

Abstract
Human papillomavirus (HPV) has developed strategies to escape eradication by innate and adaptive immunity. Immune response evasion has been considered an important aspect of HPV persistence, which is the main contributing factor leading to HPV-related cancers. HPV-induced cancers expressing viral oncogenes E6 and E7 are potentially recognized by the immune system. The major histocompatibility complex (MHC) class I molecules are patrolled by natural killer cells and CD8+ cytotoxic T lymphocytes, respectively. This system of recognition is a main target for the strategies of immune evasion deployed by viruses. The viral immune evasion proteins constitute useful tools to block defined stages of the MHC class I presentation pathway, and in this way HPV avoids the host immune response. The long latency period from initial infection to persistence signifies that HPV evolves mechanisms to escape the immune response. It has now been established that there are oncogenic mechanisms by which E7 binds to and degrades tumor suppressor Rb, while E6 binds to and inactivates tumor suppressor p53. Therefore, interaction of p53 and pRb proteins can give rise to an increased immortalization and genomic instability. Overexpression of NF-κB in cervical and penile cancers suggests that NF-κB activation is a key modulator in driving chronic inflammation to cancer. HPV oncogene-mediated suppression of NF-κB activity contributes to HPV escape from the immune system. This review focuses on the diverse mechanisms of the virus immune evasion with HPV that leads to chronic inflammation and cancer.
Key words: human papillomavirus, immune evasion, cervical cancer, penile cancer.
Introduction
The link between genital human papillomavirus (HPV) infection and cervical cancer was first reported in the early 1970s by Harold zur Hausen.1 His laboratory was the first to demonstrate that genital warts (condyloma) contain HPV genes,2,3 and also discovered the relationship between HPV infection and cervical cancer.4 He was awarded the Nobel Prize for Medicine in 2008 for the detection and isolation of HPV types 16 and 18 DNA from cervical cancer.4,5 HPV infection is associated with a broad spectrum of benign and malignant neoplastic epithelial changes. Epidemiological, clinical and pathological studies have indicated that the virus causes sexually transmitted infections. HPV infection is commonly found in the genital organs of both men and women. More than 40 HPV genotypes are able to infect the genital organs of females and males, including the vulva, vagina, cervix and penis.6–8 HPV-DNA has also been identified in head and neck cancers in the oral cavity, the oropharynx and the larynx in both sexes. The association of HPV with neoplastic transformation has been investigated most extensively in lesions of the uterine cervix and the role of HPV in malignant transformation of cervical epithelium has been well established. HPV can also affect the squamous epithelium of the male genitalia in a similar way to that of the female genital tract; however, its association with penile cancer is not clearly understood:9–13
The immune system plays a pivotal role in determining the outcome of HPV infection. The important role of host cellular and humoral immune responses protects from established HPV infections. Defense mechanisms against HPV infections are regulated in concert with innate and adaptive immunity. The innate immune response in particular is essential to initiate these antiviral immune activities in both peripheral and lymphoid tissues. The immune responses to HPV infections are production of neutralizing antibodies and induction of natural killer (NK) cells.14–18 Anti-viral CD4+ T-cell responses in healthy individuals presumed to have had transient HPV 16 infections have been reported, while no response in some individuals with pre-cancer or cancerous lesions has been observed.17
The HPV-16 is one of the most common high-risk HPV genotypes associated with cervical and penile cancers. The HPV-16 E6 and E7 oncoproteins are constitutively expressed in cancer lesions as putative targets for the immune response against HPV.19 The expressions of CD4+ helper T-cell (Th)1 and Th2-type cytokines are present in the subepithelial cervical tissue.20 Significantly lower percentages of Th1 cells with higher proportions of IL4+ and IL6+ cells were observed in high-grade squamous intraepithelial lesions compared to normal tissues. Predominant Th2 type cytokines in CD4+ T cells were also found in high-grade cervical lesions.20 CD4+ T cells are important for launching an efficient immune response against HPV. For inhibitory signals, the cytolytic functions of CD8+ T cells are inhibited by the up-regulated expression of NK cell receptors on tumor infiltrating lymphocytes (TILs) derived from cervical cancer.21
The immune evasion target is the major histocompatibility complex (MHC) class I antigen presentation pathway. The cells are under constant control from cytotoxic T lymphocytes (CTLs) that continuously scan the somatic cell surface for MHC class I molecules presenting non-self peptides, such as viral antigens. It has been suggested that HPV and HPV-infected cells have evolved mechanisms to avoid immune attack. T-cell mediated immune responses against oncogenic HPV are believed to play a central role in cervical carcinogenesis. HPV-specific CTLs infiltrating cervical cancer may play an important role in restricting disease progression. Cell mediated immunity is thought to be important in the control of HPV infection. High-risk HPV specific CTLs have been demonstrated in the peripheral blood of cervical cancer patients. HPV infections initiate and progress to genital cancers after more than decades without any effective immune response to resolve the initial HPV infection. The regional immune escape may play an important role in cancer formation arising from the HPV infected area.22,23 CD8+ CTLs play an important role in the elimination of HPV infected cells. CD8+ CTLs detect viral antigen peptides presented by MHC class I molecule on the cell surface. This pathway is designed to sample the intracellular milieu and present the information to the CTLs trafficking the area. DNA viruses with large genome coding capacity have been proven to be particularly adept at preventing CTL recognition through the action of dedicated immune evasion proteins. In addition, viral immune evasion proteins contribute to viral defense and replication.24–27 The viral immune evasion leads to the development from chronic inflammation to cancer associated with HPV infection.
HPV must be detected by pattern-recognition receptors (PRRs) before innate immune responses can be triggered. PRRs specifically recognize microbe-specific pathogen-associated molecular patterns (PAMPs). PRRs activate specific signaling cascades to induce the gene expression of targets, including the activation of type I interferons (IFNs) and proinflammatory cytokines.28–31 Type I IFNs play a critical role in anti-viral innate immunity to eradicate viruses and are secreted by macrophages or dendritic cells (DCs). IFNs regulate the activation of adaptive immunity, such as the maturation of DCs, activation of NK cells and differentiation of CTLs. Thus, type I IFNs play an important role in the overall regulation of anti-viral immune response.32,33 The main players in adaptive immune response are antibodies and T cells. Antibodies specific for viral surface antigens block binding and/or fusion of virus to host cells. The coat of the virus particles facilitate receptor-mediated phagocytosis and/or complemented mediated lysis of the virus. CTLs are the major component of cell mediated immunity and play an important role in the defense against viral infections.24,25,34
The causal relationship between chronic inflammation and cancer has been widely accepted. Specifically, there is a strong association between tumor viruses and the development of human cancers. Mechanisms of oncogenesis associated with infection and inflammation have been proposed. Effective host immune responses are essential for the control of HPV infection and persistence of HPV. However, failure to eliminate HPV infected cells increases the risk of developing chronic inflammation and cancers. Therefore, HPV associated cancers are the most common outcome of high-risk HPV infection and are preceded by a phase of persistent HPV infection due to host immune system failure to eradicate HPV. Innate and adaptive immune responses are induced in most individuals infected with HPV but are insufficient to eradicate the virus. HPV oncoproteins E7 and E6 are essential factors in HPV oncogenesis. E7 and E6 react with the tumor suppressor gene products pRb and p53 in host cell proteins, respectively. E7 and E6 induced genetic instability leading to the activation of oncogenesis and inactivation of tumor suppressor genes.6–8,35
Although many investigators have proposed oncogenesis due to inflammation to be associated with cancer development, the mechanisms underlying the relationship between chronic inflammation and cancer still remain undefined. This review focuses on the recent advances in the understanding of the molecular mechanisms of HPV immune evasions due to the MHC class I pathway, HPV-encoded proteins and NF-κB activation that may contribute to tumor formation.
Human papillomavirus genotypes
Approximately 130 HPV genotypes have been identified so far and these have been classified according to the sequence of the gene encoding the major capsid protein L1. About 40 genotypes infect the genital mucosa. A new HPV type is defined as showing less than 90% homology to any of those known on the basis of the L1 region. HPV is classified into low-risk and high-risk groups according to the potential for induction of malignant formation. Low-risk HPV types typically induce benign tumors and are rarely associated with malignant tumors. In contrast, high-risk HPV types induce malignancy. Thus, HPV is classified into low-risk types (ns. 6, 11, 26, 31, 34, 40, 42, 43, 44, 53, 54, 55, 57, 61, 62, 64, 67, 70, 71, 72, 73, 74, 79, 81, 82, 83, and 84) and high-risk types (ns. 16, 18, 31, 33, 35, 39, 45, 51, 52, 54, 56, 58, 58, 66, 68, and 69). The high-risk types are associated with more than 90% of cervical cancers. Furthermore, three main groups of HPV have been classified according to their localization: skin HPV types (ns. 1, 5, 8, 14, 20, 21, 25, and 47), skin and mucosal types (ns. 2 and 57), and mucosal types (ns. 6, 11, 13, 16, 31, 33, 35, 39, 42, 44, 45, 49, 51, 52, 56, 58, and 68).36–39 The most important players are HPV-16, found in 50–70% of cases, and HPV-18, found in 7–20% of cases.9,40–42 Therefore, the high-risk strains, HPV-16 and HPV-18, are the most closely associated with cervical and penile cancers (Figure 1A). HPV-6 and HPV-11 are typically found in genital warts and condyloma acuminata of the genital organs (Figure 1C).43–48 In cervical cancer, HPV-33, HPV-45 and HPV-31 are the next most frequently detected HPV, except for cases in Asia where HPV-58, HPV-33 and HPV-52 are the next most prevalent types.49 In penile cancer, HPV-16 (60.23%), HPV-18 (13.35%), HPV-6/11 (8.13%), HPV-31 (1.16%), HPV-45 (1.16%), HPV-33 (0.97%), HPV-52 (0.585%), and other HPV types (2.47%) have been reported.50–52
Figure 1.
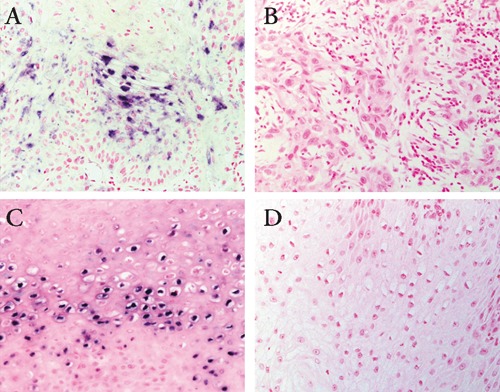
(A) Detection of human papilloma virus (HPV) DNA in penile cancer. Original magnification 200x. (B) Positive signals were not observed in uninfected control specimen of penile cancer. Original magnification 200x. (C) Detection of HPV DNA in penile condyloma. Original magnification 200x. (D) Positive signals were absent in uninfected control specimen of penile condyloma. Original magnification 200x.
Human papillomavirus genome
HPV is a member of the Papovaviridae family. HPV is a small nonenveloped DNA tumor virus, 55 nm in diameter. HPV induces hyperproliferative lesions in cutaneous and mucosal epithelia. The HPV genome is a long circular double-stranded DNA molecule of approximately 8000 base pairs. The HPV genome is organized into three regions: the late gene (L1 and L2), the early gene (E1, E2, E3, E4, E5, E6, E7, and E8), and the long control region (LCR). Viral genes are divided into early and late categories depending on the time of expression. E1 supports viral replication and control of gene transcription.53–56 E2 supports viral transcription and viral DNA replication.57,58 The E4 protein is expressed in the later stages of infection when complete virions are assembled. The E4 protein is believed to play an important role in the maturation and replication of HPV and is, therefore, involved in the induction of the release of virions from infected cells.59 The E5 protein has weak activity in maintaining the malignant transformation of the host cells.60 Proteins E6 and E7 have been shown to induce immortalization and transformation of the host cells. Therefore, E6 and E7 oncogene proteins have been shown to be the main contributors to the development of HPV-induced cancer and increased expression.61–65 The functions of E3 and E8 proteins are not clear. The late gene regions, L1 (major capsid protein) and L2 (minor capsid protein) encode for viral capsid proteins during the late stages of virion assembly. The protein encoded by L1 is highly conserved among different HPV species (Table 1). The minor capsid protein encoded by L2 has more sequence variations than those of the L1 protein.66 Early viral transcription is regulated by an element located in the non-encoding region proximal to the open reading frames called the LCR or the upstream regulatory region. The LCR has numerous binding sites for many repressors and activators of transcription. It has been suggested that the LCR plays a part in determining the characteristics of HPV types.67–70 Eighty-six complete genomes of HPV have been characterized and approximately 120 have been partially characterized.71
Table 1. Human papillomavirus gene functions.
| Gene category | Gene | Function |
|---|---|---|
| Eary genes | E1 | Viral DNA replication and control of gene transcription |
| E2 | Modulaion of replication and transcription | |
| E3 | Unknown | |
| E4 | Viral assembly | |
| E5 | Growth stimulation | |
| E6 | Oncoprotein due to interaction with p53 protein | |
| E7 | Oncoprotein due to interaction with pRb proein | |
| E8 | Unknown | |
| Late genes | L1 | Major capsid protein |
| L2 | Minor capsid protein |
Human papillomavirus life cycle
The HPV life cycle is tightly adapted to the host tissue, the differentiating epithelial cells of skin or mucosa. The strongest evidence for oncogenesis is in cervical cancer, where more than 90% of neoplastic cells contain HPV-DNA. Viral early proteins are produced in undifferentiated keratinocytes in the basal layers of stratified epithelium. Normal squamous epithelial cells grow as stratified epithelium. Amplifying stem cells in the basal layer start to differentiate, and one of the daughter cells migrates upward and begins to undergo terminal differentiation. The other daughter cells remain in the basal layers of the epithelia where HPV binds to and enters into cells through small wounds. The mechanism by which HPV actively invades cells involves its interaction with a yet unidentified receptor. HPV virions migrate to the nucleus as episomes and early HPV promoter is activated. The replication cycle within the epithelium can be divided into the latent and the lytic infections. In the latent infection, the viral genome is replicated to low copy numbers: approximately 10–200 genomes per cell within the initially infected the basal cells.72 The early HPV genes E1 and E2 are essential for DNA replication in the basal cells and for its segregation. The infected basal cells can be maintained in the lesion for a long period of time. As the viral status persists, the immune system may confine the infection to these basal cells. In the lytic infection, when infected daughter cells migrate to the upper layers of the epithelium, viral late gene products are produced to initiate the vegetative phase of the HPV life cycle, resulting in high-level amplification of the viral genome. The HPV virions are synthesized in the upper layers of stratified squamous epithelia. For multiplication of viral DNA, the early genes E5, E6, and E7 are thought to coordinate themselves to create a suitable environment in the host cells. The late promoter activates the late gene products, resulting in the vegetative phase of the HPV life cycle leading to high-level amplification of the viral genome. The HPV virions replicate in the superficial layers of the epithelium and, consequently, the progeny HPV virions are released from the cell to re-initiate infection (Figure 2).72,73
Figure 2.
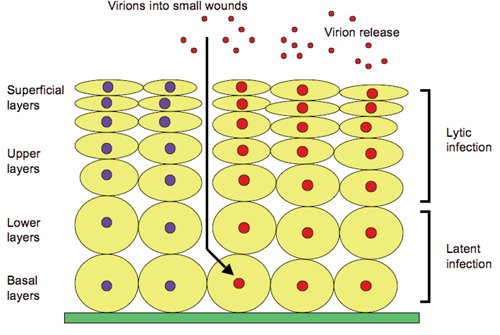
Latent infection: human papilloma virus initially infects the nucleus of basal cells. Few viral DNA are synthesized in infected cells. Latent infection is the ability of a pathogenic virus to lie dormant within cells (a type of persistent viral infection) but virus production ceases. Lytic infection: numerous viral DNA are synthesized in the nuclei of cells in the upper layer with virions released from the superficial cell layer. Lytic infection is virus beginning to produce large amounts of viral progeny and to release viral particles to destroy the infected cells.
Immune response to human papillomavirus infection
The immune system plays a central role in determining the outcome of HPV infection and the immune response essential for the clearance of HPV. Defense mechanisms include components of both the innate and adaptive immune systems. The innate immune response has an important role in anti-viral defense. The components of the innate immune response are activated complement components destroying virus infected cells and the virus itself. NK cells recognize and eliminate virus-infected cells. The adaptive immune response is made up of antibodies and T cells. IFNs induce an anti-viral state in infected cells.74–78 Antibodies specific for viral surface antigens block binding and/or fusion of virus, and coat the viral particles to facilitate receptor mediated phagocytosis and/or complemented mediated lysis of virus. Antibody production from the mucosal tissues prevents HPV infection in the mucosal surface. Cervical anti-HPV immunoglobulin (Ig)A and IgG are strongly associated with HPV-DNA and cervical diseases.79,80
TILs develop as manifestations of the recognition and defense against malignant cells by the host immune system. These play an important role in fulfilling the oncogenic process of HPV infected tissues.81 The majority of HPV infections are cleared before cancer formation begins. However, if the host immune system fails to eliminate the virus, persistent HPV infection occurs. Prolonged persistence of HPV leads to genomic instability resulting in cancer formation.
Langerhans cells are the principal DC population in the epidermis of the skin. DCs are local antigen-presenting cells (APCs) found predominantly in the skin and mucosal epithelia with high antigenic exposure. DCs are key regulatory cells in the immune system that mediate between the innate and the adaptive immune systems. DCs are clearly the most potent APCs for stimulating an effective CTL response.82–84 Generally, DCs both capture and process antigens, express lymphocyte co-stimulatory molecules, migrate to lymphoid organs and secrete cytokines to initiate immune responses. T and B lymphocytes are the key mediators of cellular immunity under the control of DCs. DCs are likely to play an important role in the progression of cervical cancer due to the activation of HPV-specific T cells. DCs bound to MHC class I or class II molecules interact with CD8+ T cells and CD4+ T cells, respectively. CD8+ CTLs facilitate the destruction of cells with CD4+ cells providing cytokines such as interleukin 2.84 Local APCs are probably recruited and secrete mediators that enhance the inflammatory response. Relatively strong T-cell proliferate responses against HPV-16 E7 are observed in patients with a persistent HPV-16 infection.85 T-cell mediated immune responses against oncogenic HPV are believed to play a central role in HPV-associated cancers. It has been suggested that HPV-16 E7 specific Th cell responses correlate with viral clearance or viral persistence in cervical cancer. Differential Th and IgG immune responses correlate with viral clearance and the development of cancer.86–88 Viruses enter permissive cells and leave the infected cells, resulting in hijack cellular metabolic pathways to generate progeny viruses. Hosts have developed an adaptive immune system that is designed to recognize and eliminate these invaders. However, viruses have also evolved strategies to combat the host defense response. Most of the DNA viruses alter the host immune response to their life-long persistent infections. The MHC class I molecules are present on almost all the nucleated cells and protect against invading HPV. MHC class I molecules at the cell surface consist of: i) a transmembrane heavy (H) chain (45,000 MW), ii) the soluble β2-microglobulin (β2m) light (l) chain (12,000 MW) subunit, and iii) eight or ten residue peptides in length.89 The assembly of MHC class I molecules is regulated by a series of interactions with endoplasmic reticulum (ER) resident chaperones and accessory molecules. The viral peptides result from proteasomal degradation of proteins in the cytosol that are translocated by the transporter associated with antigen processing in the ER. They are loaded onto newly synthesized MHC I molecules. This stage of assembly is characterized by the formation of the peptide loading complex (PLC) and the acquisition of optimal peptides.24 The PLC retains MHC class I molecules within the ER until they are loaded with high affinity peptides. Once loaded high affinity peptides have been bound, MHC class I molecules exit the ER and transit through the Golgi apparatus to the cell surface to present their cargo to CD8+ T cells. This extra level of quality control on the part of the PLC distinguishes MHC class I molecules from many other proteins that assemble within the ER. MHC class I molecules can sample both the intracellular and extracellular milieus for defective and foreign proteins by presenting peptide fragments to immune effector cells. The MHC class I peptide complexes are patrolled by cells of both the innate and the acquired immune systems, namely NK cells and CD8+ CTL, respectively. Decreased MHC class I expression on infected cells renders the cells susceptible to NK-cell mediated killing. Immune evasions selectively target the MHC class I molecules that preferentially present viral peptides (HLA-A and HLA-B) and spare the MHC class I alleles that act as dominant ligands of NK-cell inhibitory receptors (HLA-C and HLA-E).90 The HPV-16 E6 and E7 oncoproteins are expressed constitutively in the majority of pre-cancer and cancer lesions. HPV specific memory CTLs from cancer patients recognize HPV-16 E6 and E7 oncoproteins presented by MHC class 1 molecules.91 NK cells clearly respond to the viral infection through cytolysis and secreting proinflammatory cytokines, as well as the downregulation of surface MHC class I molecules in order to avoid recognition by CD8+ T cells and molecules induced by viral invasion of cells. Ligands for NK cell receptors are primary for viral immune evasion. NK cells are important components of the innate immune system directly involved in the anti-viral immune response. NK cell inhibitory receptors monitor the normal levels of MHC class I molecules, the expression of which is frequently altered by virus infection. Therefore, cells that express self-MHC class I molecules are protected from NK cells, but those that lack this self-marker are eliminated. Furthermore, NK cells are being considered for cancer treatment in anti-tumor activity (Figure 3).92–95
Figure 3.
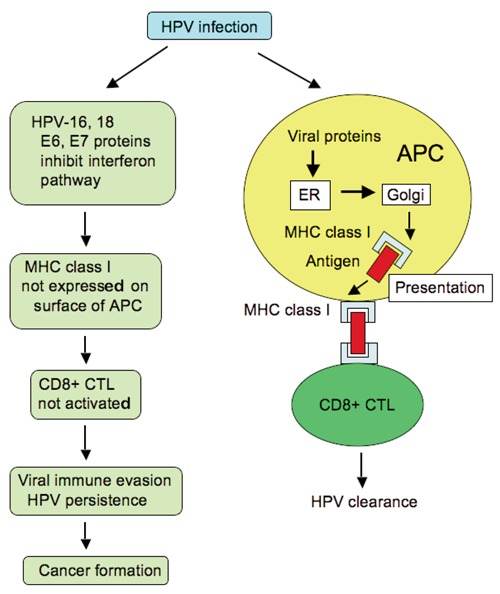
There is a strong association between human papilloma virus (HPV) infection and the development of cancer formation because of viral immune evasion. The primary mechanism of viral immune evasion for HPV is avoidance of antigen presentation. HPV-16, and -18, E6 and E7 interfere with the interferon pathway; therefore, the amount of major histocompatibility complex (MHC) class I molecules is reduced on the cell surface. CD8+ cytotoxic T lymphocytes (CTLs) play an important role in the elimination of viral-infected cells. However, MHC class I is not expressed on the cell surface of antigen-presenting cells (APCs), resulting in inactivation of CD8+ CTLs. Continuous viral immune evasion leads to viral persistence and cancer formation in the long term. ER, endoplasmic reticulum.
Host pathogen interactions are usually initiated via recognition of PAMPs by host sensors known as PRRs, such as Toll-like receptors (TLRs), RIG-l-like receptors, NOD-like receptors, and DNA receptors. PAMPs rapidly trigger host immune responses.96–99 The complex signaling pathways induce inflammatory responses and the eradication of viruses. NF-κB is a transcription factor composed of homo- or heterodomimers of Rel homology domain-containing proteins, including p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1) and p100/p52 (NF-κB2). The intranuclear translocation of activated NF-κB signaling pathway induces the expression of numerous genes involved in innate and adaptive immune regulation, inflammatory responses, and anti-apoptosis. The intracellular translocation of the activated NF-κB signaling pathway is regulated by a variety of posttranslational modifications, including phosphorylation and multiple ubiquitinations.100–105 Activation of the NF-κB and interferon regulatory factor transcription factor pathways is crucial for the first step for immune response. NF-κB is activated by a variety of diverse extracellular or intracellular stimuli, including microbial pathogens and PAMPs.106,107 Many oncogenic viruses activate NF-κB to facilitate transformation of infected cells. Some viruses maintain a delicate balance between activation and suppression of NF-κB in order to maintain long-term persistence. Impairing NF-κB activation is proposed as a viral strategy to avoid the innate response of the host. Aberrant NF-κB signaling is implicated in multiple disorders, such as chronic inflammation and cancer.103,108–111
Functions of E7 and E6 in oncogenesis
HPV oncoproteins E6 and E7 are essential factors for HPV oncogenesis. E7 and E6 react with the tumor suppressor gene products pRb and p53 in host cell proteins, respectively, resulting in induced cellular immortalization, transformation and carcinogenesis due to their interference with cell cycle and apoptosis control.35 Genetic instability induced by E7 and E6 leads to the activation of oncogenes and inactivation of tumor suppressor genes.
The HPV oncogenes, E7 and E6, have been shown to be the main contributors to the development of HPV induced cancers, probably due to integration of the viral genes in the host cell genome. Inactivation of tumor suppressors p53 and pRb is a common event in the carcinogenesis of human cells. In HPV infection, the significant interactions are with p53 and pRb proteins that are important molecules in the cell cycle and apoptosis control. Remarkably, p53 and pRb proteins are mutated in many human cancers. Both E7 and E6 HPV oncogenes interact with pRb and p53 that inhibit the activities of these tumor suppressors. S phase progression of the cell cycle would normally lead to apoptosis by the action of p53. However, in HPV-infected cells, this process is counteracted by the viral E6 protein, which targets p53 for proteolytic degradation. High-risk HPV causes persistent infections that induce the initiation and progression of malignant tumor formation. As an aberration of virus infection, the constant activity of the viral proteins E7 and E6 leads to increasing genomic instability and accumulation of oncogene mutations, resulting in cancer formation. Specifically, those involving tumor suppressor genes can give rise to immortalization associated with activation of telomerase. Extensive studies have linked the efficiency of mucosal HPV types in promoting cancer development to the viral E6 and E7 proteins. The HPV E6 and E7 genes are thought to play causative roles since E6 promotes the degradation of p53 through its interaction with E6AP, an E3 ubiquitin ligase, whereas E7 binds to pRb and disrupts its complex formation with E2F transcription factors.112,113 In addition, E6 and E7 cause degradation of the cellular genes controlling G2/M phase transition and progression.45,114
E6 and E7 proteins may immortalize various types of human cells independently. However, their cooperative interaction leads to substantially enhanced immortalization efficiency. E7 binds to cyclin-kinase complexes and cyclin-dependent kinase (CDK) inhibitors. E7 directly binds to cyclin A/CDK2. It also indirectly interacts with the cyclin E/CDK2 complexes via p107 protein of the pRb family, which acts to increase cyclins A and E in the epithelial cells. E7 also binds to the CDK inhibitors, p27 and p21.115–119
E7 is an oncogene that mediates the initiation of DNA synthesis and stimulates continuous cell growth. It hereby induces genetic abnormalities that enhance the likelihood of malignant progression. The E7 protein can interact with pRb, an important negative regulator of entry into S phase of the cell division cycle. It is thought that the high-risk HPVs have a unique ability to induce the proteolytic degradation of pRb. In the hypophosphorylated state, combined E7 and pRb activates the E2F transcription factor, which triggers the expression of proteins necessary for DNA replication and cell cycle progression. Phosphorylation of pRb by G1 cyclin-dependent kinases releases E2F leading to cell cycle progression into S phase. Because E7 is able to bind to unphosphorylated pRb, it may prematurely induce cells to enter S phase by disrupting pRb-E2F complexes.120–127
In addition to the inactivation of pRb family members, numerous functions of E7 have been reported. Histone deacetylases and transcriptional co-repressors have been reported to associate with E7 via Mi2 to promote cell growth. The interaction of E7 with cyclin E/CDK2 has also been reported. These cyclin-kinase complexes can phosphorylate the pRb proteins. Furthermore, E7 binds to the CDK-inhibitors, p27 and p21, confirming the abrogation of cell-cycle inhibition. Despite these multifunctional properties of the E7 protein, however, the expression of a mutant form of pRb that is selective for binding E7 revealed that the effects of E7 on epidermal differentiation are indeed due to pRb inactivation.120,124
The most manifest function of the E6 protein is to promote the degradation of p53 under E6 protein associated with E6AP protein, an E3 ubiquitin ligase. The affinity of E6AP for p53 is likely to be modified by the association with E6. p53 is a tumor suppressor gene involved in apoptosis after DNA damage, and regulation of both G1/S and G2/M cell cycles. HPV-infected cells induce inhibition of apoptosis by E6 inactivation of p53. In addition, E6 interferes with other proapoptotic proteins, such as Bak, FADD and procaspase 8 (Figure 4).126,127
Figure 4.
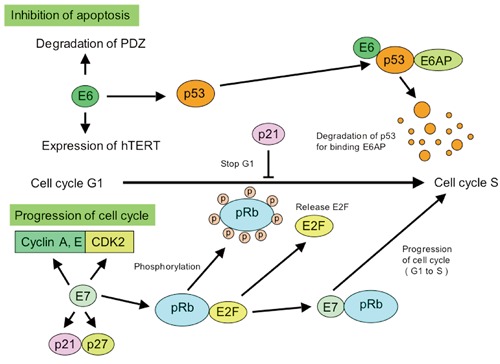
High-risk human papilloma virus (HPV) E6 and E7 proteins play a critical role in the development of HPV-associated cancers. E7 and E6 react with the tumor suppressor gene products pRb and p53 in host cell proteins, respectively, resulting in induced cellular immortalization, transformation and carcinogenesis. E7 induces hyperproliferation through inhibition of retinoblastoma family members and constitutive activation of E2F responsive genes. E6 inhibits p53-dependent growth arrest and apoptosis, resulting in the induction of genomic instability and the accumulation of cellular mutations. Degradation of p53 bypasses the normal growth arrest of the cell cycle from G1 to S phases.
Overexpression of NF-κB in human papillomavirus-related cancer
The relationship between chronic inflammation and cancer progression has long been known. Numerous investigations have identified NF-κB as an important modulator in driving chronic inflammation to cancer. This transcription factor is indispensable for the malignant progression of transformed cells associated with various inflammatory cells and a network of signaling molecules. The expression and the function of numerous cytokines, chemokines, growth factors, and survival factors are NF-κB-dependent. NF-κB activation has been implicated in a variety of processes related to transformation and oncogenesis, including proliferation, migration, angiogenesis, and prevention of apoptosis. NF-κB activity is involved in the regulation of the angiogenesis process for cancer cell growth and invasiveness. Vascular endothelial growth factor is the main member of the group of angiogenic factors and is under the transcriptional control of NF-κB.128 TLR are possible signal initiators for NF-κB activation due to inflammation inducing carcinogenesis. In most cancer cells, the activation of NF-κB plays an important role in cancer initiation, progression and metastasis. TLR are among the major activators of NF-κB and are the front-line receptors in the response to microbial infection. The activation of NF-κB through stimulated TLR in local chronic inflammation may serve as an initiator. NF-κB is constitutively activated during human cervical cancer progression13,129–131 and penile cancer progression.10–12
Increased NF-κB activity is associated with many cancers, especially cancers associated with viral infections. NF-κB dependent proliferation and protection from apoptosis are likely to have significant effects on the oncogenesis of HPV associated with cancers; HPV E6 and E7 positive cells have shown that IL-1β induces NF-κB activation and exhibits elevated levels of NF-κB components. E6 rather than E7 expression was found to be associated with the nuclear location of these components.132 It is frequently reported that HPV encoded E6 and E7 oncoproteins are important regulatory proteins in host cells that are associated with the transcriptional activity of NF-κB. A fraction of the E7 protein is found to be associated with the IkB kinase (IKK) complex and attenuates induced kinase activity of IKKα and IKKβ, thus resulting in impaired IκBα phosphorylation and degradation. While E7 obviates IKK activation in the cytoplasm, the E6 protein reduces NF-κB p65-dependent transcriptional activity within the nucleus. It has been suggested that HPV oncogene-mediated suppression of NF-κB activity contributes to HPV escape from the immune system (Figure 5).133–136 HPV E7 and E6 oncogenes are key regulatory proteins inside host cells and are associated with the transcriptional activity of NF-κB. Therefore, overexpression of NF-κB in penile and cervical cancer cases suggested that NF-κB activation is a key modulator in driving chronic inflammation to cancer.10–13
Figure 5.
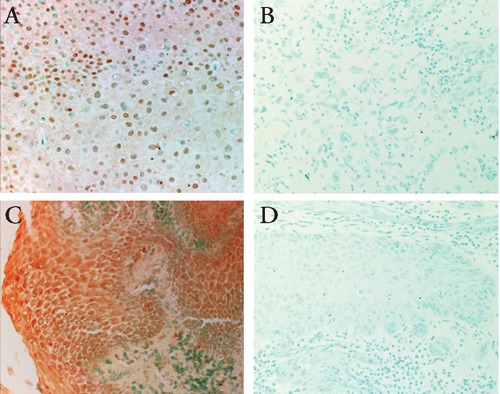
(A) Detection of NF-κB in the cytoplasms in penile cancer. Original magnification 200x. (B) Negative control section is not stained by NF-κB. Original magnification 200x. (C) Detection of NF-κB in the nuclei in penile cancer. Original magnification 200x. (D) Negative control section is not stained by NF-κB. Original magnification 200x.
PDZ domain-containing proteins have been identified as binding partners for the oncoprotein E6 of the high-risk type HPV. The PDZ binding motif of E6 is required for activation of NF-κB and the non-PDZ domain mutant from E6 is unable to activate NF-κB.137 Tumor necrosis factor (TNF), other cytokines, and growth factors stimulate the activation of NF-κB and the transcription of antiapoptotic target genes, including the inhibitor of apoptosis proteins XIAP, ciap-1, and ciap-2.138 NF-κB and TNF are functionally important molecules in inflammation-associated cancer.139,140 Prevention of DNA damage or cytokine induced apoptosis by activation of NF-κB may be a major obstacle in the treatment of HPV-associated cancers. NF-κB activation in HPV-infected cells likely plays a role in the proliferative capacity of the cells in addition to protection from apoptosis, which is reflected in the diminished ability of E6 mutants that are not able to bind PDZ domains or activate NF-κB to promote oncogenesis. Investigation of the potential role of NF-κB activation in the pathogenesis of HPV-associated cancers is of significant importance with regards to the treatment and prevention of viral cancers.137 NF-κB activity is triggered in response to infective agents and proinflammatory cytokines via the IKK complex. TNF, interleukins, chemokines, COX-2, 5-LOX, and MMP-9 are all regulated by the transcription factor NF-κB. NF-κB activation is induced by a wide variety of stimuli of inflammation and formation of cancer, because inflammatory mediators induce oncogenesis.141,142 Inflammatory biomarkers can also be exploited to develop new anti-inflammatory drugs that can be used not only for prevention but also in cancer therapy. Furthermore, inhibitors of NF-κB activation can block the cancer transformation response and this is a new approach in cancer treatment.143–146
Conclusions
In conclusion, HPV-induced cancers can evolve different immune evasion strategies according to the various immunological challenges they have to face. Viruses have evolved these diverse mechanisms to target the importance of MHC class I molecules in host defense against viruses. MHC class I molecules bind to peptides derived from endogenously synthesized proteins and present them at the cell surface for sampling by CD8+ CTLs. Viral immune evasion proteins interfere with antigen presentation and interrupt the early MHC class I assembly presentation pathway. The viral immune evasion molecules attack the ER PLC and exploit ER-associated degradation pathways. The MHC class I pathway is spared by viral immune evasion. Therefore, an antibody is not produced because no antigen is presented. Although the immune response is able to overcome the evasion mechanisms and clear infection in most cases, prolonged persistence of high-risk HPV sometimes leads to malignancy. The causal relationship between chronic inflammation and cancer is widely accepted. Therefore, there is a strong association between tumor viruses and development of cancers due to viral immune evasion.
Acknowledgements:
the authors thank Dr. Mihoko Kikuchi, Department of Immunogenetics, Institute of Tropical Medicine, Nagasaki University, Japan, for helpful advice. This work was supported in part by a Grant-in-Aid (n. 24592432) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1.zur Hausen H. Papillomaviruses in the causation of human cancers - a brief histological account. Virology. 2009;384:260–5. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Gissmann L, zur Hausen H. Partial characterization of viral DNA from human genital warts (Condylomata acuminata) Int J Cancer. 1980;25:605–9. doi: 10.1002/ijc.2910250509. [DOI] [PubMed] [Google Scholar]
- 3.de Villiers EM, Gissmann L, zur Hausen H. Molecular cloning of viral DNA from human genital warts. J Virol. 1981;40:932–5. doi: 10.1128/jvi.40.3.932-935.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dürst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983;80:3812–5. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boshart M, Gissmann L, Ikenberg H, et al. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984;3:1151–7. doi: 10.1002/j.1460-2075.1984.tb01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Senba M, Mori N, Wada A. Oncogenesis of human papillomavirus (HPV) In: Tao HE, editor. DNA tumor virus. New York: Nova Science Publishers, Inc; 2009. pp. 75–102. [Google Scholar]
- 7.Senba M, Mori N, Wada A. Oncogenesis and the link between inflammation and cancer due to human papillomavirus (HPV) infection, and the development of vaccine control strategies. Cancer Res J. 2009;2:307–38. [Google Scholar]
- 8.Senba M, Mori N, Wada A. Oncogenesis of and the link between inflammation and cancer due to human papillomavirus (HPV) infection, and the forthcoming eradication for vaccination and the development of vaccine control strategies. In: Watanabe HS, editor. Horizons in cancer research. Vol. 44. New York: Nova Science Publishers, Inc; 2011. pp. 85–115. [Google Scholar]
- 9.Senba M, Kumatori A, Fujita S, et al. The prevalence of human papillomavirus genotypes in penile cancers from northern Thailand. J Med Virol. 2006;78:1341–6. doi: 10.1002/jmv.20703. [DOI] [PubMed] [Google Scholar]
- 10.Senba M, Buziba N, Mori N, et al. Detection of human papillomavirus and cellular regulators p16INK4a, p53, and NF-κB in penile cancer cases in Kenya. Acta Virol. 2009;53:43–8. doi: 10.4149/av_2009_01_43. [DOI] [PubMed] [Google Scholar]
- 11.Senba M, Mori N, Wada A, et al. Human papillomavirus genotypes in penile cancers from Japanese patients and HPV-induced NF-κB activation. Oncol Lett. 2010;1:267–72. doi: 10.3892/ol_00000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senba M, Mori N, Fujita S, et al. Relationship among human papillomavirus infection, p16INK4a, p53 and NF-κB activation in penile cancer from northern Thailand. Oncol Lett. 2010;1:599–603. doi: 10.3892/ol_00000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senba M, Buziba N, Mori N, et al. Human papillomavirus infection induces NF-κB activation in cervical cancer: a comparison with penile cancer. Oncol Lett. 2011;2:65–8. doi: 10.3892/ol.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheu BC, Chang WC, Lin HH, et al. Immune concept of human papillomaviruses and related antigens in local cancer milieu of human cervical neoplasia. J Obstet Gynecol Res. 2007;33:103–13. doi: 10.1111/j.1447-0756.2007.00492.x. [DOI] [PubMed] [Google Scholar]
- 15.Pichlmair A, Reis Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–83. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa M, Stites DP, Patel S, et al. Persistence of human papillomavirus type 16 infection is associated with lack of cytotoxic T lymphocyte response to the E6 antigens. J Infect Dis. 2000;182:595–8. doi: 10.1086/315706. [DOI] [PubMed] [Google Scholar]
- 17.de Jong A, van Poelgeest MI, van der Hulst JM, et al. Human papillomavirus type 16-positive cervical cancer is associated with impaired CD4+ T-cell immunity against early antigens E2 and E6. Cancer Res. 2004;64:5449–55. doi: 10.1158/0008-5472.CAN-04-0831. [DOI] [PubMed] [Google Scholar]
- 18.Woo YL, van den Hende M, Sterling JC, et al. A prospective study on the natural course of low-grade squamous intraepithelial lesions and the presence of HPV16 E2-, E6- and E7-specific T-cell responses. Int J Cancer. 2010;126:133–41. doi: 10.1002/ijc.24804. [DOI] [PubMed] [Google Scholar]
- 19.Kadish AS, Ho GY, Burk RD, et al. Lymphoproliferative responses to human papillomavirus (HPV) type 16 proteins E6 and E7: outcome of HPV infection and associated neoplasia. J Natl Cancer Inst. 1997;89:1285–93. doi: 10.1093/jnci/89.17.1285. [DOI] [PubMed] [Google Scholar]
- 20.al-Saleh W, Giannini SL, Jacobs N, et al. Correlation of T-helper secretory differentiation and types of antigen-presenting cells in squamous intraepithelial lesions of the uterine cervix. J Pathol. 1998;184:283–90. doi: 10.1002/(SICI)1096-9896(199803)184:3<283::AID-PATH25>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 21.Sheu BC, Chiou SH, Lin HH, et al. Up-regulation of inhibitory natural killer receptors CD94/NKG2A with suppressed intracellular perforin expression of tumor-infiltrating CD8+ T lymphocytes in human cervical carcinoma. Cancer Res. 2005;65:2921–9. doi: 10.1158/0008-5472.CAN-04-2108. [DOI] [PubMed] [Google Scholar]
- 22.Evans EM, Man S, Evans AS, Borysiewicz LK. Infiltration of cervical cancer tissue with human papillomavirus-specific cytotoxic Tlymphocytes. Cancer Res. 1997;57:2943–50. [PubMed] [Google Scholar]
- 23.Sheu BC, Hsu SM, Ho HN, et al. Tumor immunology--when a cancer cell meets the immune cells. J Formosan Med Assoc. 1999;98:730–5. [PubMed] [Google Scholar]
- 24.Antoniou AN, Powis SJ, Elliott T. Assembly and export of MHC class I peptide ligands. Curr Opin Immunol. 2003;15:75–81. doi: 10.1016/s0952-7915(02)00010-9. [DOI] [PubMed] [Google Scholar]
- 25.Ambagala AP, Solheim JC, Srikumaran S. Viral interference with MHC class I antigen presentation pathway: the battle continues. Vet Immunol Immunopathol. 2005;107:1–15. doi: 10.1016/j.vetimm.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Antoniou AN, Powis SI. Pathogen evasion strategies for the major histocompatibility complex class I assembly pathway. Immunology. 2008;124:1–12. doi: 10.1111/j.1365-2567.2008.02804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner M, Gutermann A, Podlech J, et al. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med. 2002;196:805–16. doi: 10.1084/jem.20020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–45. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 29.Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 30.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3:920–40. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 32.Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;43:336–41. doi: 10.1016/j.cyto.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 33.Horst D, Verweij MC, Davison AJ, et al. Viral evasion of T cell immunity: ancient mechanisms offering new applications. Curr Opin Immunol. 2011;23:96–103. doi: 10.1016/j.coi.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan DH, Kissenpfennig A, Clausen BE. Insights into Langerhans cell function from Langerhans cell ablation models. Eur J Immunol. 2008;38:2369–76. doi: 10.1002/eji.200838397. [DOI] [PubMed] [Google Scholar]
- 35.Münger K, Baldwin A, Edwards KM, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muñoz N, Bosch FX, de Sanjosé S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–27. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 37.Gross G, Pfister H. Role of human papillomavirus in penile cancer, penile intraepithelial squamous cell neoplasias and in genital warts. Med Microbiol Immunol. 2004;193:35–44. doi: 10.1007/s00430-003-0181-2. [DOI] [PubMed] [Google Scholar]
- 38.de Villiers EM, Fauquet C, Broker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 39.Cogliano V, Baan R, Straif K, et al. Carcinogenicity of human papillomaviruses. Lancet Oncol. 2005;6:204–204. doi: 10.1016/s1470-2045(05)70086-3. [DOI] [PubMed] [Google Scholar]
- 40.Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006;24:1–10. doi: 10.1016/j.vaccine.2006.05.115. [DOI] [PubMed] [Google Scholar]
- 41.Parkin DM, Bray F. Chapter 2: The burden of HPV-related cancers. Vaccine. 2006;24:11–25. doi: 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- 42.Koutsky L. The epidemiology behind the HPV vaccine discovery. Ann Epidemiol. 2009;19:239–44. doi: 10.1016/j.annepidem.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 43.zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 44.Wallin KL, Wiklund F, Angström T, et al. Type-specific persistence of human papillomavirus DNA before the development of invasive cervical cancer. N Engl J Med. 1999;341:1633–8. doi: 10.1056/NEJM199911253412201. [DOI] [PubMed] [Google Scholar]
- 45.Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 46.Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst. 2005;97:1072–9. doi: 10.1093/jnci/dji187. [DOI] [PubMed] [Google Scholar]
- 47.Bernard HU. The clinical importance of the nomenclature, evolution and taxonomy of human papillomaviruses. J Clin Virol. 2005;32:1–6. doi: 10.1016/j.jcv.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Stanley M. Pathology and epidemiology of HPV infection in females. Gynecol Oncol. 2010;117:5–10. doi: 10.1016/j.ygyno.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 49.Bosch FX, Burchell AN, Schiffman M, et al. Epidemiology and natural history of human papillomavirus infections and type-specific implications in cervical neoplasia. Vaccine. 2008;26:1–16. doi: 10.1016/j.vaccine.2008.05.064. [DOI] [PubMed] [Google Scholar]
- 50.Laimins LA. The biology of human papillomaviruses: from warts to cancer. Infect Agents Dis. 1993;2:74–86. [PubMed] [Google Scholar]
- 51.zur Hausen H, de Villiers EM. Human papillomaviruses. Ann Rev Microbiol. 1994;48:427–47. doi: 10.1146/annurev.mi.48.100194.002235. [DOI] [PubMed] [Google Scholar]
- 52.Miralles-Guri C, Bruni L, Cubilla AL, et al. Human papillomavirus prevalence and type distribution in penile carcinoma. J Clin Pathol. 2009;62:870–8. doi: 10.1136/jcp.2008.063149. [DOI] [PubMed] [Google Scholar]
- 53.Mohr IJ, Clark R, Sun S, et al. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science. 1990;250:1694–9. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 54.Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 1991;10:449–57. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiang CM, Ustav M, Stenlund A, et al. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc Natl Acad Sci U S A. 1992;89:5799–803. doi: 10.1073/pnas.89.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frattini MG, Laimins LA. The role of the E1 and E2 proteins in the replication of human papillomavirus type 31b. Virology. 1994;204:799–804. doi: 10.1006/viro.1994.1596. [DOI] [PubMed] [Google Scholar]
- 57.Thierry F, Yaniv M. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J. 1987;6:3391–7. doi: 10.1002/j.1460-2075.1987.tb02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stubenrauch F, Colbert AM, Laimins LA. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J Virol. 1998;72:8115–23. doi: 10.1128/jvi.72.10.8115-8123.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doorbar J, Ely S, Sterling J, et al. Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature. 1991;352:824–7. doi: 10.1038/352824a0. [DOI] [PubMed] [Google Scholar]
- 60.Leptak C, Ramon y, Cajal S, Kulke R, et al. Tumorigenic transformation of murine keratinocytes by the E5 genes of bovine papillomavirus type 1 and human papillomavirus type 16. J Virol. 1991;65:7078–83. doi: 10.1128/jvi.65.12.7078-7083.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phelps WC, Yee CL, Münger K, Howley PM. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell. 1988;53:539–47. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- 62.Münger K, Werness BA, Dyson N, et al. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scheffner M, Werness BA, Huibregtse JM, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 64.Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–9. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 65.Klaes R, Woerner SM, Ridder R, et al. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999;59:6132–6. [PubMed] [Google Scholar]
- 66.Ozbun MA, Meyers C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J Virol. 1997;71:5161–72. doi: 10.1128/jvi.71.7.5161-5172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morris PJ, Dent CL, Ring CJ, Latchman DS. The octamer binding site in the HPV16 regulatory region produces opposite effects on gene expression in cervical and non-cervical cells. Nucleic Acids Res. 1993;21:1019–23. doi: 10.1093/nar/21.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morris PJ, Ring CJ, Lillycrop KA, Latchman DS. Transactivation of the human papilloma virus 16 octamer motif by the octamer binding protein Oct-2 requires both the N and C terminal activation domains. Nucleic Acids Res. 1993;21:4506–10. doi: 10.1093/nar/21.19.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan SH, Leong LE, Walker PA, Bernard HU. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J Virol. 1994;68:6411–20. doi: 10.1128/jvi.68.10.6411-6420.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanaya T, Kyo S, Laimins LA. The 5' region of the human papillomavirus type 31 upstream regulatory region acts as an enhancer which augments viral early expression through the action of YY1. Virology. 1997;237:159–69. doi: 10.1006/viro.1997.8771. [DOI] [PubMed] [Google Scholar]
- 71.de Villiers EM. Taxonomic classification of papillomaviruses. Papillomavirus Report. 2001;12:57–63. [Google Scholar]
- 72.Doorbar J. The papillomavirus life cycle. J Clin Virol. 2005;32:7–15. doi: 10.1016/j.jcv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 73.Fehrmann F, Laimins LA. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene. 2003;22:5201–7. doi: 10.1038/sj.onc.1206554. [DOI] [PubMed] [Google Scholar]
- 74.Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12:2061–72. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li S, Labrecque S, Gauzzi MC, et al. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-alpha. Oncogene. 1999;18:5727–37. doi: 10.1038/sj.onc.1202960. [DOI] [PubMed] [Google Scholar]
- 76.Perea SE, Massimi P, Banks L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int J Mol Med. 2000;5:661–6. doi: 10.3892/ijmm.5.6.661. [DOI] [PubMed] [Google Scholar]
- 77.Park JS, Kim EJ, Kwon HJ, et al. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem. 2000;275:6764–9. doi: 10.1074/jbc.275.10.6764. [DOI] [PubMed] [Google Scholar]
- 78.Nees M, Geoghegan JM, Hyman T, et al. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-κB-responsive genes in cervical keratinocytes. J Virol. 2001;75:4283–96. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hagensee ME, Koutsky LA, Lee SK, et al. Detection of cervical antibodies to human papillomavirus type 16 (HPV-16) capsid antigens in relation to detection of HPV-16 DNA and cervical lesions. J Infect Dis. 2000;181:1234–9. doi: 10.1086/315364. [DOI] [PubMed] [Google Scholar]
- 80.Bierl C, Karem K, Poon AC, et al. Correlates of cervical mucosal antibodies to human papillomavirus 16: results from a case control study. Gynecol Oncol. 2005;99:262–8. doi: 10.1016/j.ygyno.2005.07.100. [DOI] [PubMed] [Google Scholar]
- 81.Sheu BC, Chang WC, Lin HH, et al. Immune concept of human papillomaviruses and related antigens in local cancer milieu of human cervical neoplasia. J Obstet Gynaecol Res. 2007;33:103–13. doi: 10.1111/j.1447-0756.2007.00492.x. [DOI] [PubMed] [Google Scholar]
- 82.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nature Rev Immunol. 2008;8:935–47. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 83.Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev. 2010;234:120–41. doi: 10.1111/j.0105-2896.2009.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stoecklinger A, Eticha TD, Mesdaghi M, et al. Langerin+ dermal dendritic cells are critical for CD8+ T cell activation and IgH γ-1 class switching in response to gene gun vaccines. J Immunol. 2011;186:1377–83. doi: 10.4049/jimmunol.1002557. [DOI] [PubMed] [Google Scholar]
- 85.Kadish AS, Timmins P, Wang Y, et al. Regression of cervical intraepithelial neoplasia and loss of human papillomavirus (HPV) infection is associated with cell-mediated immune responses to an HPV type 16 E7 peptide. Cancer Epidemiol Biomarkers Prev. 2002;11:483–8. [PubMed] [Google Scholar]
- 86.de Gruijl TD, Bontkes HJ, Walboomers JM, et al. Differential T helper cell responses to human papillomavirus type 16 E7 related to viral clearance or persistence in patients with cervical neoplasia: a longitudinal study. Cancer Res. 1998;58:1700–6. [PubMed] [Google Scholar]
- 87.de Gruijl TD, Bontkes HJ, Walboomers JM, et al. Immune responses against human papillomavirus (HPV) type 16 virus-like particles in a cohort study of women with cervical intraepithelial neoplasia. I. Differential T-helper and IgG responses in relation to HPV infection and disease outcome. J Gen Virol. 1999;80:399–408. doi: 10.1099/0022-1317-80-2-399. [DOI] [PubMed] [Google Scholar]
- 88.Bontkes HJ, de Gruijl TD, Walboomers JM, et al. Immune responses against human papillomavirus (HPV) type 16 virus-like particles in a cohort study of women with cervical intraepithelial neoplasia. II. Systemic but not local IgA responses correlate with clearance of HPV-16. J Gen Virol. 1999;80:409–17. doi: 10.1099/0022-1317-80-2-409. [DOI] [PubMed] [Google Scholar]
- 89.Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer. 2002;2:59–65. doi: 10.1038/nrc700. [DOI] [PubMed] [Google Scholar]
- 90.Kloover JS, Grauls GE, Blok MJ, et al. A rat cytomegalovirus strain with a disruption of the r144 MHC class I-like gene is attenuated in the acute phase of infection in neonatal rats. Arch Virol. 2002;147:813–24. doi: 10.1007/s007050200028. [DOI] [PubMed] [Google Scholar]
- 91.Nimako M, Fiander AN, Wilkinson GW, et al. Human papillomavirus-specific cytotoxic T lymphocytes in patients with cervical intraepithelial neoplasia grade III. Cancer Res. 1997;57:4855–61. [PubMed] [Google Scholar]
- 92.Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–9. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 93.Yokoyama WM. Natural killer cell immune responses. Immunol Res. 2005;32:317–25. doi: 10.1385/IR:32:1-3:317. [DOI] [PubMed] [Google Scholar]
- 94.Pyzik M, Kielczewska A, Vidal SM. NK cell receptors and their MHC class I ligands in host response to cytomegalovirus: insights from the mouse genome. Semin Immunol. 2008;20:331–42. doi: 10.1016/j.smim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 95.Jonjić S, Babić M, Polić B, Krmpotić A. Immune evasion of natural killer cells by viruses. Curr Opin Immunol. 2008;20:30–8. doi: 10.1016/j.coi.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Ann Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 97.Ting JP, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–7. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–37. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 100.Oeckinghaus A, Ghosh S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 102.Pasparakis M. Regulation of tissue homeostasis by NF-κB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–88. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 103.Rahman MM, McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol. 2011;9:291–306. doi: 10.1038/nrmicro2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715–23. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 105.Tokunaga F, Iwai K. Linear ubiquitination: a novel NF-κB regulatory mechanism for inflammatory and immune responses by the LUBAC ubiquitin ligase complex. Endocr J. 2012;59:641–52. doi: 10.1507/endocrj.ej12-0148. [DOI] [PubMed] [Google Scholar]
- 106.Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 107.Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21:223–44. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signaling. Nat Rev Immunol. 2008;8:911–22. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Edelmann MJ, Kessler BM. Ubiquitin and ubiquitin-like specific proteases targeted by infectious pathogens: emerging patterns and molecular principles. Biochim Biophys Acta. 2008;1782:809–16. doi: 10.1016/j.bbadis.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yokota S, Okabayashi T, Fujii N. The battle between virus and host: modulation of Toll-like receptor signaling pathways by virus infection. Mediators Inflamm. 2010:e184328. doi: 10.1155/2010/184328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol. 2011;11:187–200. doi: 10.1038/nri2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huibregtse JM, Scheffner M, Howley PM. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol Cell Biol. 1993;13:4918–27. doi: 10.1128/mcb.13.8.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 114.Patel D, Incassati A, Wang N, McCance DJ. Human papillomavirus type 16 E6 and E7 cause polyploidy in human keratinocytes and up-regulation of G2-M-phase proteins. Cancer Res. 2004;64:1299–306. doi: 10.1158/0008-5472.can-03-2917. [DOI] [PubMed] [Google Scholar]
- 115.Tommasino M, Adamczewski JP, Carlotti F, et al. HPV16 E7 protein associates with the protein kinase p33CDK2 and cyclin A. Oncogene. 1993;8:195–202. [PubMed] [Google Scholar]
- 116.McIntyre MC, Ruesch MN, Laimins LA. Human papillomavirus E7 oncoproteins bind a single form of cyclin E in a complex with cdk2 and p107. Virology. 1996;215:73–82. doi: 10.1006/viro.1996.0008. [DOI] [PubMed] [Google Scholar]
- 117.Zerfass-Thome K, Zwerschke W, Mannhardt B, et al. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13:2323–30. [PubMed] [Google Scholar]
- 118.Funk JO, Waga S, Harry JB, et al. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Develop. 1997;11:2090–100. doi: 10.1101/gad.11.16.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ruesch MN, Laimins LA. Human papillomavirus oncoproteins alter differentiation-dependent cell cycle exit on suspension in semisolid medium. Virology. 1998;250:19–29. doi: 10.1006/viro.1998.9359. [DOI] [PubMed] [Google Scholar]
- 120.zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000;92:690–8. doi: 10.1093/jnci/92.9.690. [DOI] [PubMed] [Google Scholar]
- 121.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4. [PubMed] [Google Scholar]
- 122.Jones DL, Thompson DA, Münger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 123.Berezutskaya E, Yu B, Morozov A, et al. Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Diff. 1997;8:1277–86. [PubMed] [Google Scholar]
- 124.Nguyen DX, Westbrook TF, McCance DJ. Human papillomavirus type 16 E7 maintains elevated levels of the cdc25A tyrosine phosphatase during deregulation of cell cycle arrest. J Virol. 2002;76:619–32. doi: 10.1128/JVI.76.2.619-632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 126.Thomas M, Pim D, Banks L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene. 1999;18:7690–700. doi: 10.1038/sj.onc.1202953. [DOI] [PubMed] [Google Scholar]
- 127.Garnett TO, Filippova M, Duerksen-Hughes PJ. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Diff. 2006;13:1915–26. doi: 10.1038/sj.cdd.4401886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kiriakidis S, Andreakos E, Monaco C, et al. VEGF expression in human macrophages is NF-κB-dependent: studies using adenoviruses expressing the endogenous NF-κB inhibitor IkBα and a kinase-defective form of the IκB kinase 2. J Cell Sci. 2003;116:665–74. doi: 10.1242/jcs.00286. [DOI] [PubMed] [Google Scholar]
- 129.Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 130.Chen R, Alvero AB, Silasi DA, et al. Cancers take their Toll--the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27:225–33. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- 131.El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene. 2008;27:244–52. doi: 10.1038/sj.onc.1210912. [DOI] [PubMed] [Google Scholar]
- 132.Havard L, Delvenne P, Fraré P, et al. Differential production of cytokines and activation of NF-κB in HPV-transformed keratinocytes. Virology. 2002;298:271–85. doi: 10.1006/viro.2002.1468. [DOI] [PubMed] [Google Scholar]
- 133.Nees M, Geoghegan JM, Hyman T, et al. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-κB-responsive genes in cervical keratinocytes. J Virol. 2001;75:4283–96. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Spitkovsky D, Hehner SP, Hofmann TG, et al. The human papillomavirus oncoprotein E7 attenuates NF-κB activation by targeting the IκB kinase complex. J Biol Chem. 2002;277:25576–82. doi: 10.1074/jbc.M201884200. [DOI] [PubMed] [Google Scholar]
- 135.Havard L, Rahmouni S, Boniver J, Delvenne P. High levels of p105 (NFKB1) and p100 (NFKB2) proteins in HPV16-transformed keratinocytes: role of E6 and E7 oncoproteins. Virology. 2005;331:357–66. doi: 10.1016/j.virol.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 136.Mishra A, Bharti AC, Varghese P, et al. Differential expression and activation of NF-κB family proteins during oral carcinogenesis: Role of high risk human papillomavirus infection. Int J Cancer. 2006;119:2840–50. doi: 10.1002/ijc.22262. [DOI] [PubMed] [Google Scholar]
- 137.James MA, Lee JH, Klingelhutz AJ. Human papillomavirus type 16 E6 activates NF-κB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J Virol. 2006;80:5301–7. doi: 10.1128/JVI.01942-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66:1403–8. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 139.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pikarsky E, Porat RM, Stein I, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004:431, 461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 141.Kanodia S, Fahey LM, Kast WM. Mechanisms used by human papillomaviruses to escape the host immune response. Curr Cancer Drug Targets. 2007;7:79–89. doi: 10.2174/156800907780006869. [DOI] [PubMed] [Google Scholar]
- 142.Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet. 2008;371:771–83. doi: 10.1016/S0140-6736(08)60241-X. [DOI] [PubMed] [Google Scholar]
- 143.Wang CY, Cusack JC, Jr, Liu R, Baldwin AS., Jr Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-κB. Nature Med. 1999;5:412–7. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 144.Kim HJ, Hawke N, Baldwin AS. NF-κB and IKK as therapeutic targets in cancer. Cell Death Diff. 2006;13:738–47. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- 145.Karin M. The IκB kinase - a bridge between inflammation and cancer. Cell Res. 2008;18:334–2. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- 146.Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harbor Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]