

Abstract
Based on over three decades of pre-clinical data, insulin-like growth factor-1 receptor (IGF-1R) signaling has gained recognition as a promoter of tumorogenesis, driving cell survival and proliferation in multiple human cancers. As a result, IGF-1R has been pursued as a target for cancer treatment. Early pioneering efforts targeting IGF-1R focused on highly selective monoclonal antibodies, with multiple agents advancing to clinical trials. However, despite some initial promising results, recent clinical disclosures have been less encouraging. Moreover, recent studies have revealed that IGF-1R participates in a dynamic and complex signaling network, interacting with additional targets and pathways thereof through various crosstalk and compensatory signaling mechanisms. Such mechanisms of bypass signaling help to shed some light on the decreased effectiveness of selective IGF-1R targeted therapies (e.g. monoclonal antibodies) and suggest that targeting multiple nodes within this signaling network might be necessary to produce a more effective therapeutic response. Additionally, such findings have led to the development of small molecule IGF-1R inhibitors which also co-inhibit additional targets such as insulin receptor and epidermal growth factor receptor. Such findings have helped to guide the design rationale of numerous drug combinations that are currently being evaluated in clinical trials.
Key words: insulin-like growth factor-1 receptor, insulin receptor, inhibitor, drug combination, cancer
Biology around type I insulin-like growth factor receptor signaling and its role in cancer
Type I insulin-like growth factor receptor (IGF-1R) is a receptor tyrosine kinase that plays an important role in promoting tumor cell survival and growth. Early evidence demonstrated that IGF-1R is required for cellular transformation, and expression of IGF ligands is linked to a number of tumor types including breast and colon.1-3 During the past two decades, the importance of IGF-1R in cancer has been reinforced by pre-clinical studies showing that IGF-1R is an important driver of tumorogenesis, cell survival and proliferation in multiple human cancers including hepatocellular carcinoma (HCC), Ewing’s sarcoma, multiple myeloma, non-small cell lung carcinoma (NSCLC), and ovarian carcinoma.4-6 Overexpression of the IGF-1R ligands IGF-I and IGF-II has been observed in various human cancers and is associated with increased disease incidence, progression and poor prognosis.7,8 IGF-1R mediates these cancerpromoting properties through activation of two well established oncogenic pathways: the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt/PKB) pathway and the mitogen-activated protein kinases (MAPK) pathway.9 The importance of IGF-1R signaling for the treatment of cancer is further underscored by the potential for IGF-1R signaling to promote resistance to both cytotoxic chemotherapies and radiation, as well as other molecular targeted therapies (Figure 1).10-12
Figure 1.
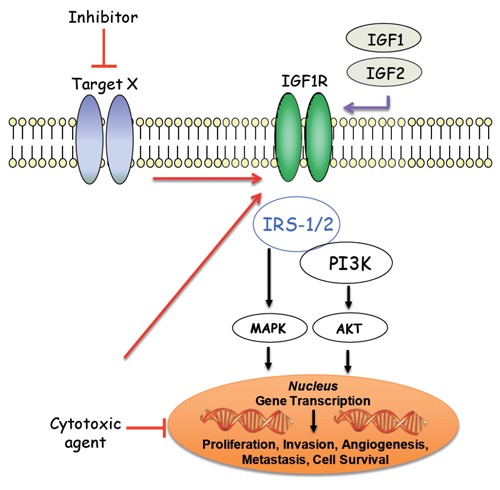
The insulin-like growth factor 1 receptor (IGF-1R), a transmembrane receptor tyrosine kinase, is activated by its cognate ligands, insulin-like growth factor-1 (IGF-1) and -2 (IGF-2), resulting in receptor auto-transphosphorylation and signaling cascades that progress through the IRS-l/PI3K/AKT and MAPK pathways. Stimulation of both pathways ultimately leads to various cancer promoting phenotypes including cellular proliferation, survival, invasion, metastasis and angiogenesis. Specific IGF-1R mediated activation of the PI3K/AKT pathway results in a pro-survival signal that has been shown to promote resistance to both chemotherapy-induced apoptosis or to molecular targeted therapies (Target X).
Based on pre-clinical data highlighting the critical role that IGF-1R plays in human cancer, major research efforts have been undertaken during the last decade, focused on discovering and developing therapeutic agents to modulate IGF-1R activity. These efforts have resulted in IGF-1R inhibitors that operate through several distinct modes of inhibition. These include monoclonal antibodies (mAbs) that neutralize the IGF-1R ligands IGF-1 and/or IGF-2 (e.g. dusigitumab, MEDI-573, an agent in this class that is currently being evaluated in clinical trials. ClinicalTrial.gov Identifiers: NCT01498952, NCT01446159), monoclonal antibodies that specifically target the IGF-1R extracellular ligand-binding domain, and small molecule inhibitors that target the IGF-1R intracellular catalytic domain. The third approach is further stratified into adenosine triphosphate (ATP) competitive inhibitors that bind to the ATP pocket of the IGF-1R kinase domain, and non-ATP competitive inhibitors that bind to an allosteric pocket (e.g. substrate binding site). Several excellent reviews have been published in recent years describing the historical developments in these areas.13-15 This review will cover recently disclosed pre-clinical agents and provide an update on the current clinical landscape targeting IGF-1R. Furthermore, this review will discuss the evolution that has taken place in cancer biology surrounding IGF-1R signaling, from the initial static view of a single target in an isolated pathway to the current view of a dynamic landscape encompassing multiple targets and pathways interacting through a complex signaling network. Such a dynamic signaling network highlights the need for combination therapies for IGF-1R inhibitors in order to provide maximal benefit in the oncology setting.
Initial effort to develop selective type I insulin-like growth factor receptor inhibitors over insulin receptor
Type I insulin-like growth factor receptor specific monoclonal antibodies
One of the major concerns during the initial discovery and development of IGF-1R inhibitors centered around potential toxicity which was speculated to arise from targeting the highly homologous insulin receptor (IR). The IR supports critical physiological functions in regulating glucose homeostasis and, therefore, inhibition of IR could potentially lead to undesired toxicities such as hyperglycemia and hyperinsulinemia. Due to these concerns, mAbs that target the IGF-1R extra-cellular ligand-binding domain became a major focus since this recognition epitope is differentiated between IGF-1R and IR. Multiple IGF-1R mAb agents have been developed and have entered into clinical trials, including figitumumab (CP-751,871). Although the early disclosures for figitumumab reported encouraging clinical response,16 two phase III trials evaluating figitumumab in NSCLC, NCT00596830 (figitumumab in combination with paclitaxel and carboplatin) and NCT00673049 (figitumumab in combination with erlotinib) were discontinued in 2010 as independent data analysis indicated both trials were unlikely to meet the primary end points. It should be pointed out that insulin resistance and hyperglycemia are observed as side effects in human clinical trials with figitumumab, presumably through a growth hormone-related mechanism, indicating that disturbances in glucose homeostasis can occur even through IGF-1R specific targeting.17,18 Some data emerging from clinical studies with other IGF-1R neutralizing antibodies are also discouraging. For example, Amgen recently disclosed that a phase III trial evaluating their IGF-1R antibody AMG479 (ganitumab) in pancreatic cancer did not meet the primary end point. We are currently awaiting the results from a number of other clinical trials, single agent and combination, evaluating IGF-1R mAbs. These ongoing clinical activities are summarized in Table 1.
Table 1.
Insulin-like growth factor 1 receptor monoclonal antibodies: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R mAbs | In combination with | Phase | Indication | Trial ID |
|---|---|---|---|---|
| AMG 479 (Ganitumab) |
— | II | Epithelial ovarian cancer | NCT00718523 |
| — | II | Recurrent platinum sensitive ovarian cancer | NCT00719212 | |
| — | II | Ewing’s family tumor and desmoplastic small round cell tumors | NCT00563680 | |
| — | II | Neuroendocrine tumor, carcinoid tumor, pancreatic neuroendocrine tumor | NCT01024387 | |
| Gemcitabine, radiation, capecitabine | I | Locally advanced cancer of the pancreas | NCT01298401 | |
| Everolimus | I | Advanced solid tumors | NCT01122199 | |
| Everolimus, panitumumab | I | Advanced solid tumors | NCT01061788 | |
| Platinum-based chemotherapy | Ib/II | Extensive stage small cell lung cancer | NCT00791154 | |
| MEK162 | Ib/II | Selected advanced solid tumors | NCT01562899 | |
| Panitumumab | II | Colorectal cancer | NCT00788957 | |
| Panitumumab | II | Wt K-Ras metastatic colorectal cancer | NCT00891930 | |
| Metformin | II | Breast cancer | NCT01042379 | |
| FOLFIRI | II | KRAS-mutant metastatic colorectal cancer | NCT00813605 | |
| IMC-A12 (Cixutumumab) |
— | I | Solid tumors | NCT01007032 |
| — | II | Solid tumors | NCT00831844 | |
| — | II | Mesothelioma | NCT01160458 | |
| — | II | Thymoma, thymic carcinoma | NCT00965250 | |
| — | II | Metastatic melanoma of the eye | NCT01413191 | |
| — | II | Metastatic prostate cancer | NCT00520481 | |
| Sorafenib | I | Hepatocellular carcinoma | NCT01008566 | |
| Temozolomide, multi-agent chemotherapy | I | Metastatic rhabdomyosarcoma | NCT01055314 | |
| Everolimus, octreotide acetate | I | Neuroendocrine carcinoma | NCT01204476 | |
| Gemcitabine, Erlotinib | I/II | Metastatic pancreatic cancer | NCT00617708 | |
| Temsirolimus | I/II | Metastatic prostate cancer | NCT01026623 | |
| Temsirolimus | I/II | Locally recurrent or metastatic breast cancer | NCT00699491 | |
| Doxorubicin | I/II | Soft tissue sarcoma | NCT00720174 | |
| Sorafenib | II | Hepatocellular carcinoma | NCT00906373 | |
| Temsirolimus | II | Metastatic sarcomas | NCT01016015 | |
| Temsirolimus | II | Advanced cancers | NCT00678769 | |
| Bicalutamide, goserelin, leuprolide acetate | II | Metastatic prostate cancer | NCT01120236 | |
| Paclitaxel, carboplatin, bevacizumab | II | Non-small cell lung cancer | NCT00955305 | |
| Cisplatin, pemetrexed | II | Non-small cell lung cancer | NCT01232452 | |
| Carboplatin, pemetrexed | II | Non-small cell lung cancer | NCT01263782 | |
| Antiestrogens | II | Breast cancer | NCT00728949 | |
| Octreotide acetate | II | Metastatic neuroendocrine tumors | NCT00781911 | |
| Mitotane | II | Adrenocortical carcinoma | NCT00778817 | |
| Capecitabine, lapatinib | II | HER2 positive breast cancer | NCT00684983 | |
| Cisplatin, etoposide | II | Extensive stage small cell lung cancer | NCT00887159 | |
| Temsirolimus | II | Pediatric patients with recurrent or refractory solid tumors | NCT01614795 | |
| Cisplatin, etoposide | II | Extensive stage small cell lung cancer | NCT00887159 | |
| Cetuximab | II | Head and neck squamous cell carcinoma | NCT00957853 | |
| MK-0646 (Dalotuzumab) |
Ridaforolimus | I | Advanced cancers | NCT01243762 |
| Ridaforolimus | I | Advanced solid tumors | NCT01431547 | |
| Gemcitabine, erlotinib | I/II | Advanced pancreatic cancer | NCT00769483 | |
| Pemetrexed, cisplatin | II | Metastatic non-squamous lung cancer | NCT00799240 | |
| Ridaforolimus | II | Estrogen receptor positive breast cancer | NCT01234857 | |
| Irinotecan | II | Metastatic rectal carcinoma | NCT01609231 | |
| Ridaforolimus, exemestane | II | Breast cancer | NCT01605396 | |
| R1507 | — | II | Ewing’s sarcoma, osteosarcoma, synovial sarcoma, rhabdomyosarcoma and other sarcomas | NCT00642941 |
| 12 standard chemo drugs | I | Advanced malignancies | NCT00811993 | |
| Everolimus | I | Advanced solid tumors | NCT00985374 | |
| SCH 717454 (Robatumumab) |
— | II | Osteosarcoma sarcoma, Ewing’s peripheral neuroectodermal tumor |
NCT00617890 |
| MM-141 | — | I | Advanced solid tumors | NCT01733004 |
IGF-1R, insulin-like growth factor 1 receptor; mAbs, monoclonal antibodies.
Efforts to develop small molecule type I insulin-like growth factor receptor inhibitors selective over insulin receptor
Discovery of small molecule inhibitors selectively targeting the IGF-1R intracellular kinase domain over IR has proven to be challenging, due to an overall protein sequence homology of 84% and an ATP binding pocket sequence homology of 100% between these two family members. Nonetheless, several compounds have been reported with more than 10-fold selectivity for IGF-1R over IR in specific mechanistic assays. As highlighted in Figure 2, compound 1a (NVP-AEW541), a pyrrolo[2,3-d]pyrimidine derivative, was reported to inhibit IGF-IR in cells (cellular IC50=0.086 M) with ~27-fold selectivity versus IR (cellular IC50=2.3 M), despite the observed equal potency versus IGF-1R and IR in biochemical assays. The selectivity achieved was attributed to subtle conformational differences between the two receptors in the cellular setting along with the fact that the biochemical assay employed only the truncated kinase domain of each receptor. Compound 1a significantly reduces the growth of IGF-IR-driven fibrosarcomas and demonstrated in vivo tumor growth inhibition (TGI) in xenograft models.19 Very recently, it was reported that compound 1a enhances the effect of ionizing radiation in PTEN wild-type, but not in PTEN-deficient, prostate cancer cells.20 Compound 1b (NVP-ADW742), a close analog of 1a, was also disclosed with a 16-fold selectivity window for IGF-1R over IR in the cellular setting.21 Compounds 1a and 1b were the first ATP competitive small molecule IGF-1R inhibitors reported with more than 10-fold selectivity over IR. However, despite the initial claimed selectivity window over IR for this series of compounds, some recent reports refer to compound 1a as a dual IGF-1R/IR inhibitor with equal potencies against both receptors.22
Figure 2.
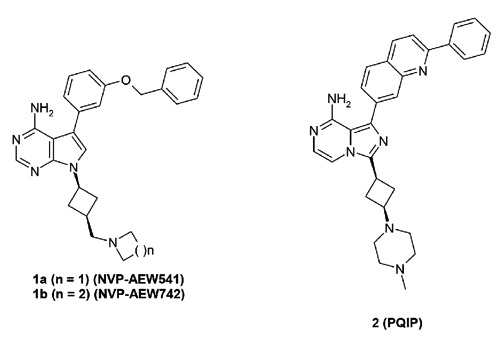
Adenosine triphosphate competitive insulin-like growth factor 1 receptor inhibitors with reported selectivity over insulin receptor.
Another reported small molecule ATP competitive IGF-1R inhibitor with selectivity over IR is compound 2 (PQIP, Figure 2).23 The cellular pIGF-1R (human) IC50 of PQIP was 19 nM as measured in 3T3/huIGF1R cells. This compound also showed a cellular pIR (human) IC50 of 261 nM in HepG2 cells and a cellular pIR (mouse) IC50 of 90 nM in Hepa-1 cells, thus an ~14-fold IGF-1R (human)/IR (human) selectivity was achieved based on comparison of IC50 values. However, a narrower window (~4-fold) was achieved when comparing potencies against IGF-1R (human) and IR (mouse). PQIP displayed potent anti-proliferative effects in a number of colorectal, breast, and pancreatic cancer cell lines, and further showed significant (>90%) TGI in multiple xenograft models including 3T3/huIGF1R and GEO (colorectal model) at an oral dose of 100 mg/kg.
In terms of non-ATP competitive IGF-1R inhibitors, cyclolignan picropodophyllin, a molecule of natural product origin (PPP, AXL1717, compound 3), was reported with potent cellular pIGF-1R activity (IC50=40 nM) without effects against pIR.24 It also uniquely induced IGF-1R degradation, which was thought to contribute to its anti-tumor activity. In pre-clinical studies, 3 is efficacious in a variety of tumor models including those derived from breast, prostate, melanoma and multiple myeloma.25-27 However, the mode of action of this compound has been called into question based on its microtubule inhibition activity and ability to induce cell death in IGF-IR–deficient cells.28 AXL1717 has advanced into clinical trials (Table 2). Currently, in a phase II randomized, open-label study, the effectiveness and safety of AXL1717 is being compared to docetaxel in patients with previously treated, locally advanced, or metastatic squamous cell carcinoma or adenocarcinoma of the lung (NCT01561456). Additionally, AXL1717 is being evaluated in a phase I trial in combination with gemcitabine and carboplatin against previously untreated, locally advanced, or metastatic squamous non-small-cell lung cancer (NCT01466647).
Table 2.
PPP/AXLl717: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R inhibitor | Phase | Indication | Trial ID |
|---|---|---|---|
 |
I | Advanced malignant tumors | NCT01725555 |
| I/II | Recurrent malignant astrocytomas | NCT01721577 | |
| II | Previously treated, locally advanced, or metastatic squamous cell carcinoma or adenocarcinoma of the lung | NCT01561456 |
IGF-1R, insulin-like growth factor 1 receptor.
It should be noted that in such cases where cellular selectivity is reported for a respective IGF-1R inhibitor, it is unclear as to how this selectivity translates over to different cell lines as well as to the in vivo setting. The selectivity of IGF-1R over IR for an inhibitor might be further validated by studies that demonstrate that the selectivity is not just specific to a particular cell line and is, therefore, translatable across multiple cell lines and ideally into the in vivo setting. The availability of IGF-1R and IR dependent xenograft models offers the opportunity to determine whether the observed in vitro selectivity is transferable to the in vivo setting where PK/PD/Efficacy relationships can be established and linked to IGF-1R selectivity over IR.
Development of type I insulin-like growth factor receptor inhibitors based on compensatory signaling/pathway cross talks
In recent years, the biology around IGF-1R has continued to advance and has revealed that IGF-1R signaling engages cross-talk interactions with various feedback, compensatory and redundant signaling pathways in cancer cells. This phenomenon provides cancer cells with additional survival pathways when IGF-1R signaling is suppressed, and may help to explain the marginal clinical success achieved to date by agents that selectively target IGF-1R, through either mAbs or small molecules. The field has since migrated toward a multi-targeting strategy in order to achieve enhanced efficacy through either an additive or synergistic effect. This strategy can be realized by two approaches: i) combination of an IGF-1R inhibitor with other molecular targeted agents (multi-drugs multi-targets); or ii) design of specific multi-targeting activities [(i.e. IGF-1R and IR or IGF-1R and epidermal growth factor receptor (EGFR)] into a single molecule (single-drug multi-targets). Both approaches, in theory, can be empirically guided by established biological rationale; however, each is presented with unique challenges. When combining agents, the potential for drug-drug interactions must be considered. Along with enhanced efficacy, the potential is introduced for enhanced toxicities and subsequent reduced tolerability. In addition, the feasibility of combining more than two agents, which includes a tolerability as well as cost component, must be taken into consideration. The latter approach, the design of multi-targeted and ideally rationally designed selectively non-selective inhibitors remains a challenge for drug discoverers.29 Achieving the right balance between building in preferred targets and building out dose-limiting off-targets to afford a truly selective multi-targeted inhibitor and not a highly promiscuous inhibitor remains a challenge due to the structural differences or lack thereof between targets within and outside a target class (e.g. kinases, G protein coupled receptors, ion channels). As various cross-talk interactions engaging IGF-1R have been reported,30-32 this review will focus on interactions of IGF-1R with kinases of therapeutic significance in cancer, including IR, EGFR, the mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase kinase (MEK). Additionally, the multi-drugs multi-target and single-drug multi-target approaches employed to target the IGF-1R cross-talk network will be discussed within.
Type I insulin-like growth factor receptor and insulin receptor signaling interactions
Pre-clinical research has demonstrated that IR signaling may also promote tumor growth and in this manner share redundancies with IGF-1R signaling. IR has been shown to promote tumor cell survival and proliferation both in vitro and in vivo.33-36 Detailed studies further demonstrate that IR is able to drive cell growth upon activation by IGF-2 through autocrine/paracrine mechanisms.37,38 While IR can be further classified into two isoforms (namely IR-A and IR-B), IR-A appears to be the primary contributor in cancer cell biology since this isoform is the predominant isoform of IR expressed in many tumor cells and is activated potently by both insulin and IGF-2.39 Epidemiological data also support a role for IR in cancer, where elevated levels of insulin are a poor prognostic indicator for prostate and breast cancer patients. In addition, overexpression of IR has been observed in several types of cancers such as breast, ovarian, and thyroid.40-42
More recent studies have established that there is reciprocal cross talk between IR and IGF-1R. IR can form a heterodimer with IGF-1R, and this heterodimer can be activated by the growth factor ligands IGF-1, IGF-2 and insulin (Figure 3). In normal cells that co-express both IGF-1R and IR, IR activity may be up-regulated in a compensatory manner upon inhibition of IGF-1R. For example, disruption of IGF-1R in osteoblasts enhances insulin signaling and action,43 and insulin-stimulated IRS-1 phosphorylation is elevated in IGF-1R knockout cells.44 Recent studies have shown that cross-talk resonates between IGF-1R and IR in human tumor cells. For example, treatment with an IGF-1R specific antibody is associated with elevated signaling through IR, which may limit sustained inhibition of AKT signaling (Figure 4).34 In contrast, the dual IGF-1R/IR inhibitor OSI-906 is capable of reducing phosphorylation of both receptors. In head-to-head studies comparing these two agents, OSI-906 displays superior efficacy compared to an IGF-1R specific antibody in human tumor xenograft models in which both IGF-1R and IR were phosphorylated (Figure 5).34 Taken together, these data strongly suggest that co-targeting both IGF-1R and IR is critical to achieve superior and broader anti-tumor efficacy as compared to targeting IGF-1R alone.
Figure 3.
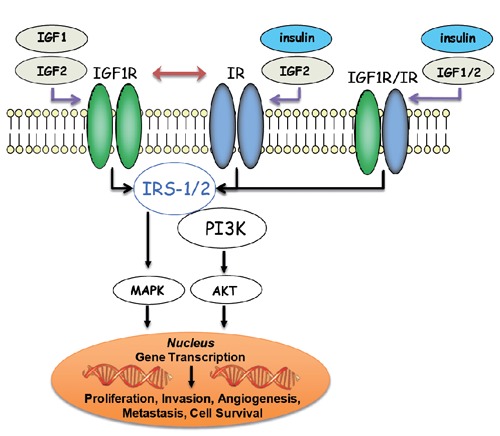
Insulin-like growth factor 1 receptor (IGF-1R)-insulin receptor (IR) reciprocal interactions: signal transduction from extracellular stimuli such as IGF-1, IGF-2 and insulin to intracellular pathways is mediated through IGF-1R, IR and IGF-1R/IR heterodimers. Furthermore, IR activity may be up-regulated in a compensatory manner upon inhibition of IGF-1R.
Figure 4.
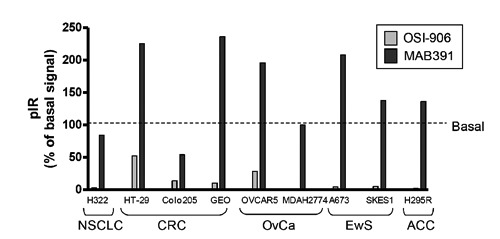
Compensatory activation of pIR observed upon treatment with MAB-391 but not OSI-906 across multiple cancer cell lines.34 NSCLC, non-small cell lung carcinoma; CRC, colorectal cancer; OvCa, ovarian carcinoma; EwS, Ewing’s sarcoma; ACC, adrenocortical carcinoma.
Figure 5.
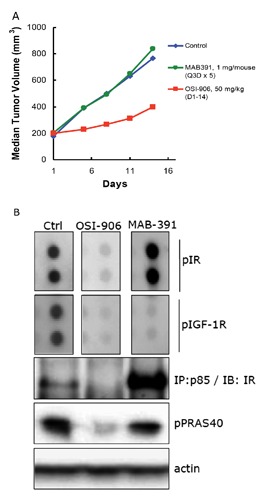
(A) Tumor growth inhibition observed in GEO colorectal xenograft model with OSI-906 but not IGF-1R mAb, MAB392. (B) Compensatory activation of pIR and restoration of pAKT signaling observed following 24-h treatment with MAB-391 but not OSI-906.34
Recent developments in small molecule type I insulin-like growth factor receptor/insulin receptor dual inhibitors
The discovery and clarification of IGF-1R/IR compensatory signaling provided a rationale for co-targeting IGF-1R and IR. As a result, a variety of potent, bioavailable dual IGF-1R/IR inhibitors derived from more than 10 distinct chemotypes have been reported. Several review articles covering the historical developments of dual IGF-1R/IR inhibitors have been published in recent years, where the major chemotypes, as represented by compounds 4-13 in Figure 6 and Table 3, their potencies, and binding interactions have been summarized and discussed.13-15 Here we will review and up-date the recent developments of the two most advanced clinical agents, OSI-906 (compound 4) and BMS-754807 (compound 6), as well as highlight very recent disclosures of pre-clinical compounds which have not been covered by previous reviews (compounds 14-15).
Figure 6.
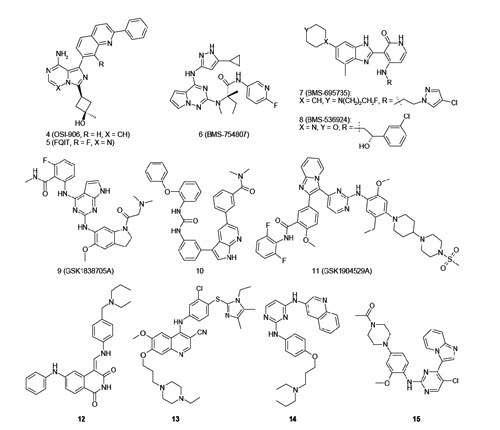
Selected sdenosine triphosphate competitive dual insulin-like growth factor 1 receptor/insulin receptor inhibitors.
Table 3.
A summary of the current status of disclosed adenosine triphosphate competitive dual insulin-like growth factor 1 receptor/insulin receptor inhibitors.
| Compound | Company | Current status |
|---|---|---|
| 4 (OSI-906) |
OSI | Phase-III |
| 5 (FQIT) |
OSI | Pre-clinical |
| 6 (BMS-754807) |
BMS | Phase II |
| 7 (BMS-695735) |
BMS | Pre-clinical |
| 8 (BMS-536924) |
BMS | Pre-clinical |
| 9 (GSK1838705A) |
GSK | Pre-clinical |
| 10 | GSK | Pre-clinical |
| 11 (GSK1904529A) |
GSK | Pre-clinical |
| 12 | Wyeth | Pre-clinical |
| 13 | Wyeth | Pre-clinical |
| 14 | Amgen | Pre-clinical |
| 15 | AZ | Pre-clinical |
OSI-906
OSI-906 is a potent, dual IGF-1R/IR inhibitor derived from an imidazo[1,5-a]pyrazine scaffold. As shown in Figure 7, compound 16 represented an unoptimized early hit from the series with a benzyloxyphenyl moiety at the C1 position with moderate IGF-1R potency (cellular pIGF-1R IC50=1.2 M). Binding interaction optimizations driven by structure-based drug design (SBDD) with both IGF-1R and IR co-crystal structures led to the identification of a 2-phenylquinoline sub-series as exemplified by compound 17 which showed a significant improvement in potency (cellular pIGF-1R IC50=86 nM) but suboptimal drug metabolism and pharmacokinetic (DMPK) properties as a result of metabolism at the C3 position of the cyclobutyl ring. Further optimization of potency and DMPK properties led to more advanced compounds such as 18 (AQIP),45 2 (PQIP)23 and eventually to the clinical agent, OSI-906 (linsitinib, compound 4).46 OSI-906 potently inhibits IGF-1R and IR in the cellular setting. It demonstrates good oral bioavailability across multiple pre-clinical species as well as significant tumor growth inhibition in multiple tumor xenograft models, including NIH-3T3/LISN, GEO [colorectal cancer (CRC)], H295R [adrenocortical carcinoma (ACC)], and IGF-1R and IR direct complementary (DC) models.
Figure 7.
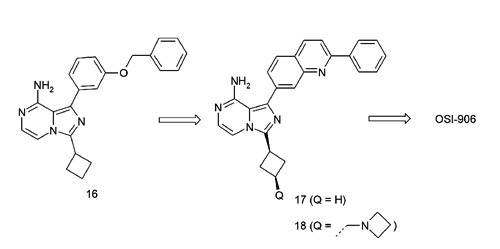
Evolution of dual insulin-like growth factor 1 receptor/insulin receptor inhibitors derived from an imidazo[1,5-a]pyrazine scaffold and structure-based drug design efforts.
Compared to other disclosed small molecule dual IGF-1R/IR inhibitors, OSI-906 has uniquely demonstrated exquisite selectivity when screened against a panel of 442 kinases (KinomeScanTM, DiscoveRx Corporation, Fremont, CA, USA), only inhibiting the IGF axis receptors; selectivity score (S-score) 0.008. The high degree of selectivity is attributed to unique binding interactions of OSI-906 with both IGF-1R and IR which stabilize both receptors in an intermediate conformation between active and inactive states.46 OSI-906 completed dose-escalating phase I clinical trials in humans.47,48 OSI-906 is orally bioavailable and noted to have an acceptable safety profile in humans. A maximum tolerated dose of 150 mg bid was achieved on a continuous dosing schedule where sustained plasma concentrations above the threshold required for anti-tumor efficacy in pre-clinical models were exceeded. Although hyperglycemia was evident in patients treated with OSI-906, a PK/PD relationship was observed where OSI-906 inhibited both p-IGF-1R and p-IR in peripheral blood mononuclear cells of cancer patients, with the degree of inhibition correlating with OSI-906 plasma levels. Encouraging disease control rate for response in evaluable patients was observed. One patient with ACC had a confirmed partial response at 16 weeks of treatment with OSI-906 and remained on the study for over 90 weeks with no drug-related toxicities observed. This response, in conjunction with pre-clinical efficacy in ACC xenograft models, led to the design of a randomized phase III study in ACC with OSI-906 as a single agent. Major emphasis for the ongoing clinical development plan is centered around combination therapy, and OSI-906 is being evaluated in multiple advanced clinical studies including combination with paclitaxel for the treatment of ovarian cancer and in combination with erlotinib for the treatment of NSCLC (Table 4).
Table 4.
OSI-906: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R inhibitor | In combination with | Phase | Indication | Trial ID |
|---|---|---|---|---|
| OSI-906 | — | I | Advanced solid tumors | NCT01529684 |
| — | II | Relapsed small cell lung cancer | NCT01533181 | |
| — | II | Asymptomatic or mildly symptomatic metastatic prostate cancer | NCT01533246 | |
| — | II | Gastrointestinal stromal tumors | NCT01560260 | |
| — | II | Relapsed small cell lung cancer | NCT01387386 | |
| Irinotecan | I | Advanced cancer | NCT01016860 | |
| Everolimus | I | Metastatic colorectal cancer | NCT01154335 | |
| Paclitaxel | I/II | Recurrent epithelial ovarian cancer (and other solid tumors) | NCT00889382 | |
| Erlotinib, radiation therapy | I/II | Advanced-stage cutaneous squamous cell carcinoma of the head and neck | NCT01465815 | |
| Erlotinib | II | Advanced NSCLC with activating mutations in EGFR | NCT1221077 | |
| Erlotinib | II | Maintenance therapy in patients with non-progression following chemotherapy in NSCLC | NCT01186861 | |
| Cetuximab | II | Platinum-refractory, recurrent/metastatic head and neck squamous cell carcinoma | NCT0142720 |
IGF-1R, insulin-like growth factor 1 receptor; NSCLC, non-small cell lung carcinoma; EGFR, epidermal growth factor receptor.
BMS-754807
BMS-754807 (compound 6) is a pyrrolotriazine-based IGF-1R inhibitor which evolved from an earlier pyrazolopyrimidine series (e.g. compound 19) through medicinal chemistry optimization (Figure 8).49 This molecule inhibited both IGF-1R and IR with equal potency (enzyme biochemical IC50=1.7 nM (IGF-1R); 1.8 nM (IR), measured at [ATP]=Km for each kinase). It also inhibited the phosphorylation of IGF-1R potently in the IGF-Sal cell line with an IC50 of 13 nM. This compound showed potent in vitro anti-proliferative effects and demonstrated significant TGI effects in multiple human tumor xenograft models when dosed orally. BMS-754807, a type I kinase inhibitor, has been reported to have off-target activities against other kinases, some of which have been implicated in cancer growth and survival. It is not clear as to whether these off-target activities are significantly inhibited in humans and offer a benefit or present dose limiting toxicities. BMS-754807 is currently in multiple advanced clinical trials, including a phase II study against hormone receptor-positive breast cancer (resistance to non-steroidal aromatase inhibitors), as well as in advanced or metastatic solid tumors (colorectal cancer, head and neck cancer, neoplasm metastasis) as a single agent in combination with cetuximab (Table 5). BMS-754807 was also evaluated in combination with trastuzumab against advanced or metastatic Her-2-positive breast cancer (NCT00788333, completed, data not disclosed to date), and in combination with paclitaxel and carboplatin against advanced or metastatic solid tumors (NCT00793897, completed, data not disclosed to date).
Figure 8.
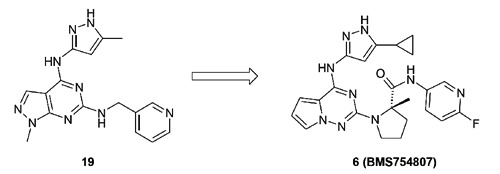
Pyrrolotriazine-based dual insulin-like growth factor 1 receptor/insulin receptor inhibitors 6 (BMS-754807) evolved from pyrazolopyrimidine scaffold.
Table 5.
BMS-754807: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R inhibitor | In combination with | Phase | Indication | Trial ID |
|---|---|---|---|---|
| BMS-754807 | — | I | Advanced or metastatic solid tumors | NCT00569036 |
| Cetuximab | I/II | Advanced or metastatic solid tumors | NCT00908024 | |
| Letrozole | II | Hormone receptor-positive breast cancer | NCT01225172 |
IGF-1R, insulin-like growth factor 1 receptor.
Most recent preclinical disclosure of type I insulin-like growth factor receptor/insulin receptor inhibitors
In 2011, a series of 2,4-bis-arylamino-1,3-pyrimidines were disclosed by Amgen as dual IGF-1R and IR inhibitors.50 As shown in Figure 9, medicinal chemistry efforts initiated from triazine-derived screening hit 20 (cellular pIGF-1R IC50=175 nM). Progression to a pyrimidine core afforded compound 21 and a nearly 10-fold potency improvement (cellular pIGF-1R IC50=19 nM) as a result of increased binding interactions and a decrease in the desolvation energy from the binding process. Further analoging around the aniline pharmacophore led to the identification of 14 with a cellular pIGF-1R IC50 value of 81 nM, ~4-fold less potent than that of 21. Both compounds 14 and 21 were further assessed in vivo in a Calu-6 tumor xenograft model. Interestingly, the less potent analog 14 demonstrated tumor growth inhibition of 56% at an oral dose of 100 mg/kg bid, while the more potent analog 21 failed to show any efficacy (0% TGI) at the same dose and dosing regimen. The authors attributed the efficacy of 14 to the achievement of high levels of tumor drug concentrations. In a single dose mouse PK study, compound 14 displayed a large volume of distribution (Vss) of 28 L/kg that translated into very high tumor drug concentrations (Cmax=142.9 M at 100 mg/kg dose) despite a much lower plasma drug exposure (Cmax=2.2 M at 100 mg/kg dose). In contrast, compound 21 displayed a lower Vss of 0.3 L/kg, which in turn resulted in lower tumor drug concentrations (Cmax=2.7 M at 100 mg/kg dose) although it showed a higher plasma drug exposure (Cmax=13.4 M at 100 mg/kg dose). Strong PD inhibition (~80%) of pIGF-1R was observed for compound 14 from the aforementioned xenograft study at the 100 mg/kg dose, but not at 30 mg/kg which may be the result of significantly lower tumor drug concentrations (Cmax=1.36 M). Compound 14 was selective against cell cycle regulators Aurora A, CDK2 and several kinases involved in tumor cell growth and differentiation, including MET, EGFR, FGFR, KDR and Tie2. Selectivity of this compound against a larger panel of kinases has not been disclosed.
Figure 9.
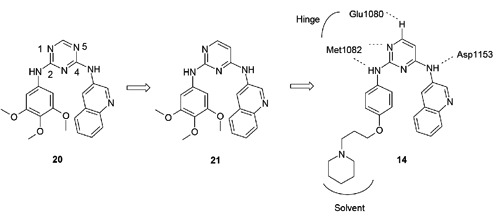
Optimization of 2,4-bis-arylamino-1,3-pyrimidines based dual insulin-like growth factor 1 receptor/insulin receptor (IGF-1R/IR) inhibitors led to compound 14. The proposed binding mode and major hydrogen bond interactions of 14 with IGF-1R based on a co-crystal structure of a related analog (PDB code: 3QQU).
In early 2012, AstraZeneca reported on an imidazo[1,2-a]pyridine series of IGF-1R/IR inhibitors.51,52 As shown in Figure 10, compound 22, which emerged from a historical CDK2 program, was identified from an IGF-1R cell-based HTS campaign as a moderate IGF-1R inhibitor. The CDK2 activity was successfully dialed out with the retention of IGF-1R activity as a result of modifications to the aniline pharmacophore (compound 23). Substitutions on the pyrimidine moiety especially with halogens significantly increased potency. For example, in the case where a bromine atom was implemented (compound 25), a remarkable ~150-fold improvement was achieved in both biochemical and cellular potencies when compared to the non-substituted analog 23. Further medicinal chemistry analoging efforts on the imidazopyridine pharmacophore did not yield any additional improvements. Compound 15 was selected for further profiling and was shown to be orally bioavailable in multiple pre-clinical species including mouse, rat and dog (F%>30%); however, no in vivo efficacy data has been disclosed. The kinase selectivity of 15 was evaluated in a panel of 69 kinases at a compound concentration of 1 M, with 3 kinases, JNK1 (MAPK8), JNK2 (MAPK9) and ERK8 (MAPK15) being inhibited at over 80%.
Figure 10.
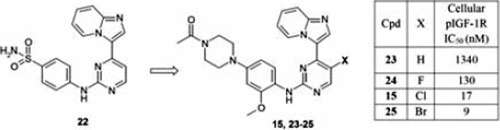
Optimization of imidazo[1,2-a]pyridine-based dual insulin-like growth factor 1 receptor (IGF-1R) and insulin receptor (IR) inhibitors.
Type I insulin-like growth factor receptor and epidermal growth factor receptor signaling interactions
Although both small molecule and antibody EGFR inhibitors have demonstrated clinical utility,53 resistance often develops in the clinic following longer term treatment. In pre-clinical models, interaction between IGF-1R and EGFR signaling has been shown to be a contributing mechanism to resistance to anti-EGFR therapies (Figure 11). For example, in early stage NSCLC patients, IGF-1R protein expression was shown to be associated with EGFR protein expression and elevated coexpression of both IGF-1R and EGFR correlates with poor prognosis, as evidenced by shorter disease-free survival.54 Reciprocal interaction between IGF-1R and EGFR has also been described in a variety of other tumor types, including breast cancer, CRC, pancreatic cancer (PaCa) and HCC, where inhibition of either receptor can promote the activation of the other receptor.55-58 Multiple mechanisms for IGF-1R/EGFR interaction have been proposed, including direct association and activation between the two receptors, modulation of the availability of reciprocal ligands, or through interactions with common partners or downstream effectors.59
Figure 11.
Reciprocal interaction between insulin-like growth factor 1 receptor (IGF-1R) and epidermal growth factor receptor (EGFR) where inhibition of either receptor can promote the activation of the other receptor.
In pre-clinical studies, co-targeting EGFR and IGF-1R was shown to produce superior efficacy compared to treatment with the respective monotherapies in multiple models.60,61 For example, the combination of erlotinib and an IGF-1R TKI, such as PQIP or OSI-906, leads to a synergistic increase in apoptosis in multiple cell lines including H292 (Figure 12) as well as synergistic tumor growth inhibition, regressions and enhanced cure rates in xenograft models of NSCLC and pancreatic cancer (Figure 13).62 Collectively, these pre-clinical studies strongly support the hypothesis of co-targeting IGF-1R and EGFR signaling as a novel approach to achieve superior clinical response and prevent/minimize drug resistance due to IGF-1R/EGFR reciprocal cross talk. This hypothesis is currently being evaluated in multiple clinical trials (multi-drug multi-target approach) as highlighted in Table 6.
Figure 12.
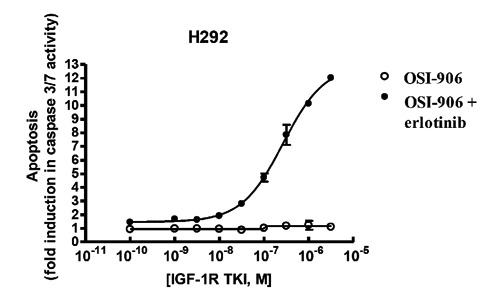
Synergistic increase in apoptosis observed in vitro with the combination of insulin-like growth factor 1 receptor (IGF-1R)/insulin receptor dual inhibitor OSI-906 and epidermal growth factor receptor inhibitor erlotinib (10 uM). Either erlotinib or OSI-906 alone had minimal (<2-fold) effect on caspase 3/7 activity; however, the combination results in more than 12-fold increased caspase 3/7 activity (E. Buck, M.J. Mulvihill, et al., 2013, unpublished results).
Figure 13.
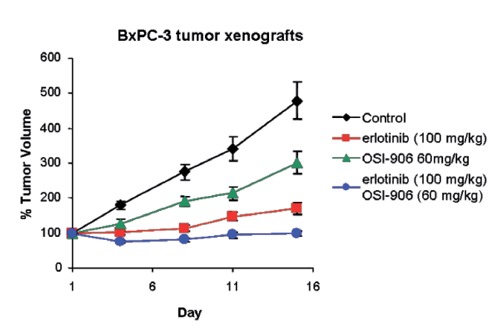
Enhanced tumor growth inhibition observed with the combination of insulin-like growth factor 1 receptor/insulin receptor dual inhibitor OSI-906 and epidermal growth factor receptor inhibitor erlotinib E. Buck, M.J. Mulvihill, et al., 2013, unpublished results).
Table 6.
Insulin-like growth factor 1 receptor inhibitor in combination with epidermal growth factor receptor/HER2 inhibitor: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R inhibitor | In combination with EGFR or HER2 inhibitor | Phase | Indication | Trial ID |
|---|---|---|---|---|
| AMG479 | Panitumumab | II | Wt K-Ras metastatic colorectal cancer | NCT00891930 |
| Panitumumab | II | Colorectal cancer | NCT00788957 | |
| IMC-A12 (Cixutumumab) |
Cetuximab | II | Head and neck squamous cell carcinoma | NCT00957853 |
| OSI-906 | Cetuximab | II | Platinum-refractory, recurrent/metastatic head and neck squamous cell carcinoma | NCT01427205 |
| Erlotinib | I/II | Advanced-stage cutaneous squamous cell carcinoma of the head and neck | NCT01465815 | |
| Erlotinib | II | Advanced NSCLC with EGFR activating mutations | NCT01221077 | |
| Erlotinib | II | Maintenance therapy in patients with non-progression following chemotherapy in NSCLC | NCT01186861 | |
| BMS-754807 | Cetuximab | I/II | Advanced or metastatic solid tumors | NCT00908024 |
IGF-1R, insulin-like growth factor 1 receptor; EGFR, epidermal growth factor receptor; NSCLC, non-small cell lung carcinoma.
Recent developments in small molecule inhibitors co-targeting both type I insulin-like growth factor receptor and epidermal growth factor receptor
The strong rationale previously described for co-targeting IGF-1R/IR and EGFR through the combination of selective inhibitor provided the scientific basis to pursue a single molecule that could selectively target both pathways. Developments in this area utilizing the single-drug multi-target approach has been reported by Abbott Laboratories around a series of imidazo[2,1-b]thiazole and pyrazolo[3,4-d]pyrimidine derived IGF-1R inhibitors (Table 7).63-65 The imidazo[2,1-b]thiazole series, as highlighted by compound 26, displayed significant potency versus IGF-1R in A431cells with an IC50 value of 41 nM and modest EGFR and ErbB2 potencies of 350 and 310 nM, respectively. From the pyrazolopyrimidine class, compound 27a was reported to inhibit all three targets with a cellular IC50 of ~100 nM. Unfortunately, the overall kinase selectivity profile of this compound was not reported and, therefore, these cellular activities are unable to be placed into a proper context. Compound 27a displayed moderate mouse PK properties (oral bioavailability 12%) and demonstrated significant PD inhibition of both pIGF-1R and pEGFR in a mouse model. Unfortunately, no in vivo efficacy or tolerability results were reported. From the same pyrazolopyrimidine chemotype, compound 27b was highlighted as an advanced analog, displaying cellular potencies (IC50 values) versus IGF-1R, pEGFR and pErbB2, of 207, 2300 and 340 nM, respectively. The oral bioavailability was modest, at 20% in the mouse. Compound 27 was evaluated in an in vivo PD model where intravenous dosing at 30 mg/kg demonstrated 50% inhibition of pIGF-1R; however, PD versus EGFR or ErbB2 was not reported. The kinase selectivity of compound 27 was assessed against 80 kinases and the results showed potent inhibition of several other kinases including the Aurora family. No in vivo tumor growth inhibition data were reported. Collectively, these efforts highlight the challenges as noted by Morphy29 associated with achieving highly selective nonselective kinase inhibitors, when targeting structurally distinct kinase family members.
Table 7.
Enzyme and cellular IC50 values for select pyrazolo[3,4-d]pyrimidine and imidazo[2,1-b]thiazole-derived insulin-like growth factor 1 receptor/epidermal growth factor receptor/ErbB-2 inhibitors.
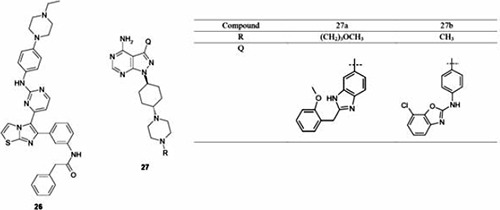
| Compound | Enzyme (cellular) IC50 nM | ||
|---|---|---|---|
| IGF-1R | EGFR2* | Erb-B2 | |
| 26° | 52 (41) | 16 (350) | 170 (310) |
| 27a# | 81 (115) | 58 (85) | 54 (94) |
| 27b§ | 12.5 (207) | 31.5 (2300) | 7.5 (340) |
IGF-1R, insulin-like growth factor 1 receptor; EGFR, epidermal growth factor receptor.
*EGFR construct with L858R mutation;
°ELISA assays of receptor auto-phosphorylation, in A431cells for pIGF-1R and pEGFR and N87 cells for pErbB2;
#ELISA assays of receptor auto-phosphorylation, in MiaPaCa cells for pIGF-1R and pEGFR and N87 cells for pErbB2;
§ELISA assays of receptor auto-phosphorylation, in MiaPaCa cells for pIGF-1R and pEGFR and BT474 cells for pErbB2.
Ranbaxy Laboratories recently disclosed compound 28 as a dual IGF-1R/EGFR inhibitor derived from a bis-arylamino-pyrimidine scaffold.66 This compound has balanced and potent IGF-1R and EGFR activities, and showed superior anti-proliferative effects in multiple cell lines, such as A431, HT29, and A549, when compared to EGFR inhibitor erlotinib alone or IGF-1R inhibitor NVP-AEW541 (compound 1a) alone (Table 8). Compound 28 also showed anti-proliferative sensitivity in H1975 cells with a GI50 of 1.3 M, a cell line that harbors an EGFR mutation (T790M) and reported to be resistant to erlotinib (GI50 of 47 M). The authors attributed the observed efficacy to the unique multi-targeted profile of compound 28, although it is not clear if compound 28 directly inhibits EGFR with the T790M mutation. The kinase selectivity of this compound was evaluated in a focused kinase panel at a concentration of 10 M, where no significant inhibition (<50%) of kinases such as FGFR, Aurora A, GK, Wee-1 and Erk-1, was observed. Interestingly, only moderate inhibition of ErbB-2 (55% inhibition) was observed at this concentration, providing a potential point of differentiation to IGF-1R/EGFR/ErbB-2 inhibitors 26 and 27. Unfortunately, poor mouse PK properties associated with this compound as indicated by high plasma clearance and low T1/2 prevented it from being further evaluated in animal studies (e.g. mouse xenograft efficacy and tolerability studies).
Table 8.
Small molecule inhibitor co-targeting insulin-like growth factor 1 receptor and epidermal growth factor receptor.
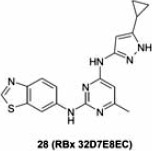
| Compound | Enzymatic IC50 (nM) | Anti-proliferative IC50 (nM) | |||
|---|---|---|---|---|---|
| IGF-1R | EGFR | A431 | HT29 | A549 | |
| Erlotinib | >10,000 | 45 | 1080 | 2275 | >10,000 |
| 28 | 74 | 45 | 170 | 274 | 111 |
| 1a | 291 | >10,000 | 899 | 9540 | 615 |
IGF-1R, insulin-like growth factor 1 receptor; EGFR, epidermal growth factor receptor.
Within the field of dual IGF-1R/EGFR inhibitors, what appears to be lacking is a head-to-head in vivo comparison of selective dual IGF-1R/EGFR inhibitors to selective compounds only targeting IGF-1R and/or EGFR in respect to both efficacy and tolerability. Furthermore, it would be of interest to determine if such specifically designed multitargeted inhibitors display a superior or differentiated profile (e.g. efficacy, tolerability) to that of the combination of a selective IGF-1R/IR inhibitor with a selective EGFR/Erb2 inhibitor. Unfortunately, no such comparative data have been disclosed to date.
Type I insulin-like growth factor receptor and mammalian target of rapamycin signaling interactions
Studies of the PI3K/AKT/mTOR pathway have revealed that inhibition of mTORC1 by a rapamycin analog led to upregulation of AKT activity via the S6K-IRS2 negative feedback loop and thus rendering the mTORC1 inhibitor less effective.67 Since inhibition of IGF-1R leads to downregulation of AKT signaling, it was then hypothesized that inhibiting both mTORC1 and IGF-1R may counteract this feedback loop and produce a superior therapeutic response compared to mTORC1 inhibition alone. This hypothesis has been confirmed in pre-clinical studies by using a combination of rapamycin and the IGF-1R inhibitor NVP-AEW541 (compound 1a).67 A recent study assessing the combination of ridaforolimus (MK-8669, mTORC1 inhibitor) and dalotuzumab (MK-0646, IGF-1R mAb) also showed similar results.68 In this study, a panel of over 60 breast cancer cell lines were screened with ridaforolimus to test this hypothesis. It was found that ridaforolimus treatment up-regulated multiple key nodes of the IGF-1R pathway and provided the rationale for the mTORC1/IGF-1R combination. Eventually, Lum-B breast cancer was determined to be the most suitable for this combination study due to its ER+ characteristic (correlates with sensitivity of ridaforolimus) and highest IGF-1R mRNA level. Based on encouraging pre-clinical results, the combination of ridaforolimus and dalotuzumab is currently being evaluated in multiple phase I and phase II trials (Table 9). Based on similar biological rationale, R1507, an IGF-1R mAb is also being evaluated in a phase I study in combination with mTORC1 inhibitor everolimus.
Table 9.
Insulin-like growth factor 1 receptor inhibitor in combination with mTORC1 inhibitors: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R inhibitor | In combination with mTORC1 inhibitor | Phase | Indication | Trial ID |
|---|---|---|---|---|
| R1507 | Everolimus | I | Advanced solid tumors | NCT00985374 |
| AMG479 (Ganitumab) |
Everolimus | I | Advanced solid tumors | NCT01122199 |
| IMC-A12 (Cixutumumab) |
Everolimus | I | Neuroendocrine carcinoma | NCT01204476 |
| Temsirolimus | I/II | Metastatic prostate cancer | NCT01026623 | |
| Temsirolimus | I/II | Locally recurrent or metastatic breast cancer | NCT00699491 | |
| Temsirolimus | II | Advanced cancers | NCT00678769 | |
| Temsirolimus | II | Metastatic sarcomas | NCT01016015 | |
| Temsirolimus | II | Pediatric patients with recurrent or refractory solid tumors | NCT01614795 | |
| MK-0646 (Dalotuzumab) |
Ridaforolimus | I | Advanced cancers | NCT01243762 |
| Ridaforolimus | I | Advanced solid tumors | NCT01431547 | |
| Ridaforolimus | II | Estrogen receptor positive breast cancer | NCT01234857 | |
| OSI-906 | Everolimus | I | Metastatic colorectal cancer | NCT01154335 |
IGF-1R, insulin-like growth factor 1 receptor.
It has recently been shown that the tumor suppressor gene, LKB1 is lost in ~35% of NSCLC. LKB1 plays a critical role in energy sensing and regulates the PI3K/mTOR pathway.69 Loss of LKB1 leads to activation of the mTOR and IGF-1R pathways.70 Dual inhibition of IGF-1R and mTOR with dual IGF-1R/IR TKI OSI-906 and dual mTORC1/2 inhibitor OSI-02771 synergistically inhibits proliferation in several pre-clinical models with LKB1 loss (mutation) including H1437, A427, H2126 and H1395. The combination of IGF-IR + mTOR inhibition with OSI-906 and OSI-027 was more effective than either monotherapy in the H1437 mouse xenograft model.70
Type I insulin-like growth factor receptor and mitogen-activated protein kinases signaling interactions
IGF-1R signaling has also been implicated in drug resistance developed following treatment with a myriad of other anti-tumor agents including inhibitors acting within the MAPK pathway. Treatment with an MEK kinase inhibitor has been shown to result in elevated IGF-driven AKT signaling, and the combination of the dual IGF-1R/IR inhibitor OSI-906 with an MEK inhibitor has been shown to inhibit tumor cell proliferation in a synergistic manner.62 Furthermore, MEK inhibitors in combination with an IGF-1R inhibitor (AXL1717) induce cell death in tumor cells with acquired resistance to a RAF inhibitor.72 There is also clinical data highlighting the potential for IGF-1R to play a role in resistance to MAPK pathway inhibitors. For example, resistance following long-term treatment with the B-RAF inhibitor vemurafenib in melanoma patients is associated with elevated IGF-1R expression.73 These data suggest a combination of a MAPK pathway inhibitor and an IGF-1R inhibitor may overcome certain clinically observed drug resistance to vemurafenib. Currently, IGF-1R monoclonal antibody AMG479 is being evaluated in a clinical trial in combination with MEK1/2 inhibitor MEK162 against advanced solid tumors (Table 10).
Table 10.
Insulin-like growth factor 1 receptor inhibitor in combination with mitogen-activated protein kinases pathway inhibitor: ongoing clinical trials as of January 2013 (www.clinicaltrials.gov).
| IGF-1R inhibitor | MAPK pathway inhibitor | Phase | Indication | Trial ID |
|---|---|---|---|---|
| AMG479 (Ganitumab) |
MEK162 | lb/II | Selected advanced solid tumors | NCT01562899 |
IGF-1R, insulin-like growth factor 1 receptor; MAPK, mitogen-activated protein kinases.
Promiscuous kinase inhibitors with significant type I insulin-like growth factor receptor activities
XL228 is a multikinase inhibitor with potent activities against IGF-1R, SRC family kinases, BCR-ABL, FGFR1-3 (fibroblast growth factor receptors 1-3) and the aurora kinases. This compound was derived from a 2,4-disubstituted pyrimidine series although detailed information about its structure has not been disclosed. XL228 was progressed into two phase I clinical trials (NCT00464113, NCT00526838) and early clinical data showed evidence of IGF-1R and SRC kinase pathway inhibition, as supported by phospho-protein analysis in circulating leukocytes, hair follicles and skin biopsy samples, and by transient upregulation of plasma glucose and insulin. It was generally well tolerated at its maximum tolerated dose of 6.5 mg/kg, and showed evidence of single agent anti-tumor activity which included a confirmed partial response in a NSCLC patients.74 However, both trials have been terminated and no new clinical activity of this compound has been disclosed to date.
A class of catechol derivatives was reported as IGF-1R inhibitors, most likely operating through a non-ATP competitive manner. Some representative compounds are shown in Figure 14. From this class, natural product 30 (NDGA, INSM-18)75 has progressed into phase-I/II clinical trials against non-metastatic hormone-sensitive prostate cancer. Compound 30 was dosed at 2000 mg/day orally without any reported significant toxicities. However, it was recently reported that analysis showed no significant decline in prostate-specific antigen after three cycles of treatment among the first 12 patients enrolled.76 It should be pointed out that compound 30 also has activities against lipoxygenase and other kinases such as ErbB-2 and FGFR3.77 Both compounds 31 and 32 are synthetic analogs with only moderate IGF-1R potencies and remain to date in the pre-clinical stage. There are additional small molecule IGF-1R inhibitors that have been reported to have progressed into clinical trials, such as KW2450 (NCT00921336, NCT01199367) and PL2258 (NCT01779336). While these are certainly interesting developments, it is difficult to make any further comments on these agents since no information as to the structure or any pharmacological details of these compounds have been disclosed.
Figure 14.
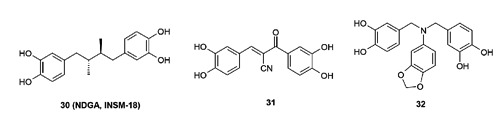
Catechol derivatives as non-adenosine triphosphate competitive insulin-like growth factor 1 receptor inhibitors.
Conclusions
Since the initial discovery of the involvement of IGF-1R signaling in human cancer in the 1980s,1-3 pre-clinical studies have provided a foundation for targeting IGF-1R signaling as a potential treatment for cancer. Consequently, numerous approaches have been undertaken to modulate IGF-1R activity, resulting in the progression of multiple agents into human clinical trials for the ultimate validation of IGF-1R as a cancer target. The plethora of drug discovery efforts around IGF-1R was pioneered by monoclonal antibodies which specifically target the ligand binding extracellular domain of this receptor,78 and this was then followed by small molecule IGF-1R kinase inhibitors with both ATP competitive and non-ATP competitive binding modes. Unlike monoclonal antibodies that are highly specific for the designated target, small molecule kinase inhibitors can be tuned to include additional therapeutically beneficial off-target activities. The understanding of cancer biology around IGF-1R, and especially its role in various stages of cancer and its interactions with other signaling pathways, clearly influenced the design of small molecule IGF-1R kinase inhibitors. Historically, selective IGF-1R inhibitors that did not inhibit IR were pursued, due to concerns about potential toxicity that were suggested to arise from the inhibition of IR. Thus, initial efforts focused around creating selective IGF-1R agents and, therefore, avoiding IR inhibition through various strategies such as targeting allosteric binding sites. Although compounds with various degrees of selectivity (IGF-1R vs IR) have been reported and agents from this class have since progressed into clinical trials, success from this approach as single agents appears to be limited.
Recent research on IGF-1R signaling has focused on compensatory signaling as a resistance mechanism utilized by cancer cells for survival when IGF-1R activity is inhibited. Researchers now realize that resistance mechanisms involve a dynamic network of multiple intertwining signaling pathways. This includes interactions of IGF-1R with other kinases such as IR, EGFR, mTOR and MEK, as well as other receptors, such as the estrogen receptor.79 These studies have suggested various possibilities of multi-drug multi-target and/or single-drug multi-target approaches to simultaneously block IGF-1R/IR signaling and other compensatory survival mechanisms as cancer treatments. Generally speaking, the multi-drugs multi-target approach can be successful when the combination of drugs are tolerated and provide greater efficacy. The risks and challenges associated with this approach are potential drug-drug interactions, complications in designing dose schedules and overall complications in drug combination clinical trial design. Nonetheless, drug combinations have become a common approach in clinical development, with examples in the oncology kinase field including the ongoing clinical evaluation of drug combinations co-targeting IGF-1R and EGFR, as well as MEK and EGFR, just to name a few.
Compared to the multi-drugs multi-target approach, the single-drug multi-target approach has the advantage of no risks related to drug-drug interactions and simplifies clinical trial design. This approach is particularly attractive when targeting multiple proteins that share relatively conserved binding sites, such as the ATP binding pocket of kinases. Kinases are also an example of a preferred target class for this approach because binding interactions are relatively well understood within ATP pockets and selectivity optimization can be greatly facilitated by taking advantage of available structural information via SBDD. The major challenge associated with this single-drug multi-target approach is engineering a molecule that possesses an optimal potency and selectivity profile against multiple targets. The compound optimization process could become a daunting task based upon the targets of interest when steering through multiple undesired off-targets while maintaining desired drug-like properties. Nonetheless, it is encouraging to see drug discovery efforts that have adopted the single-drug multi-target concept in recent years.29,80,81 This approach has also been pursued in the IGF-1R field, as exemplified by small molecule dual IGF-1R/IR inhibitors and IGF-1R/EGFR inhibitors reviewed within. There is excitement in the oncology community around the multiple approaches and various drug combinations that are being actively pursued in the clinical setting based upon compelling and supporting pre-clinical data. It is our hope that ongoing clinical trials centered around IGF-1R will provide proof-of-concept for this target in humans and beneficial therapies for cancer patients around the world.
Acknowledgments
OSI Pharmaceuticals LLC is a wholly owned subsidiary of Astellas US LLC, 1 Bioscience Park Drive, Farmingdale, NY 11735, USA.
References
- 1.Myal Y, Shiu RP, Bhaumick B, et al. Receptor binding and growth-promoting activity of insulin-like growth factors in human breast cancer cells (T-47D) in culture. Cancer Res 1984;44:5486-90. [PubMed] [Google Scholar]
- 2.Pollak MN, Perdue JF, Margolese RG, et al. Presence of somatomedin receptors on primary human breast and colon carcinomas. Cancer Lett 1987;38:223-30. [DOI] [PubMed] [Google Scholar]
- 3.Sell C, Rubini M, Rubin R, et al. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A 1993;90:11217-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubin R, Baserga R. Insulin-like growth factor-I receptor. Its role in cell proliferation, apoptosis, and tumorigenicity. Lab Invest 1995;73:311-31. [PubMed] [Google Scholar]
- 5.Kondo M, Suzuki H, Ueda R, et al. Frequent loss of imprinting of the H19 gene is often associated with its overexpression in human lung cancers. Oncogene 1995;10:1193-8. [PubMed] [Google Scholar]
- 6.Kaiser U, Schardt C, Brandscheidt D, et al. Expression of insulinlike growth factor receptors I and II in normal human lung and in lung cancer. J Cancer Res Clin Oncol 1993;119:665-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma J, Giovannucci E, Pollak M, et al. A prospective study of plasma C-peptide and colorectal cancer risk in men. J Natl Cancer Inst 2004;96:546-53. [DOI] [PubMed] [Google Scholar]
- 8.LeRoith D, Roberts CT., Jr.The insulin-like growth factor system and cancer. Cancer Lett 2003;195:127-37. [DOI] [PubMed] [Google Scholar]
- 9.Ceresa BP, Pessin JE. Insulin regulation of the Ras activation/inactivation cycle. Mol Cell Biochem 1998;182:23-9. [PubMed] [Google Scholar]
- 10.Gooch JL, Van Den Berg CL, Yee D. Insulin-like growth factor (IGF)-I rescues breast cancer cells from chemotherapy-induced cell death--proliferative and anti-apoptotic effects. Breast Cancer Res Treat 1999;56:1-10. [DOI] [PubMed] [Google Scholar]
- 11.Turner BC, Haffty BG, Narayanan L, et al. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res 1997;57:3079-83. [PubMed] [Google Scholar]
- 12.Jerome L, Alami N, Belanger S, et al. Recombinant human insulinlike growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res 2006;66:7245-52. [DOI] [PubMed] [Google Scholar]
- 13.Wittman M, Velaparthi U, Vyas D. Recent progress in the development of small molecule inhibitors of insulin-like growth factor-I receptor kinase. Annu Rep Med Chem 2009;44:281-99. [Google Scholar]
- 14.Li R, Pourpak A, Morris SW. Inhibition of the insulin-like growth factor-1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J Med Chem 2009;52:4981-5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin M, Wang J, Buck E, Mulvihill MJ. Small-molecule ATP-competitive dual IGF-1R and insulin receptor inhibitors: structural insights, chemical diversity and molecular evolution. Future Med Chem 2012;4:315-28. [DOI] [PubMed] [Google Scholar]
- 16.Karp DD, Paz-Ares LG, Novello S, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol 2009;27:2516-22. [DOI] [PubMed] [Google Scholar]
- 17.Haluska P, Shaw H, Batzel GN, et al. Phase I dose escalation study of the anti-IGF-1R monoclonal antibody CP-751,871 in patients with refractory solid tumors. ASCO Annual Meeting Abstracts 2007;25:3586. [DOI] [PubMed] [Google Scholar]
- 18.Jassem J, Langer CJ, Karp DD, et al. Randomized, open label, phase III trial of figitumumab in combination with paclitaxel, carboplatin versus paclitaxel, carboplatin in patients with non-small cell lung cancer (NSCLC). J Clin Oncol 2010;28:7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garría-Echeverría C, Pearson MA, Marti A, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-1R kinase. Cancer Cell 2004;5:231-9. [DOI] [PubMed] [Google Scholar]
- 20.Isebaert SF, Swinnen JV, McBride WH, Haustermans KM. Insulin-like growth factor-type 1 receptor inhibitor NVP-AEW541 enhances radiosensitivity of PTEN wild-type but not PTEN-deficient human prostate cancer cells. Int J Radiat Oncol Biol Phys 2011;1:239-47. [DOI] [PubMed] [Google Scholar]
- 21.Warshamana-Greene GS, Litz J, Buchdunger E, et al. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin Cancer Res 2005;11:1563-71. [DOI] [PubMed] [Google Scholar]
- 22.Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res 2012;72:3372-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ji Q, Mulvihill MJ, Rosenfeld-Franklin M, et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol Cancer Ther 2007;6:2158-67. [DOI] [PubMed] [Google Scholar]
- 24.Girnita A, Girnita L, Del PF, et al. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res 2004;64:236-42. [DOI] [PubMed] [Google Scholar]
- 25.Menu E, Jernberg-Wiklund H, Stromberg T, et al. Inhibiting the IGF-1 receptor tyrosine kinase with the cyclolignan PPP: an in vitro and in vivo study in the 5T33MM mouse model. Blood 2006;107:655-60. [DOI] [PubMed] [Google Scholar]
- 26.Karasic TB, Hei TK, Ivanov VN. Disruption of IGF-1R signaling increases TRAIL-induced apoptosis: A new potential therapy for the treatment of melanoma. Exp Cell Res 2010;316:1994-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan Z, Choy E, Harmon D, et al. Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol Cancer Ther 2009;8:2122-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linder S, Shoshan MC, Gupta RS. Picropodophyllotoxin or podophyllotoxin does not induce cell death via insulin-like growth factor-I receptor. Cancer Res 2007;67:2899-900. [DOI] [PubMed] [Google Scholar]
- 29.Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem 2010;53:1413-37. [DOI] [PubMed] [Google Scholar]
- 30.Buck E, Mulvihill M. Small molecule inhibitors of the IGF-1R/IR axis for the treatment of cancer. Expert Opin Investig Drugs 2011;20:605-21. [DOI] [PubMed] [Google Scholar]
- 31.Carapancea M, Cosaceanu D, Budiu R, et al. Dual targeting of IGF-1R and PDGFR inhibits proliferation in high-grade gliomas cells and induces radiosensitivity in JNK-1 expressing cells. J Neurooncol 2007;85:245-54. [DOI] [PubMed] [Google Scholar]
- 32.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res 2010;16:2505-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giorgino F, Belfiore A, Milazzo G, et al. Overexpression of insulin receptors in fibroblast and ovary cells induces a ligand-mediated transformed phenotype. Mol Endocrinol 1991;5:452-9. [DOI] [PubMed] [Google Scholar]
- 34.Buck E, Gokhal PC, Koujak S, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther 2010;9:2652-64. [DOI] [PubMed] [Google Scholar]
- 35.Heuson JC, Legros N. Effect of insulin and of alloxan diabetes on growth of the rat mammary carcinoma in vivo. Eur J Cancer 1970;6:349-51. [DOI] [PubMed] [Google Scholar]
- 36.Heuson JC, Legros N. Influence of insulin deprivation on growth of the 7, 12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Res 1972;32:226-32. [PubMed] [Google Scholar]
- 37.Sciacca L, Costantino A, Pandini G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 1999;18:2471-9. [DOI] [PubMed] [Google Scholar]
- 38.Vella V, Pandini G, Sciacca L, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab 2002;87:245-54. [DOI] [PubMed] [Google Scholar]
- 39.Sciacca L, Prisco M, Wu A, et al. Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate-1. Endocrinology 2003;144:2650-8. [DOI] [PubMed] [Google Scholar]
- 40.Cox ME, Gleave ME, Zakikhani M, et al. Insulin receptor expression by human prostate cancers. Prostate 2009;69:33-40. [DOI] [PubMed] [Google Scholar]
- 41.Pollak M. Insulin and insulin-like growth factor signaling in neoplasia. Nat Rev Cancer 2008;8:915-28. [DOI] [PubMed] [Google Scholar]
- 42.Pollak M. Targeting insulin and insulin-like growth factor signaling in oncology. Curr Opin Pharmacol 2008;8:384-92. [DOI] [PubMed] [Google Scholar]
- 43.Fulzele K, DiGirolamo DJ, Liu Z, et al. Disruption of the insulin-like growth factor type 1 receptor in osteoblasts enhances insulin signaling and action. J Bio Chem 2007;282:25649-58. [DOI] [PubMed] [Google Scholar]
- 44.Entingh-Pearsall A, Kahn CR. Differential roles of the insulin and insulin-like growth factor-I (IGF-I) receptors in response to insulin and IGF-I. J Biol Chem 2004;279:38016-24. [DOI] [PubMed] [Google Scholar]
- 45.Mulvihill MJ, Ji Q, Coate H, et al. Novel 2-phenylquinolin-7-yl-derived imidazo[1,5-a]pyrazines as potent insulin-like growth factor-I receptor (IGF-1R) inhibitors. Bioorg Med Chem 2008;16:1359-75. [DOI] [PubMed] [Google Scholar]
- 46.Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, et al. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem 2009;1:1153-71. [DOI] [PubMed] [Google Scholar]
- 47.Lindsay CR, Chan E, Evans TR, et al. Phase I dose escalation study of the continuous oral dosing of OSI-906, an insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase inhibitor, in patients with advanced solid tumors. J Clin Oncol 2009;27:Abstract 2559. [Google Scholar]
- 48.Mulvihill MJ. OSI-906, a selective, orally available dual inhibitor of IGF-1R and IR currently undergoing clinical testing in a phase III clinical trial in ACC patients. ACS National Meeting Abstracts 2010; MEDI-8. [Google Scholar]
- 49.Wittman MD, Carboni JM, Yang Z, et al. Discovery of a 2,4-disubstituted pyrrolo-[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. J Med Chem 2009;52:7360-3. [DOI] [PubMed] [Google Scholar]
- 50.Buchanan JL, Newcomb JR, Carney DP, et al. Discovery of 2,4-bisarylamino-1,3-pyrimidines as insulin-like growth factor-1 receptor (IGF-IR) inhibitors. Bioorg Med Chem Lett 2011;21:2394-9. [DOI] [PubMed] [Google Scholar]
- 51.Ducray R, Simpson I, Jung FH, et al. Discovery of novel imidazo[1,2-a]pyridines as inhibitors of the insulin-like growth factor-1 receptor tyrosine kinase. Bioorg Med Chem Lett 2011;21:4698-701. [DOI] [PubMed] [Google Scholar]
- 52.Ducray R, Jones CD, Jung FH, et al. Novel imidazo[1,2-a]pyridine based inhibitors of the IGF-1 receptor tyrosine kinase: optimization of the aniline. Bioorg Med Chem Lett 2011;21:4702-4. [DOI] [PubMed] [Google Scholar]
- 53.Seshacharyulu P, Ponnusamy MP, Haridas D, et al. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Target 2012;16:15-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ludovini V, Bellezza G, Pistola L, et al. High coexpression of both insulin-like growth factor receptor-1 (IGFR-1) and epidermal growth factor receptor (EGFR) is associated with shorter diseasefree survival in resected non-small-cell lung cancer patients. Ann Oncol 2009;20:842-9. [DOI] [PubMed] [Google Scholar]
- 55.Desbois-Mouthon C, Cacheux W, Blivet-Van Eggelpoël MJ, et al. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int J Cancer 2006;119:2557-66. [DOI] [PubMed] [Google Scholar]
- 56.Hu YP, Patil SB, Panasiewicz M, et al. Heterogeneity of receptor function in colon carcinoma cells determined by cross-talk between type I insulin-like growth factor receptor and epidermal growth factor receptor. Cancer Res 2008;68:8004-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ueda S, Hatsuse K, Tsuda H, et al. Potential crosstalk between insulin-like growth factor receptor type 1 and epidermal growth factor receptor in progression and metastasis of pancreatic cancer. Mod Pathol 2006;19:788-96. [DOI] [PubMed] [Google Scholar]
- 58.Riedemann J, Takiguchi M, Sohail M, et al. The EGF receptor interacts with the type 1 IGF receptor and regulates its stability. Biochem Biophys Res Commun 2007;13:707-14. [DOI] [PubMed] [Google Scholar]
- 59.Denley A, Cosgrove LJ, Booker GW, et al. Molecular interactions of the IGF system. Cytokine Growth Factor Rev 2005;16:421-39. [DOI] [PubMed] [Google Scholar]
- 60.Goetsch L, Gonzalez A, Leger O, et al. Combination of IGF-1R and EGFR antibodies. Int J Cancer 2005;113:316-28. [DOI] [PubMed] [Google Scholar]
- 61.Wilsbacher JL, Zhang Q, Tucker LA, et al. Combinations of gefitinib and NVP-ADW742 caused synergistic decreases in cellular proliferation across a diverse set of cancer cell lines. J Biol Chem 2008;283:23721-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buck E, Eyzaguirre A, Rosenfeld-Franklin M, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res 2008;68:8322-32. [DOI] [PubMed] [Google Scholar]
- 63.Hubbard RD, Bamaung NY, Fidanze SD, et al. Development of multitargeted inhibitors of both the insulin-like growth factor receptor (IGF-IR) and members of the epidermal growth factor family of receptor tyrosine kinases. Bioorg Med Chem Lett 2009;19:1718-21. [DOI] [PubMed] [Google Scholar]
- 64.Wang GT, Mantei RA, Hubbard RD, et al. Substituted 4-amino-1Hpyrazolo[3,4-d]pyrimidines as multi-targeted inhibitors of insulin-like growth factor-1 receptor (IGF1R) and members of the ErbB-family receptor kinases. Bioorg Med Chem Lett 2010;19:6067-71. [DOI] [PubMed] [Google Scholar]
- 65.Fidanze SD, Erickson SA, Wang GT, et al. Imidazo[2,1-b]thiazoles: Multi-targeted inhibitors of both the insulin-like growth factor receptor and members of the epidermal growth factor family of receptor kinases. Bioorg Med Chem Lett 2010;20:2452-5. [DOI] [PubMed] [Google Scholar]
- 66.Tandon R, Kapoor S, Vali S, et al. Dual epidermal growth factor receptor (EGFR)/insulin-like growth factor-1 receptor (IGF-1R) inhibitor: a novel approach for overcoming resistance in anti-cancer treatment. Eur J Pharm 2011;67:56-65. [DOI] [PubMed] [Google Scholar]
- 67.O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66:1500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang T, Feldman I, Sathyanarayanan S, et al. Preclinical and molecular profiling data suggest LumB breast cancer as a potential indication for co-targeting IGF1R and mTOR with MK-0646 and MK-8669. Clin Cancer Res 2010;16:PR3. [Google Scholar]
- 69.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004;6:91-9. [DOI] [PubMed] [Google Scholar]
- 70.Byers LA, Cortez MA, Yang C, et al. Proteomic profiling of signaling pathways in LKB1 deficient non-small cell lung cancers (NSCLC) identifies novel therapeutic targets including IGF1R pathway. AACR Annual Meeting Abstracts 2013;2403. [Google Scholar]
- 71.Zask A, Verheijen JC, Richard DJ. Recent advances in the discovery of small-molecule ATP competitive mTOR inhibitors: a patent review. Expert Opin Ther Pat 2011;21:1109-27. [DOI] [PubMed] [Google Scholar]
- 72.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by co-targeting MEK and IGF-IR/PI3K. Cancer Cell 2010;18:683-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aplin AE, Kaplan FM, Shao Y. Mechanisms of resistance to RAF inhibitors in melanoma. J Invest Dermatol 2011;131:1817-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith DC, Britten CD, Clary DO, et al. A phase 1 study of XL228, a potent IGF-1R / AURORA / SRC inhibitor. J Clin Oncol 2009;27: Abstract 3512. [Google Scholar]
- 75.Blecha JE, Anderson MO, Chow JM, et al. Inhibition of IGF-1R and lipoxygenase by nordihydroguaiaretic acid (NDGA) analogs. Bioorg Med Chem Lett 2007;17:4026-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Friedlander TW, Weinberg VK, Huang Y, et al. A phase II study of insulin-like growth factor receptor inhibition with nordihydroguaiaretic acid in men with non-metastatic hormone-sensitive prostate cancer. Oncol Rep 2012;27:3-9. [DOI] [PubMed] [Google Scholar]
- 77.Meyer AN, McAndrew CW, Donoghue DJ. Nordihydroguaiaretic acid inhibits an activated fibroblast growth factor receptor 3 mutant and blocks downstream signaling in multiple myeloma cells. Cancer Res 2008;68:7362-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arteaga CL, Kitten LJ, Coronado EB, et al. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J Clin Invest 1989;84:1418-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bartella V, De Marco P, Malaguarnera R, et al. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signal 2012;24:1515-21. [DOI] [PubMed] [Google Scholar]
- 80.Apse B, Blair JA, Gonzalez B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Bio 2008;4:691-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 2010;10:130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]