THF ligand formed strong hydrogen bonds with the backbone amides of Asp30 and Asp29. Interestingly, both fluorine atoms on the bis-THF ligand formed strong interactions with the carbonyl oxygen of Gly48 as shown in the figure. For the major conformation, these interactions show short distances of 2.2 Å and 2.6 Å. For the minor conformation, it was 2.6 Å and 2.8 Å. Similar interactions of fluorine atom with carbonyl oxygen of various amino acids have been previously observed in various protein structures with a frequency of over 9%.[31] Since Gly48 is located in the flap, for inhibitor 3, these F….OC interactions with Gly48 may tend to stabilize the flexible flap region as well as improve the binding affinity for HIV-1 protease complex. The structure also shows hydrogen bonding interactions of urethane NH with the backbone carbonyl oxygen of Gly27. The amine functionality of the P2′-ligand formed a strong hydrogen bond with Asp30′ NH, as well as with the side chain carboxylic acid of Asp30′. Furthermore, the inhibitor formed water mediated interactions with the urethane carbonyl oxygen, sulfonamide oxygen, and the amides of Ile50 and Ile50′ in the flaps of HIV-1 protease. Similar interactions are inherent to the darunavir-bound HIV-1 protease X-ray structure. Both P1 and P2’ aromatic rings, as well as the P1’-isobutyl side chain effectively fill the hydrophobic pockets of HIV-1 protease in S1, S2’ and S1’ subsites respectively. These extensive molecular interactions including F….OC interactions may be responsible for inhibitor ‘s’ high affinity for HIV-1 protease and its robust potency against drug resistant HIV-1 variants.
Figure 2. A. GRL-05010A.

-bound X-ray structure of HIV-1 protease. The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in green, water molecules are red spheres, and the hydrogen bonds are indicated by dotted lines. B. Nonbonded interactions of gem-difluorides with the carbonyl of Gly48 in the protease flap region are shown. These interactions are indicated with respective distances.
Conclusions
We have investigated fluorine containing inhibitors to improve brain penetration. In this context, we have incorporated gem-difluorides based upon the X-ray structure of darunavir-bound HIV-1 protease. The corresponding gem-difluoro-bis-THF ligand was synthesized stereoselectively utilizing a Reformatskii-Claisen reaction as the key step. Both enantiomers of bis-THF ligand were synthesized in the optically active form. These fluoro-derivatives were then incorporated in the (R)-hydroxyethylsulfonamide isostere. Absolute stereochemistry of the P2-fluoro-bis-THF ligand was critical to the potency of the inhibitors. Consistent with our previous studies, the ligand containing 3(R),3a(S), 6a(R)-bis-THF displayed enhanced enzyme inhibitory and antiviral potency compared to the enantiomeric ligand. Both inhibitors 3 and 4 maintained excellent antiviral activity against a variety of multidrug-resistant clinical HIV-1 variants with EC50 values ranging from 0.021 μM to 0.002 μM. These values are significantly better than those seen for amprenavir and comparable or better than for darunavir. Of particular note, both inhibitors have shown much improved blood-brain-barrier permeability coefficients compared to darunavir in an in vitro model. A high resolution X-ray crystal structure of inhibitor 4-bound HIV-1 protease revealed that the fluoro-bis-THF ligand is involved in extensive interactions in the S2 subsite including a number of critical hydrogen bonding interactions with Asp29 and Asp30 backbone NHs. Also, both gem-difluorides formed strong interactions with the Gly48 carbonyl oxygen located in the flap region of the HIV-1 protease. These interactions are likely responsible for inhibitor ‘s’ exceptional activity, particularly activity against a wide spectrum of drug-resistant HIV-1 variants. Our current results warrant further investigation.
Experimental Section
General
All moisture sensitive reactions were carried out in an oven dried flask under an argon atmosphere. Anhydrous solvents were obtained as follows: THF, diethyl ether and benzene, distilled from sodium and benzophenone; dichloromethane, pyridine, triethylamine, and diisopropylethylamine, distilled from CaH2. All other solvents were HPLC grade. 1H NMR and 13C NMR spectra were recorded on Varian INOVA300-1 and Bruker Avance ARX-400 spectrometers. NMR data were resolved with Mestrec software. Optical rotations were recorded on a Perkin Elmer 341 polarimeter. Mass spectra were obtained at the Purdue University Campus-wide Mass Spectrometry Center. Column chromatography was performed with Whatman 240–400 mesh silica gel under a low pressure of 3–5 psi. TLC was carried out with E. Merck silica gel 60-F-254 plates. HPLC was performed on an Agilent 1100 instrument. All test inhibitors showed purity >96% by HPLC analysis.
Ethyl (R,E)-4,5-bis(benzyloxy)pent-2-enoate (6)
Methyl ester 5 (3.01 g, 10 mmol) was dissolved in CH2Cl2 (50 mL) and cooled to −78 °C. To the solution was added DIBAL (1.0 M in CHCl2, 15 mL, 15 mmol), and the mixture was stirred for 1.5 h. The reaction was quenched with saturated aqueous Rochelle salt, and the mixture was stirred overnight until the aqueous phase became clear. The organic phase was then washed with brine, dried over sodium sulfate, filtered, and evaporated in vacuo to give the corresponding crude aldehyde (2.85 g). The aldehyde was immediately subjected to the next Horner-Emmons reaction.
A suspension of sodium hydride (60% dispersion in mineral oil, 1.48 g, 37.0 mmol) in THF (30 mL) was cooled to 0 °C. Triethyl phosphonoacetate (7.95 mL, 40.1 mmol) was added dropwise to the above suspension. After 30 mins, the reaction mixture was neutralized with saturated aqueous ammonium chloride and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and evaporated in vacuo. The residue was purified by silica gel chromatography (Hexanes/ethyl acetate 20/1) to give unsaturated ethyl ester 6 (2.99 g, 8.78 mmol, 88% for 2-steps) as colorless oil. 1H-NMR spectrum was consistent with the spectrum reported ([α]D23 −23.4 (c 1.39, CHCl3, lit.[20] [α]D23 +19.2 (c 1.45, CHCl3)).
(R, E)-4, 5-Bis(benzyloxy)pent-2-en-1-yl 2-chloro-2,2-difluoroacetate (7)
α,β-Unsaturated ester 6 (2.96 g, 8.7 mmol) was dissolved in dry, distilled CH2Cl2 (50 mL) and the solution was cooled to −78 °C. To this cold reaction mixture, DIBAL-H (1.0 M in CH2Cl2, 34.8 mL) was added dropwise. The resulting mixture was stirred for 4 h. After this period, the reaction was quenched with saturated aqueous Rochelle salt at −78°C and the mixture was warmed up to 23 °C and was continued to stir for 12 h until the aqueous phase became clear. The organic phase was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered and evaporated in vacuo to yield the crude allylic alcohol which was used directly without further purification.
The crude allylic alcohol was dissolved in chloroform (50 mL) and chlorodifluoroacetic acid (3.67 mL, 43.45 mmol) was added. The resulting mixture was stirred at reflux for 10 h. After this period, the reaction mixture was cooled to 23 °C and extracted with CH2Cl2. The organic phase was washed with brine, dried with anhydrous sodium sulfate, filtered and evaporated in vacuo. The crude residue was purified by silica gel chromatography (hexanes: ethyl acetate, 15:1) to provide the title compound 7 (3.2 g, 90% over two steps). [α]D23 - 15.4 (c 1.08, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.35-7.30 (m, 10H), 5.89-5.87 (m, 2H), 4.83 (d, J = 3.92 Hz, 1H), 4.63 (d, J = 11.84 Hz, 1H), 4.51 (s, 2H), 4.50 (d, J = 11.84 Hz, 1H), 4.10-4.08 (m, 1H), 3.59 (dd, J = 10.2 Hz, 6.1 Hz, 1H), 3.52 (dd, J = 10.2 Hz, 4.7 Hz, 1H ). 13C-NMR (75.5 MHz, CDCl3): δ 158.89, 137.91, 137.83, 134.56, 132.72, 129.75, 128.95, 128.35, 127.71, 127.66, 124.91, 116.80, 78.22, 76.68, 73.39, 72.83, 72.43, 71.01, 70.64, 67.58.
Ethyl (R)-4,5-bis(benzyloxy)-2,2-difluoropentanoate (8)
Above chlorodifluoroacetate derivative 7 (1.68 g, 4.45 mmol) and chlorotrimethylsilane (0.76 mL, 6 mmol) were dissolved in dry distilled acetonitrile (40 mL). To this mixture activated Zn dust (2.9 g, 44.6 mmol) was added and the resulting mixture was heated at 105 °C for 24 h. After this period, the reaction mixture was cooled to 23 °C and filtered through a pad of Celite. The solvent was removed in vacuo and the residue was dissolved in ethanol (40 mL), concentrated H2SO4 (1.1 mL) was added and the mixture was stirred for 36 h at 50 °C. After this period, the reaction mixture was concentrated and the residue was diluted with water. It was then extracted with hexane, dried over sodium sulfate, filtered, evaporated to dryness. The residue was purified by silica gel chromatography (Hexanes/ethyl acetate, 30:1) to afford compound 8 (1.44 g, 80 %) as a diastereomeric mixture (2:1 by 1H-NMR). 1H-NMR (400 MHz, CDCl3): δ 7.34-7.25 (m, 15H), 5.92 (dt, J = 17.2 Hz, 9.9 Hz, 1H) major, 5.66 (m, 1H) minor, 5.32 (dd, J = 8.8 Hz, 0.9 Hz, 2H) major, 5.23-5.19 (m, 2H) minor, 4.68-4.63 (m, 2H) minor, 4.56-4.42 (m, 2H) major, 4.10-4.04 (m, 3H), 3.89-3.86 (m, 1H) major, 3.59-3.56 (m, 1H) minor, 3.52-3.51 (m, 1H) minor, 3.44 (dd, J = 9.5 Hz, 6.9 Hz, 1H) major, 1.20 (t, J = 7.28, 3H) major, 1.09 (t, J = 7.08, 3H) minor. 13C-NMR (75.5 MHz, CDCl3): δ 138.09, 137.85 minor, 128.64, 128.34, 128.14, 128.07, 127.59, 122.78, 122.57, 74.94, 73.22, 72.82, 72.46, 69.75, 62.56, 62.18, 51.29, 13.74, 13.607.
(R)-3-((R)-1,2-Bis(benzyloxy)ethyl)-2,2-difluoro-N-methoxy-N-methylpent-4-enamide (9a) and (S)-3-((R)-1,2-bis(benzyloxy)ethyl)-2,2-difluoro-N-methoxy-N-methylpent-4-enamide (9b)
To a suspension of N, O-dimethylhydroxylamine hydrochloride (1.7 g, 17.5 mmol) in THF (40 mL) at −78 °C, n-BuLi (22 mL, 1.6 M in hexane) was added. The resulting reaction mixture was stirred for 1 h at this temperature. To this reaction mixture, solution of compound 8 (1.42 g, 3.51 mmol) in THF (10 mL) was added via a cannula. The reaction was monitored by TLC and after 2 h, the reaction was quenched at −78 °C using saturated aqueous NH4Cl solution. The resulting mixture was warmed to 23 °C. The reaction mixture was extracted with ethyl acetate, washed with brine, dried over sodium sulfate and concentrated in vacuo. The crude residue was purified by silica gel chromatography (Hexanes/ethyl acetate, 10:1) to afford 9a (0.80 g, 1.88 mmol) and 9b (0.40 g, 0.94 mmol) in 2:1 ratio in 81% overall yield.
9a
[α]D23 −25.1 (c 1.07, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.35-7.25 (m, 10H), 5.92 (dt, J = 17.2 Hz, 9.9 Hz, 1H), 5.31 (dd, J = 10.3 Hz, 0.9 Hz, 1H), 5.19 (d, J = 17.3 Hz, 1H), 4.65 (dd, J = 15.2 Hz, 11.3 Hz, 2H), 4.54 (d, J = 11.8 Hz, 1H), 4.45 (d, J = 11.9 Hz, 1H), 4.03 (bs, 1H), 3.68 (s, 3H), 3.60 (dt, J = 11.5 Hz, 4.45 Hz, 1H), 3.50 (dd, J = 9.7 Hz, 6.4 Hz, 1H), 3.42-3.37 (m, 1H), 3.13 (s, 3H).
9b
[α]D23 −18.0 (c 0.55, CHCl3); 1H-NMR (400 MHz, CDCl3): δ 7.36-7.24(m, 10H), 5.71 (dt, J = 16.7 Hz, 10.5 Hz, 1H), 5.33 (s, 1H), 5.30 (d, J = 11.2 Hz, 1H), 4.66 (d, J = 10.8 Hz, 1H), 4.50 (dd, J = 12.1 Hz, 5.5 Hz, 2H), 4.42 (d, J = 10.7 Hz, 1H), 3.79-3.75 (m, 1H), 3.69-3.65 (m, 1H), 3.62 (s, 3H), 3.59-3.51(m, 1H), 3.46 (dd, J =10.6 Hz, 4.7 Hz, 1H), 2.78 (bs, 3H).
(R)-3-((R)-1,2-Bis(benzyloxy)ethyl)-2,2-difluoropent-4-en-1-ol (10)
Weinreb amide 9a (270 mg, 0.64 mmol) was dissolved in THF (10 mL). To the solution at 0 °C, LAH (77 mg, 2.03 mmol) was added and the resulting reaction mixture was stirred for 30 min. The reaction was quenched by adding water and 3 M aqueous NaOH. After the resultant mixture was diluted with diethyl ether, NaBH4 was added to it. The reaction mixture was stirred overnight, filtered, and purified by silica gel chromatography (1% methanol in CH2Cl2) to give alcohol 10 (231 mg, 99%) as a clear oil. 1H-NMR (400 MHz, CDCl3): δ 7.40-7.31 (m, 10H), 5.92 (dt, J = 17.2, 10.0 Hz, 1H), 5.35-5.23 (m, 2H), 4.79-4.49 (m, 4H), 4.15-4.11 (m, 1H), 3.83-3.49 (m, 4H), 3.05-3.00 (m, 1H), 2.70 (br, 1H). 13C-NMR (100 MHz, CDCl3): δ 137.83, 129.95, 128.38, 127.86, 127.80, 127.73, 127.68, 122.62, 122.03, 75.72, 73.31, 73.04, 70.23, 63.27, 50.20. 19F-NMR (376 MHz): δ −109.61, −111.29. [α]D23 −17.9 (c 0.66, CHCl3). LRMS (CI): 361 (M−H)+.
(S)-3-((R)-1,2-Bis(benzyloxy)ethyl)-2,2-difluoropent-4-en-1-ol (13)
Alcohol 13 was obtained in 96% yield from Weinreb amide 9b through the same sequence of reactions as for alcohol 10. 1H-NMR (400 MHz, CDCl3): δ 7.40-7.32 (m, 10H), 5.78 (dt, J = 17.4, 9.8 Hz, 1H), 5.36-5.29 (m, 2H), 4.83 (d, J = 11.1 Hz, 1H), 4.60-4.52 (m, 3H), 3.96-3.61 (m, 5H), 3.23-3.10 (m, 1H), 3.00 (br, 1H). 13C-NMR (100 MHz, CDCl3): δ 137.95, 137.41, 130.37, 128.51, 128.38, 128.06, 128.00, 127.69, 122.77, 121.46, 77.20, 73.38, 72.80, 70.49, 63.87, 49.99. 19F-NMR (376 MHz): δ −104.01, −113.05. [α]D23 [+9.9 (c 1.06, CHCl3). LRMS (CI): 363 (M+H)+.
(3R,3aS,6aS)-4,4-Difluorohexahydrofuro[2,3-b]furan-3-ol (12)
Alcohol 10 Yield for alcohol 10 was 231 mg (0.63 mmol) was dissolved in CH2Cl2 (30 mL), and ozone was bubbled through the solution for 5 mins at −78 °C. After bubbling with argon, triphenylphosphine (510 mg, 1.94 mmol) was added to the solution. The resulting mixture was stirred for 2 h at −78 °C and then 3 h at 23 °C. The mixture was concentrated under vacuo, and the residue was passed through a silica gel column (hexanes/ethyl acetate, 4/1 to 2/1) to give crude lactol 11 (332 mg) as a clear oil.
Above crude lactol (332 mg) was then dissolved in ethyl acetate (25 mL) and palladium hydroxide (20% on activated carbon, 180 mg) was added to the solution. The resulting suspension was stirred under a hydrogen-filled balloon for overnight. The reaction mixture was filtered through a pad of Celite and the filtrate was evaporated in vacuo to give crude triol (150 mg) as a pale-yellow oil. This crude triol (150 mg) was dissolved in a mixture of CH2Cl2 and THF (30 mL and 5 mL), and camphorsulfonic acid (130 mg) was added to the solution. The reaction mixture was stirred overnight and neutralized with sodium bicarbonate (100 mg). The resulting mixture was stirred further for 4 h. After filtration and evaporation, the residue was purified by silica gel chromatography (1% methanol in CH2Cl2) to provide the title difluoro-bis-THF ligand 12 (110 mg, 68% over 3-steps) as an amorphous soild. 1H-NMR (400 MHz, CDCl3): δ 5.79 (d, J = 5.3 Hz, 1H), 4.65-4.59 (m, 1H), 4.30-4.20 (m, 1H), 4.08-3.99 (m, 3H), 3.03-2.96 (m, 1H), 2.60 (t, J = 5.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3): δ 128.31, 108.31, 74.71, 72.85, 70.65, 51.77. 19F-NMR (376 MHz): δ −91.68, −122.38. [α]D23 +7.21 (c 0.68, CHCl3). LRMS (CI): 167 (M+H)+.
(3R,3aR,6aR)-4,4-Difluorohexahydrofuro[2,3-b]furan-3-ol (14)
Difluoro-bis-THF 14 was obtained in 73% yield from alcohol 13 by following the same sequence of reactions as for compound 12. 1H-NMR (400 MHz, CDCl3): δ 6.00 (d, J = 5.2 Hz, 1H), 4.66 (d, J = 2.8 Hz, 1H), 4.07-3.88 (m, 4H), 3.09-3.02 (m, 1H), 2.69 (br, 1H). 13C-NMR (100 MHz, CDCl3): δ 126.74, 108.33, 76.25, 72.05, 71.86, 58.41. 19F-NMR (376 MHz): δ −97.60, −115.49. [α]D23 +10.8 (c 1.59, CHCl3). LRMS (CI): 167 (M+H)+.
(3S,3aR,6aR)-4,4-Difluorohexahydrofuro[2,3-b]furan-3-ol (ent-12)
Difluoro alcohol 14 (38 mg, 0.23 mmol) was dissolved in CH2Cl2 (5 mL) and Dess-Martin periodinane (146 mg, 0.34 mmol) was added to the solution at 23 °C. The mixture was continued to stir for 2 h. After this period, the reaction mixture was filtered through a short silica gel column using CH2Cl2 as the eluent to give crude ketone (35 mg). The crude ketone (35 mg) was dissolved in THF (4 mL) and the mixture was cooled to −78 °C. To the solution was dropwise added L-Selectride (1 M in THF, 320 μL, 0.32 mmol) and the mixture was stirred for 30 mins. The reaction was quenched with methanol. After removing the solvent in vacuo, the residue was purified over silica gel chromatography (hexanes/ethyl acetate, 3/1) to give difluoro-bis-THF ligand ent-12 (32 mg, 84% over 2-steps) as an amorphous solid. 1H-NMR (400 MHz, CDCl3): δ 5.79 (d, J = 5.3 Hz, 1H), 4.65-4.59 (m, 1H), 4.30-4.20 (m, 1H), 4.08-3.99 (m, 3H), 3.03-2.96 (m, 1H), 2.60 (t, J = 5.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3): δ 128.31, 108.31, 74.71, 72.85, 70.65, 51.77. 19F-NMR (376 MHz): δ −91.68, −122.38. [α]D23 −7.9 (c 0.66, CHCl3). LRMS (CI): 167 (M+H)+.
3R,3aS,6aS)-4,4-Difluorohexahydrofuro[2,3-b]furan-3-yl (4-nitrophenyl) carbonate 15
A solution of alcohol 12 (14.3 mg, 0.09 mmol) and pyridine (35 μL, 0.43 mmol) in CH2Cl2 (1 mL) was cooled to 0 °C, and 4-nitrophenyl chloroformate (54 mg, 0.26 mmol) was added to the solution in one portion. The reaction temperature was raised to 23 °C and the mixture was stirred for 2 h. The reaction was quenched with ethanol, and the solvent was removed in vacuo. The residue was passed through a short silica gel column using hexanes/ethyl acetate 5/1 to 2/1 as the eluent to give carbonate 15 (25 mg).
Activated carbonate ent-15 and 16 were prepared following the procedure for carbonate 15. These unstable mixed activated carbonates were directly employed for the inhibitor synthesis.
Inhibitor 3
Activated mixed carbonate 15 (12.5 mg) was added to a solution of amine 17 (40 mg) and triethylamine (150 μL) in CH2Cl2 (2 mL). The resulting reaction mixture was stirred for 3 days until all of the carbonate was consumed. After this period, solvents were evaporated and the residue was purified by silica gel chromatography (1% methanol in CH2Cl2) to give inhibitor 3 (18 mg, 83% for 2-steps from 12). 1H-NMR (400 MHz, CDCl3): δ 7.69 (d, J = 8.8 Hz, 2H), 7.30-7.20 (m, 5H), 6.99-6.96 (m, 2H), 5.77 (d, J = 5.1 Hz, 1H), 5.35-5.29 (m, 1H), 5.00 (d, J = 8.3 Hz, 1H), 4.08-3.68 (m, 10H), 3.15-2.75 (m, 7H), 1.86-1.79 (m, 1H), 0.91-0.85 (m, 6H). 13C-NMR (100 MHz, CDCl3): δ 163.00, 154.84, 137.17, 129.68, 129.38, 128.48, 126.87, 126.54, 114.27, 108.07, 72.67, 72.26, 72.09, 71.10, 71.03, 58.68, 55.54, 55.13, 53.61, 50.05, 35.15, 27.18, 20.02, 19.75. 19F-NMR (376 MHz): δ −91.82, −123.21. LRMS (ESI): 599 (M+H)+. HRMS-ESI (m/z): [M + Na]+ calcd for C28H36F2N2O8SNa, 621.2059, found 621.2054.
Inhibitor 4
The title inhibitor was obtained in 58% yield from activated mixed carbonate 15 and amine 18 by following the same procedure as inhibitor 3. 1H-NMR (400 MHz, CDCl3): δ 7.53 (d, J = 8.4 Hz, 2H), 7.32-7.21 (m, 5H), 6.68 (d, J = 8.5 Hz, 2H), 5.78 (d, J = 5.1 Hz, 1H), 5.35-5.28 (m, 1H), 4.76 (d, J = 8.3 Hz, 1H), 4.23-3.70 (m, 9H), 3.15-2.72 (m, 7H), 1.86-1.73 (m, 1H), 0.93-0.87 (m, 6H). 13C-NMR (100 MHz, CDCl3): δ 154.81, 150.66, 137.22, 129.42, 128.45, 126.85, 126.50, 126.02, 114.01, 108.08, 72.67, 72.32, 72.11, 71.03, 58.77, 55.11, 53.67, 53.32, 50.08, 35.20, 27.20, 20.05, 19.78. 19F-NMR (376 MHz): δ −91.79, −123.24. LRMS (ESI): 584 (M+H)+. HRMS-ESI (m/z): [M + H]+ calcd for C27H36F2N3O7S, 584.2242, found 584.2245.
Inhibitor 19
The title inhibitor was obtained in 64% yield from activated mixed carbonate ent-15 and amine 17 by following the same procedure as inhibitor 3. 1H-NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.9 Hz, 2H), 7.32-7.22 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.77 (d, J = 5.3 Hz, 1H), 5.29-5.25 (m, 1H), 4.93 (d, J = 8.3 Hz, 1H), 4.12-3.75 (m, 10H), 3.19-2.70 (m, 7H), 1.83-1.71 (m, 1H), 0.92-0.84 (m, 6H). 13 C-NMR (100 MHz, CDCl3): δ 162.97, 154.97, 137.23, 129.66, 129.43, 129.36, 128.48, 127.09, 126.56, 114.28, 108.13, 72.58, 72.44, 71.85, 71.08, 58.85, 55.53, 55.11, 53.68, 49.70, 35.73, 27.12, 19.99, 19.67. 19F-NMR (376 MHz): δ −91.54, −123.41. LRMS (ESI): 599 (M+H)+. HRMS-ESI (m/z): [M + Na]+ calcd for C28H36F2N2O8SNa, 621.2059, found 621.2059.
Inhibitor 20
The title inhibitor was obtained in 64% yield from activated mixed carbonate 16 and amine 17 by following the same procedure as inhibitor 3. 1H-NMR (400 MHz, CDCl3): δ 7.70 (d, J = 8.8 Hz, 2H), 7.31-7.22 (m, 5H), 6.99 (d, J = 11.6 Hz, 2H), 5.87 (d, J = 5.2 Hz, 1H), 5.24 (br, 1H), 4.98 (d, J = 8.3 Hz, 1H), 4.04-3.69 (m, 10H), 3.17-2.75 (m, 7H), 1.88-1.77 (m, 1H), 0.93-0.87 (m, 6H). 13C-NMR (100 MHz, CDCl3): δ 164.04, 154.94, 137.32, 129.58, 129.42, 129.39, 128.46, 126.58, 126.29, 114.30, 108.14, 74.52, 73.89, 72.38, 72.03, 58.74, 55.67, 55.54, 55.01, 53.65, 35.45, 27.18, 20.03, 19.77. 19F-NMR (376 MHz): δ −98.62, −114.71. LRMS (ESI): 599 (M+H)+. HRMS-ESI (m/z): [M + H]+ calcd for C28H37F2N2O8S 599.2238, found 599.2227. HRMS-ESI (m/z): [M + Na]+ calcd for C28H36F2N2O8SNa, 621.2059, found 621.2059.
Inhibitor 21
The title inhibitor was obtained in 70% yield from activated mixed carbonate 16 and amine 18 by following the same procedure as inhibitor 3. 1H-NMR (400 MHz, CDCl3): δ 7.53 (d, J =7.4 Hz, 2H), 7.32-7.21 (m, 5H), 6.68 (d, J = 8.6 Hz, 2H), 5.87 (d, J = 5.2 Hz, 1H), 5.24 (br, 1H), 4.94 (d, J = 8.3 Hz, 1H), 4.22-3.68 (m, 9H), 3.17-2.74 (m, 7H), 1.85-1.76 (m, 1H), 0.93-0.86 (m, 6H). 13C-NMR (100 MHz, CDCl3): δ 154.91, 150.74, 137.39, 129.43, 128.43, 126.55, 126.29, 125.80, 114.02, 108.15, 74.48, 73.93, 72.40, 72.02, 58.83, 55.65, 54.98, 53.72, 35.50, 27.19, 20.06, 19.82. 19F-NMR (376 MHz): δ −98.63, −114.70. LRMS (ESI): 584 (M+H)+. HRMS-ESI (m/z): [M + Na]+ calcd for C27H35F2N3O7SNa, 606.2062, found 606.2061.
Determination of X-ray structures of HIV-1 protease-inhibitor 4 complexes
The optimized HIV-1 protease was expressed and purified as described.[32] The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions of 1.0 M NaCl, 0.1M sodium acetate buffer (pH 4.8). Diffraction data were collected on a single crystal cooled to 90 K at the SER-CAT (22-BM beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelength of 1.0 Å, and processed by HKL-2000[33] with an Rmerge of 6.2%. Using the isomorphous structure[34] in the crystal structure was solved by PHASER[35] in the CCP4i Suite[36, 37] and refined by SHELX-97[38, 39] with 1.3 Å resolution data. COOT[40] was used for manual modification of the atomic structure. PRODRG-2[41] was used to construct the inhibitor and the restraints for refinement. Alternative conformations were modeled, anisotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules, and hydrogen atoms were added in the final round of refinement. The final refined solvent structure comprised one Na+ ion, three Cl− ions, two acetate and 129 water molecules. The crystallographic statistics are listed in Table 1 [please see, supporting Information]. The coordinates and structure factors of the PR with GRL-050-10A structure have been deposited in Protein Data Bank[42] with ID: 4U8W.
Supplementary Material
Acknowledgments
The research was supported by grants from the National Institutes of Health (GM53386, AKG and GM62920, IW). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and in part by a Grant-in-aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan (Kosei Rohdosho: H15-AIDS-001), and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Kumamoto University) of Monbu-Kagakusho. We also thank Ms. Heather Osswald (Purdue University) for helpful disscussions.
Footnotes
Supporting information for this article is available on the WWW underInstitution http://dx.doi.org.
References
- 1.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 2.Bohm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, Obst-Sander U, Stahl M. Chembiochem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 3.Park BK, Kitteringham NR, O’Neill PM. Annu Rev Pharmacol Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- 4.Smart BE. J Fluorine Chem. 2001;109:3–11. [Google Scholar]
- 5.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. Bioorg Med Chem Lett. 1998;8:687–690. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]
- 8.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh AK, Dawson ZL, Mitsuya H. Bioorg Med Chem. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh AK, Ramu Sridhar P, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh AK, Martyr CD. In: Modern Drug Synthesis. Li JJ, Johnson DS, editors. Wiley; 2010. pp. 29–44. [Google Scholar]
- 12.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 13.McArthur JC, Brew BJ, Nath A. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 14.Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Tan IL, McArthur JC. CNS Drugs. 2012;26:123–134. doi: 10.2165/11597770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 16.Langford D, Marquie-Beck J, de Almeida S, Lazzaretto D, Letendre S, Grant I, McCutchan JA, Masliah E, Ellis RJ. J Neurovirol. 2006;12:100–107. doi: 10.1080/13550280600713932. [DOI] [PubMed] [Google Scholar]
- 17.Vulpetti A, Schiering N, Dalvit C. Proteins. 2010;78:3281–3291. doi: 10.1002/prot.22836. [DOI] [PubMed] [Google Scholar]
- 18.Zhou P, Zou J, Tian F, Shang Z. J Chem Inf Model. 2009;49:2344–2355. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- 19.Fujita K, Nakai H, Kobayashi S, Inoue K, Nojima S, Ohno M. Tetrahedron Lett. 1982;23:3507–3510. [Google Scholar]
- 20.Yang YY, Xu J, You ZW, Xu XH, Qiu XL, Qing FL. Org Lett. 2007;9:5437–5440. doi: 10.1021/ol7023955. [DOI] [PubMed] [Google Scholar]
- 21.Yang YY, Meng WD, Qing FL. Org Lett. 2004;6:4257–4259. doi: 10.1021/ol0482947. [DOI] [PubMed] [Google Scholar]
- 22.Greuter H, Lang RW, Romann AJ. Tetrahedron Lett. 1988;49:2344–2355. [Google Scholar]
- 23.Ghosh AK, Chapsal BD, Parham GL, Steffey M, Agniswamy J, Wang YF, Amano M, Weber IT, Mitsuya H. J Med Chem. 2011;54:5890–5901. doi: 10.1021/jm200649p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh AK, Leshchenko S, Noetzel M. J Org Chem. 2004;69:7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]
- 25.Toth MV, Marshall GR. Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 26.Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2007;51:2143–2155. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. J Virol. 2002;76:1349–1358. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salcedo Gomez PM, Amano M, Yashchuk S, Mizuno A, Das D, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2013;57:6110–6121. doi: 10.1128/AAC.01420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, Niwa M. Cell Mol Neurobiol. 2007;27:687–694. doi: 10.1007/s10571-007-9195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, Fenart L. Nat Rev Drug Discov. 2007;6:650–661. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 31.Vulpetti A, Schiering N, Dalvit C. Proteins. 2010;78:3281–91. doi: 10.1002/prot.22836. [DOI] [PubMed] [Google Scholar]
- 32.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W, Carter CW, Sweet RM. Academic Press; New York: 1997. pp. 307–326. [Google Scholar]
- 34.Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. J Med Chem. 2007;50:4509–4515. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collaborative Computational Project, Number 4, The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Potterton E, Briggs P, Turkenburg M, Dodson E. Acta Crystallogr, Sect D: Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 38.Sheldrick GM. Acta Crystallogr, Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 39.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Schuettelkopf AW, van Aalten DMF. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 42.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.