

Abstract
Most of the pheochromocytomas (PCCs) are benign neoplasms, but when they are malignant, they can be difficult to treat. Despite advances in diagnosis and imaging, it remains an untreatable tumor, when metastases develop. A deeper understanding of the alteration of the specific molecular pathways causing malignant PCCs might hopefully lead in the future to the development of multiple molecular-targeted therapies to treat it successfully. Clinical experience and the use of murine models of metastatic PCCs have helped introduce new experimental treatment options which will significantly help the PCCs community explore novel targeted therapies that have already shown promising results in many other types of tumors.
Key words: pheochromocytoma, metastatic, target therapy
Introduction
Pheochromocytoma (PCC) is a rare tumor of catecholamine-secreting chromaffin cells, 80-90% of which are located in the adrenal medulla and approximately 10-20% in other sites, usually throughout the sympathetic chain in the thorax, abdomen, and pelvis. Extra-adrenal pheochromocytomas are referred to as paragangliomas.1
Approximately 10% of PCCs are malignant. In particular, extra-adrenal PCCs are more likely to be malignant, are characterized by metastases and spread in sites where chromaffin tissue is normally absent, such as lymph nodes, liver, lungs, and bones.2
Most cases are sporadic, but up to 25% are inherited, usually as part of multiple endocrine type 2A and 2B neoplasia (MEN2A and MEN2B), von Hippel-Lindau disease, type 1 neurofibromatosis (von Recklinghausen disease), and familial paragangliomas.3
They may occur at any age and their peak incidence is between the third and the fifth decades of life with an almost equal female/male ratio.4
PCCs may cause challenges to patients in two ways: firstly due to adverse effects caused by the autonomous production of catecholamines and secondly due to malignancy and metastatic spread. Associated symptoms are variable. The triad including tachycardia, diaphoresis and cephalalgia is encountered in 40-80% of patients and is highly sensitive and specific for a presumptive diagnosis of PCC. Hypertension (newly diagnosed or an exacerbation of known hypertension, most often paroxysmal) is very common (in over 90% of patients), but non-specific.5
Despite progress in diagnosis and imaging, PCCs remain untreatable tumors for which no cure exists when metastases are present. The prognosis of benign and malignant tumors differs significantly, since benign tumors are generally amenable to complete surgical resection and have a 10-year survival in up to 94% of cases.6 Conversely, no curative treatment exists for the metastatic form in which the 10-year survival rate drops to only 20%. Many different types of systemic chemotherapy for malignant PCC have been tried. One of the challenges in understanding the role of chemotherapy is that only few prospective trials have investigated different types of chemotherapeutic options. Also, most knowledge about the effectiveness of chemotherapy is based on anecdotal evidence and individual cases.7
Conventional chemotherapy is usually based on cyclophosphamide, vincristine and doxorubicin, but even with this treatment, it is difficult to demonstrate any effect on overall survival. Since these tumors show an uptake of radiopharmaceutical I123-metaiodobenzylguanidine (I123-MIBG), a therapy with I131-MIBG is possible and may have positive therapeutic effects, although tumor regression is not frequently seen.8,9 Therefore there is a considerable clinical need for new effective therapies, hence novel therapeutic approaches to treat metastatic malignant PCCs are urgently required.
Genetics of pheochromocytoma
A gene profiling study demonstrated a genetic germ-line or somatic cause in 45.5% of these tumors.10 Mutations of ten different genes are known to cause hereditary PCCs and paragangliomas: the von Hippel-Lindau gene (VHL), the RET proto-oncogene, the neurofibromatosis type 1 gene (NF1), the succinate dehydrogenase B subunit genes (SDHA, SDHB, SDHC, SDHD, and SDHAF2), MYC associated factor X (MAX), and transmembrane protein 127 (TMEM127).11 It has been suggested that these mutations can be divided into distinct molecular pathways causing errors in the hypoxia-inducible factor (HIF) hypoxia-driven pathway (cluster 1) and errors in RNA synthesis and metabolism (cluster 2), while a kinesin family member (KIF1B ) is thought to impact both pathways by controlling the apoptotic pathway. Also its missense mutations have been identified in PCCs suggesting that KIF1B is a pathogenic target of these deletions.12
Cluster 1
Cluster 1 is related to the Krebs cycle modulation of hypoxia signaling, i.e. mutations in succinate dehydrogenase subunit A (AF2)/B/C/D (SDHA(AF2)/B/C/D), the gene associated with VHL or 2-oxoglutarate-dependent prolyl hydroxylase (PHD2).
The HIF is a transcription factor responsible for regulating the cell response to hypoxia. It is expressed and regulated by the oxygen tension in the cell. Under normal normoxic conditions, HIF is hydroxylated by PHDs allowing it to be degraded by a VHL-mediated process. Mutations of VHL prevent HIF degradation, causing increased HIF accumulation, unregulated angiogenesis, and tumor formation. Mutations of SDHB and SDHD genes cause succinate to accumulate. Excess succinate prevents PHDs domains from hydroxylating HIF, which also leads to HIF accumulation, activation, and tumor formation (Figure 1).13
Figure 1.
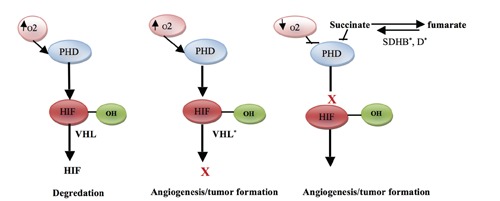
Three hypoxia-inducible factor (HIF) pathway mutations are associated with pheochromocytoma. *Mutation. (VHL, von Hippel-Lindau; PHD, prolyl hydroxylase domain; SDHB, succinate dehydrogenase B subunit gene; SDHD, succinate dehydrogenase D subunit gene).
Stable HIF acts therefore as a transcription factor that causes the up regulation of several genes involved in the hypoxia response. Many of these genes, such as the vascular endothelial growth factor (VEGF), are responsible for angiogenesis as well as cellular growth and metabolism.14 As HIF is the downstream effector of mTORC1, the signaling pathways converging on mTORC1/2 are attractive candidates for therapeutic manipulation.
Cluster 2
Cluster 2 is associated with kinase signaling pathways (PI3K/AKT, RAS/RAF/ERK and mTORC1/p70S6K), neuroendocrine differentiation and comprises mutations in the rearrangement during transfection of genes RET, NF1, KIF1B, TMEM127 and MAX. It remains unclear exactly how these mutations cause the abnormal gene expression profiles that lead to tumor formation. However, RET, NF1, and KIF1B participate all in the molecular cascade that leads to apoptosis.15 Mutations of these genes are thought to decrease apoptosis, leading to abnormal cell growth. RET and NF1 have both shown to activate the MAPK pathway of cellular signaling via abnormal RAS activation, which is involved in a wide variety of human tumors (Figure 2). Progress in the treatment of malignant PCCs has been limited, in part because of the rarity of this malignancy, the lack of sensitivity to cytotoxic therapy, and the anecdotal nature of responses to therapy reported in small case studies or very limited numbers of patients in larger clinical trials.16,17
Figure 2.
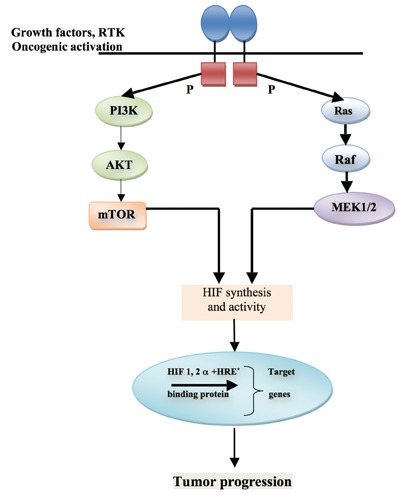
Clusters 2 associated with pheochromocytoma. *Hypoxia related product.
Experimental and targeted therapies
The recent insights in the understanding of the molecular pathways that cause malignant PCCs formation are leading to the development of new drug therapies. Most of them have cytostatic properties in that not only do they kill cancer cells, but they can also interfere with the cytotoxic activity of other chemotherapeutic drugs. In addition, they can modulate the activity of key enzymes responsible for the specific signaling pathways capable of transforming benign PCCs into their malignant counterparts. The cytostatic properties of these anticancer therapies are derived from the specific molecular targets found along the oncogenic signaling pathways that promote carcinogenesis and tumor growth.18 Specific receptors and proteins have been discovered and reflect the different genetic clusters that categorize the malignant transformation of these adrenal neoplasms. Our better understanding of the biology of these tumors offers the opportunity for using more selective targeted therapeutic options, with the promise of superior efficacy and substantially reduced adverse effects.
Therapeutic targets
Heat shock protein 90
This multichaperone ATP-dependent complex is responsible for folding therapeutically relevant proteins, and plays an important role in the stability and function of a host of oncoproteins (BCR-ABL, ERBB2, EGFR, CRAF, BRAF, AKT, MET, VEGFR, FLT3, AR, ER, HIF, and telomerase).19 These oncoproteins are responsible for many molecular processes usually attributed to the malignant phenotype, including growth factor independence, resistance to antigrowth signals, cell replication, tumor invasion and metastases, angiogenesis, and lack of apoptosis.20 Heat shock protein 90 (Hsp90) may represent a potential therapeutic target, as its high protein expression level has been found in malignant compared to benign PCC.21 Because of the many oncogenic signaling pathways regulated by Hsp90, inhibitors of Hsp90 can target several oncogenic proteins in parallel.22
In a xenograft mouse model, the use of two different Hsp90 inhibitors, 17-AAG and ganetespib have demonstrated a significant antitumor activity against both subcutaneous and metastatic tumor growth. As a result several Hsp90 inhibitors have been developed in the last decade, and several clinical trials are ongoing.23
mTOR inhibitors
The PI3K/AKT/mTOR pathway is responsible for regulating cell growth and survival. If this pathway becomes dysfunctional, mTOR becomes upregulated, leading to increased cell proliferation, angiogenesis, and evasion of apoptosis.24 Everolimus (RAD001), an mTORC1 inhibitor, in combination with octreotide has shown to be effective for low- and intermediate grade neuroendocrine tumors.25 The efficacy of everolimus has been evaluated also in malignant PCCs, but all patients experienced disease progression.26 Two potential explanations for this treatment failure have been proposed. Firstly, mTORC1 repression only inhibits HIF1, but it appears to have no effect on HIF2,27 and in general it is regarded as the more oncogenic of the two isoforms.28 Secondly, a compensatory activation of a parallel signaling pathway involved in cell growth, RAS/RAF/ERK, was noted, when mTORC1 was inhibited.29 To address these concerns, an agent that inhibits both mTORC-1 and -2 (e.g. AZD-8055) can potentially be a rewarding option. Alternatively, given the compensatory ERK activation through PI3K, a combination with an ERK inhibitor (e.g. AEZS-131) to prevent any feedback loop that the transformed cells could use to their advantage can be a viable choice. Combination therapies, like the ones mentioned, can also potentially make cancer cells less likely to develop resistance over time.
Anti-angiogenesis
Angiogenesis is a major player in the development of several tumors, and in particular in the formation of tumor metastases. VEGF family is a major player in angiogenesis, and its role in several cancers is well established. PCCs are highly vascularized tumors, and angiogenesis in these tumors has been associated with a malignant phenotype.30 Takekoshi et al. have analyzed the expression of VEGF and its receptors in PCCs.31 Compared with normal adrenomedullary tissue, these tumors expressed higher levels of Flk-1/KDR and Flt-1, suggesting that the expression of both the receptors and the ligands has an important role in the pathogenesis of these tumors. Interestingly, using antibodies against VEGF was able to reduce tumor angiogenesis and tumor proliferation in a xenograft mouse model of PCCs.32
Hypoxia-inducible factor inhibitors
HIF is a key regulator of the tumor environment and has spawned the research and development of several targeted therapies. Some agents being investigated are PX-12 (1-methylpropyl 2-imidazolyl disulfide) and PX-478 (S-2-amino-3-[4’-N,N,-bis (2-chloroethyl)amino]-phenyl propionic acid N-oxide dihydrochloride). HIF activity is decreased indirectly by PX-12 and is directly inhibited by PX-478. Although no data have been reported for malignant PCCs, these agents have shown marked antitumor activity in various human tumor xenografts in mice and seem to be promising also for malignant PCCs, but conclusive data are missing.33-35
Prolyl hydroxylase activators
Evidence has now indicated that activators of prolyl hydroxylase, such as R59949 and KRH102053, result in increased hydroxylation of HIF, this ultimately decreases the expression of proteins that regulate angiogenesis and those that resist apoptosis.36 As our understanding of the association between the HIF pathway and the role of prolyl hydroxylases in PCCs continues to expand, it is likely to lead to the discovery of new antineoplastic agents.
HER-2/neu inhibitors
HER2 is a receptor tyrosine kinase involved in cell growth and differentiation. When activated, it leads to the synthesis of HIF, which subsequently leads to the overexpression of VEGF.37 HER2 was reported to induce bilateral PCCs in mice.38 It was significantly overexpressed in malignant PCCs compared to benign or extra-adrenal PCCs and associated with tumor metastasis and resistance to treatment.39,40 To date, no trials have been conducted using HER-2/neu inhibitors for patients diagnosed with malignant PCCs.
Specific drugs
Thalidomide
Initially introduced in the 1950s as a sedative, thalidomide was quickly removed from the market when its teratogenic effects became known. At the turn of this century, thalidomide, in combination with dexamethasone, gained popularity, when it showed to increase survival in patients diagnosed with multiple myeloma.41 Thalidomide is an anti-angiogenic agent, specifically targeting VEGF and the basic fibroblast growth factor. To date, only one phase II trial has been conducted to determine the efficacy of thalidomide in combination with temozolomide (TMZ), as an alternative to streptozocin-based therapy in neuroendocrine tumors. In the PCCs group, which included three patients, a partial radiologic response to the therapy occurred in one patient. Due to the limited number of cases, it is impossible to draw any conclusions about the efficacy of thalidomide in PCCs patients. However, the combination of thalidomide and TMZ may offer some benefits to patients with PCCs and carcinoid and/or pancreatic neuroendocrine tumors.42
Imatinib mesylate
This tyrosine kinase inhibitor specifically inhibits the protein tyrosine kinase activity of the BCL-ABL, c-KIT, and PDGF-R enzymes. Imatinib, as a targeted therapy against the BCL-ABL gene and the tyrosine kinase protein c-KIT, became important as an effective treatment for hematologic malignancies (e.g., chronic myelogenous leukemia) as well as solid tumors (e.g., gastrointestinal stromal tumors). Several studies were designed to determine whether other solid endocrine malignancies expressed either PDGF-R or c-KIT.43 Yet the data concerning patients with malignant PCCs (n=2) indicated a progression of the disease. Interestingly, the immune-histochemical profiles of these tumors (PDGF-R+ and c-KIT+; PDGF-R+ and c-KIT–) made no difference in the tumors’ response to imatinib.44
Sunitinib
Sunitinib is a receptor tyrosine kinase inhibitor that acts on several targets (VEGF, PDGF, c-KIT) and demonstrates anti-angiogenic and anti-tumor activity. The mechanism through which sunitinib acts on malignant PCCs is associated with the mutation of the VHL gene. This gene, as previously mentioned, leads to the activation of HIF and upregulates both VEGF and PDGF. Sunitinib can inhibit both the VEGF and PDGF receptors, as was previously shown in renal cell malignancies. Case studies have reported a partial or near-complete tumor regression after treatment with sunitinib by measuring tumor size, performance status, patient symptoms, and reduction of biochemical tumor marker levels.45 The preliminary results of the effectiveness of sunitinib as a molecular-targeted therapy for malignant PCCs are promising. We expect growth in the number of clinical trials using receptors tyrosine kinase and their ligands as targets, either alone or in combination with traditional chemotherapy or radiotherapy.
Future therapeutic options
Carboxypeptidase E is another molecular target that could be used in the treatment of metastatic PCCs.46 The increased expression of an N-terminal splice isoform in various human metastatic tumor cell lines with high copy numbers of its mRNA were observed in metastatic PCCs, and interestingly this high copy numbers in benign tumors are able to predict recurrence or metastatic disease. Suppression of this gene in these cell lines with siRNA led to inhibition of growth and invasion in xenograft mouse models.47 It appears that the N-terminal splice isoform increases neural precursor expressed, developmentally down-regulated 9 (NEDD9) by interacting with histone deacetylase 1 and -2 (HDAC 1 and -2), another potential molecular target for metastatic PCCs. As a side note, NEDD9 has been associated with tumor progression in various types of cancer.48
Many different types of experimental therapeutic agents, either alone or in combination with other therapeutic options, have shown to have a significant effect on PCCs cell apoptosis. For example, the HDAC inhibitors romidepsin and trichostatin A were studied to assess their ability to enhance apoptosis induced by [123I]-MIBG uptake in a mouse model of metastatic PCCs and found to profoundly increase the expression of the norepinephrine transporter system (NET) and the uptake of [123I]-MIBG in vitro and in vivo.49 So the use of HDAC inhibitors can enhance the therapeutic efficacy of [131I]-MIBG treatment in patients with metastatic PCCs.
The ability of LB1, a small molecule inhibitor of serine/threonine protein phosphatase 2A (PP2A) had been investigated to inhibit mouse PCCs cells in vitro and in vivo in a mouse model of metastatic state alone and in combination with TMZ. When LB1 or TMZ were used separately or in combination, they inhibited cell proliferation in vitro only modestly, however in vivo the combination of both drugs markedly reduced the rate of increase of the metastatic (hepatic) tumor volume in all animals. There was a slight or no inhibitory effect on tumor growth in vivo when these drugs were used separately. The transient pharmacologic inhibition of PP2A could represent a new approach for enhancing the efficacy of non-specific cancer chemotherapy regimens against a broad spectrum of low growth fraction of PCCs tumors commonly resistant to cytotoxic drugs.50
Another study demonstrated that NFkB inhibition using either trip-tolide or capsaicin led to NET upregulation and apoptosis induction in the three available PCC cell lines.51 Moreover, NFkB inhibition with triptolide resulted in a significant reduction of liver metastases in an animal model of metastatic PCCs. Apoptosis can also be induced in PCCs/PGLs via the inhibition of TNF receptor-associated protein 1 (TRAP-1).52
Somatostatin analogs are also promising agents in the treatment of PCCs, not only because of their established use in neuroendocrine tumors, but more importantly because it has been shown that PCC tissues express more than one type of somatostatin receptor.53 A study reported a more potent inducer of apoptosis, SOM230, in comparison to octreotide. SOM230 also markedly suppressed intracellular catecholamine levels. The superior response profile of SOM230 is attributed to its ability to block 4 out of 5 somatostatin receptors compared to octreotide or lanreotide, which are known to have high affinity to somatostatin receptor 2 (sst2).54
Nutritional therapy for cancer has also been gaining momentum. Eicosapentanoic acid (EPA), a polyunsaturated fatty acid sourced from aquatic animals, has been found to induce apoptosis in the rat PCCs cell line.55 It was postulated that lipid peroxidation with subsequent cellular and DNA damage could account for the anticancer properties of EPA. It is important to outline that, unlike the more widely used and approved chemotherapeutic agents, EPA was reported to cause only negligible side effects,56 outlining the importance of exploring this class of therapy in greater depth.
Highly proliferative cells such as cancer cells are dependent on topoisomerases that release torsional stress on the DNA double strand during replication. This presents a potential drug target, whereby the inhibition of this enzyme (e.g. with compounds such as camptothecin) leads to irreversible DNA strand breaks and subsequent activation of apoptotic pathways.57
Lastly, increased dependence on glycolysis, which has been observed in several tumors and constitutes another important hallmark of cancer, is currently under investigation, with the hope to select a few molecular targets for future therapy.
Clinical trials
Several phase I and II clinical trials are currently recruiting patients who have been diagnosed with either malignant or refractory PCCs and paragangliomas or with other metastatic neuroendocrine tumors expressing somatostatin receptors. The spectrum of trials ranges from the discovery of novel diagnostic assays for PCCs/paragangliomas to novel targeted therapeutic agents and newly radiolabeled somatostatin analogs for the treatment of these malignant tumors.
Table 1 lists the clinical trials that are recruiting new patients. If successful, the new agents under study in these trials might improve the treatment of malignant PCCs.
Table 1.
Clinical trials based on experimental or targeted therapies in malignant pheochromocytoma/paraganglioma.
| Protocol IDs | Trial aim | Type | Status | Phase |
|---|---|---|---|---|
| MAW002 NCT00978211 | Investigate the role of [90Y-DOTA]-TOC and [177LuDOTA]-TOC therapy in advanced neuroendocrine cancer | Treatment | Active | II |
| SNIPP NCT00843037 | Investigate the role of sunitinib malate in patients with advanced malignant paraganglioma or pheochromocytoma cancer | Treatment | Active | II |
| NCI-2011-02588 CDR0000699430, MAYO-MC107B, MC107B, 8783, N01CM00039, N01CM00071, P30CA015083, N01CM00099, NCT01340794 | Investigate the mechanism of action of pazopanib hydrochloride in patients with advanced or progressive malignant pheochromocytoma or paraganglioma | Biomarker/laboratory analysis, treatment | Active | II |
| IGR2010/1715 2010-024621-20, MSI/A110356-31, NCT01371201 | Determine of the efficacy of sunitinib on progression-free survival at 12 months in subjects with progressive malignant pheochromocytoma and paraganglioma treated with sunitinib at a starting dose of 37.5 mg daily | Biomarker/laboratory analysis, treatment | A ctive | II |
| I206 NCT01396408 | Identify the effects of sunitinib or temsirolimus on this type of cancer; the study will begin by finding out if sunitinib can shrink the tumor; if sunitinib does not work, temsirolimus will be tested next | Treatment | Active | II |
| UPCC 23811 NCT01635907 | Evaluate the potential ability of dovitinib to shrink the tumor or slow down cancer growth in patients with certain types of neuroendocrine tumors; this study will also evaluate safety in greater detail | Treatment | Active | II |
| 140001 14-C-0001, NCT01967576 |
Evaluate the activity of axitinib (AG-013736) | Biomarker/laboratory analysis, treatment | Active | II |
| UCL/12/0499 NCT01941849 |
This phase I trial aims to determine the recommended phase II dose (RP2D) of vandetanib in combination with standard radiation therapy, 131I-mIBG, in patients with advanced pheochromocytoma and paraganglioma by assessing the safety and tolerability of the combination treatment | Treatment | Approved-not yet active | I |
NCT ID, National Clinical Trials identifier. Information based on clinical trials listed on clinicaltrials.gov (search terms: pheochromocytoma and/or paraganglioma).
Conclusions
Malignant PCCs are rare cancers, and both surgeons and oncologists are still confronted with the ambiguity of the available treatment strategies. Because any chemotherapy for malignant PCCs needs to be given long-term without survival benefit, it has to be considered on a case-by-case basis. Despite a vast interest and effort to develop new therapeutic approaches to treat metastatic PCCs, data is either limited or still at an experimental level, although there are some promising results and observations. So far the treatments are basically palliative. Molecular targeted therapies are promising strategies, but, due to the complexity of the pathogenesis of these tumors, further studies of tumor biology, discovery of novel targeted drugs, and new trials are still needed to develop more effective treatments.
Ethical consideration
The ethical approval to conduct the study was sought from the Institutional Review Board before its commencement.
References
- 1.Lenders J, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005;366:665-75. [DOI] [PubMed] [Google Scholar]
- 2.Chrisoulidou A, Kaltsas G, Ilias I, Grossman A. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer 2007;14:569-85. [DOI] [PubMed] [Google Scholar]
- 3.Gimenez-Roqueplo A, Dahia P, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 2012;44:328-33. [DOI] [PubMed] [Google Scholar]
- 4.Plouin F, Gimenez P. Pheochromocytomas and secreting paragangliomas. Orphanet J Rare Dis 2006;1:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan NM. Kaplan’s clinical hypertension. Pheochromocytoma with a preface about incidental adrenal masses. Philadelphia, PA: Lippincott, Williams & Wilkins; 2006. pp 389-433. [Google Scholar]
- 6.Manger WM. An overview of pheochromocytoma: history, current concepts,vagaries, and diagnostic challenges. Ann N Y Acad Sci 2006;1073:1-20. [DOI] [PubMed] [Google Scholar]
- 7.Nomura K, Kimura H, Shimizu S, et al. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab 2009;94:2850-6. [DOI] [PubMed] [Google Scholar]
- 8.Pacak K, Linehan M, Eisenhofer G, et al. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 2001;134:315-29. [DOI] [PubMed] [Google Scholar]
- 9.Scholz T, Eisenhofer G, Pacak K, et al. Clinical review: current treatment of malignant pheochromocytoma. J Clin Endocrinol Metab 2007;92:1217-25. [DOI] [PubMed] [Google Scholar]
- 10.Burnichon N, Vescovo L, Amar L, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet 2011;20:3974-85. [DOI] [PubMed] [Google Scholar]
- 11.Galan R, Kann H. Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin Endocrinol (Oxf) 2013;78:165-75. [DOI] [PubMed] [Google Scholar]
- 12.Raymon H, Grogan M, Elliot J, et al. Changing paradigms in the treatment of malignant pheochromocytoma. Cancer Control 2011;18:104-12. [DOI] [PubMed] [Google Scholar]
- 13.Haase VH. The VHL tumor suppressor: master regulator of HIF. Curr Pharm Dis 2009;15:3895-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahia P, Ross K, Wright M, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 2005;1:72-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlisio S, Kenchappa RS, Vredeveld LC, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev 2008;22:884-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grogan RH, Mitmaker EJ, Duh QY. Changing paradigms in the treatment of malignant pheochromocytoma. Cancer Control 2011;18:104-12. [DOI] [PubMed] [Google Scholar]
- 17.Pacak K. Phaeochromocytoma: a catecholamine and oxidative stress disorder. Endocr Regul 2011;45:65-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adjallé R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res 2009;41:687-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res 2009;15:9-14. [DOI] [PubMed] [Google Scholar]
- 20.Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer 2006;13:125-35. [DOI] [PubMed] [Google Scholar]
- 21.Boltze C, Mundschenk J, Unger N, et al. Expression profile of the telomeric complex discriminates between benign and malignant pheochromocytoma. J Clin Endocrinol Metab 2003;88:4280-6. [DOI] [PubMed] [Google Scholar]
- 22.Workman P. Strategies for treating cancers caused by multiple genome abnormalities: from concepts to cures? Curr Opin Investig Drugs 2003;4:1410-5. [PubMed] [Google Scholar]
- 23.Giubellino A, Sourbier C, Lee MJ, et al. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PLoS ONE 2013;8:e56083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan R, Kay A, Berg W, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. J Hematol Oncol 2009;2:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao J, Phan A, Chang D, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol 2008;26:4311-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Druce M Kaltsas G, Fraenkelet al. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001). Horm Metab Res 2009;41:697-702. [DOI] [PubMed] [Google Scholar]
- 27.Sparks C, Guertin D. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 2010;29:3733-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nölting S, Grossman A. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol 2012;23:21-33. [DOI] [PubMed] [Google Scholar]
- 29.Svenja N, Edwin G, Ghassan A, et al. Combined blockade of signalling pathways shows marked anti-tumour potential in phaeochromocytoma cell lines. J Mol Endocrinol 2012;49:79-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Q, Djuricin G, Staren ED, et al. Tumor angiogenesis in pheochromocytomas and paragangliomas. Surgery 1996;120:938-42; discussion 942-3. [DOI] [PubMed] [Google Scholar]
- 31.Takekoshi K, Isobe K, Yashiro T, et al. Expression of vascular endothelial growth factor (VEGF) and its cognate receptors in human pheochromocytomas. Life Sci 2004;74:863-71. [DOI] [PubMed] [Google Scholar]
- 32.Zielke A, Middeke M, Hoffmann S, et al. VEGF-mediated angiogenesis of human pheochromocytomas is associated to malignancy and inhibited by anti-VEGF antibodies in experimental tumors. Surgery 2002;132:1056-63; discussion 1063. [DOI] [PubMed] [Google Scholar]
- 33.Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today 2007;20:853-59. [DOI] [PubMed] [Google Scholar]
- 34.Welsh S, Williams R, Kirkpatrick L, et al. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1. Mol Cancer Ther 2004;3:233-44. [PubMed] [Google Scholar]
- 35.Welsh S, Williams R, Birmingham A, et al. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther 2003;2:235-43. [PubMed] [Google Scholar]
- 36.Choi H, Song B, Gong Y, et al. Rapid degradation of hypoxiainducible factor-1alpha by KRH102053, a new activator of prolyl hydroxylase 2. Br J Pharmacol 2008;154:114-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santarpia L, Habra M, Jiménez C. Malignant pheochromocytomas and paragangliomas: molecular signaling pathways and emerging therapies. Horm Metab Res 2009;41:680-6. [DOI] [PubMed] [Google Scholar]
- 38.Burnichon N, Lepoutre-Lussey C, Laffaire J, et al. A novel TMEM127 mutation in a patient with familial bilateral pheochromocytoma. Eur J Endocrinol 2011;164:141-5. [DOI] [PubMed] [Google Scholar]
- 39.Tavangar S, Shojaee A, Moradi H, et al. Immunohistochemical expression of Ki67, c-erbB-2, and c-kit antigens in benign and malignant pheochromocytoma. Pathol Res Pract 2010;206:305-9. [DOI] [PubMed] [Google Scholar]
- 40.Yuan W, Wang W, Cui B, et al. Overexpression of ERBB-2 was more frequently detected in malignant than benign pheochromocytomas by multiplex ligation-dependent probe amplification and immunohistochemistry. Endocr Relat Cancer 2008;15:343-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med 1999;341:1565-71. [DOI] [PubMed] [Google Scholar]
- 42.Kulke M, Stuart K, Enzinger P, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 2006;24:401-6. [DOI] [PubMed] [Google Scholar]
- 43.Gross D, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer 2006;13:535-40. [DOI] [PubMed] [Google Scholar]
- 44.Yao J, Zhang J, Rashid A, et al. Clinical and in vitro studies of imatinib in advanced carcinoid tumors. Clin Cancer Res 2007;13:234-40. [DOI] [PubMed] [Google Scholar]
- 45.Robert J, Thomas E, Pharm D, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115-24. [DOI] [PubMed] [Google Scholar]
- 46.Murthy S, Pacak K, Loh Y. Carboxypeptidase E: elevated expression correlated with tumor growth and metastasis in pheochromocytomas and other cancers. Cell Mol Neurobiol 2010;30:1377-81. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Lee T, Murthy S, Cawley N, et al. An N-terminal truncated carboxypeptidase E splice isoform induces tumor growth and is a bio-marker for predicting future metastasis in human cancers. J Clin Invest 2011;121:880-92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Ahn J, Sanz-Moreno V, Marshall CJ. The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J Cell Sci 2012; 125:1814-26. [DOI] [PubMed] [Google Scholar]
- 49.Martiniova L, Perera S, Brouwers F, et al. Increased uptake of 123Imeta-iodobenzylguanidine, 81Ffluorodopamine, and 3Hnorepinephrine in mouse pheochromocytoma cells and tumors after treatment with the histone deacetylase inhibitors. Endocr Relat Cancer 2011;18:143-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martiniova L, Lu J, Chiang J, et al. Pharmacologic modulation of serine/threonine phosphorylation highly sensitizes PHEO in a MPC cell and mouse model to conventional chemotherapy. PLoS ONE 2011;6:4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pacak K, Sirova M, Giubellino A, et al. NF-kappaB inhibition significantly upregulates the norepinephrine transporter system, causes apoptosis in pheochromocytoma cell lines and prevents metastasis in an animal model. Int J Cancer 2012;131:2445-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neckers L, Kern A, Tsutsumi S. Hsp90 inhibitors disrupt mitochondrial homeostasis in cancer cells. Chem Biol 2007;14:1204-6. [DOI] [PubMed] [Google Scholar]
- 53.Mundschenk J, Unger N, Schulz S, et al. Somatostatin receptor subtypes in human pheochromocytoma: subcellular expression pattern and functional relevance for octreotide scintigraphy. J Clin Endocrinol Metab 2003;88:5150-7. [DOI] [PubMed] [Google Scholar]
- 54.Pasquali D, Rossi V, Conzo G, et al. Effects of somatostatin analog SOM230 on cell proliferation, apoptosis, and catecholamine levels in cultured pheochromocytoma cells. J Mol Endocrinol 2008;40:263-71. [DOI] [PubMed] [Google Scholar]
- 55.Li M, Kong ZM, Liu ZL. Antioxidant enzyme activities and lipid peroxidation induced by eicosapentaenoic acid (EPA) in PC12 cells. Cell Biol Toxicol 2006;22:331-7. [DOI] [PubMed] [Google Scholar]
- 56.Senzaki H, Iwamoto S, Ogura E, et al. Dietary effects of fatty acids on growth and metastasis of KPL-1 human breast cancer cells in vivo and in vitro. Anticancer Res 1998;18:1621-7. [PubMed] [Google Scholar]
- 57.Sordet O, Khan QA, Kohn KW, Pommier Y. Apoptosis induced by topoisomerase inhibitors. Current medicinal chemistry. Anticancer Agents 2003;3:271-90. [DOI] [PubMed] [Google Scholar]