

Abstract
The presentations of primary and metastatic cutaneous malignant melanoma (CMM) are very diverse. Evidence increasingly indicates that single CMM cells spread to distant sites quite early during cancer progression and are soon eliminated before they become clinically detectable. However bulky metastases which appear at a later stage might derive from some of these early neoplastic cells. It seems that local CMM single cell micro-metastases commonly predict sentinel lymph node involvement without overtly reflecting CMM progression to bulky visceral metastases. This study is intended to review the current understanding of the mechanisms underlying two CMM presentations. The first is the long interval, apparently disease-free, with persistent CMM dormancy, which may precede overt metastatic growth. Immunosurveillance may induce dormancy in single CMM cells disseminated in the body by blocking their proliferation cycle. The second is the so-called CMM smoldering phenomenon, which is marked by an alternate progression and regression of CMM locally with metastases that wax and wane for long periods of time over restricted skin areas. These very diverse patterns of CMM progression are likely to be ascribable to a number of biological factors, including the activation of CMM stem cells, and the combined phenotypic heterogeneity and variability in proliferative amplification in CMM cell clusters. Furthermore an adequate stimulation of CMM immune-surveillance and the induction of a specific stromal structure and vascular response are required. In this context, most early CMM tumors are in part controlled by lymphocyte-mediated responses before they become clinically detectable. However both the role of immune-surveillance and the mechanisms underlying both persistent and smoldering CMM dormancy remain unclear.
Key words: malignant melanoma, tumor cell proliferation, smoldering phenomenon, neoplastic dormancy, melanoma stem cell
Introduction
Cutaneous malignant melanoma (CMM) accounts for about 3% of all human cancers and causes about 80% of skin cancer deaths. In Western countries, the overall incidence of CMM is raising faster than any other cancer. The lifetime risk of developing invasive CMM is currently estimated at about 1/50. The process of malignant transformation, progression and metastasis of CMM is only partially understood. Furthermore tumor staging is particularly complex due to its multifaceted nature.1-4
The primary aims of this review of the metastatic process in CMM are as follows: i) to describe the basic aspects and implications of stem cell and amplification proliferation; ii) to highlight any relationships between CMM progression and the host; iii) to consider early tumor cell dissemination and CMM microsatellites; iv) to contrast CMM persistent and smoldering dormancies.
Cutaneous malignant melanoma metastases
Despite different definitions have been coined over the years, CMM tumors are commonly regarded as heterogeneous populations of neo-plastic melanocytes, which display a temporary unrestricted growth compared with ordinary cells.5 The progressive thickening of any primary CMM is accompanied by an increased expression of various proto-oncogenes which contrasts with a decreased expression of putative tumor suppressor genes.6,7 The transition from non-metastatic to metastatic gene expression occurs with a thickening of the CMM tumor and increased cell proliferation.7-10 A number of recent studies have attempted to identify the expression of specific genes associated with CMM progression and metastasis,6,7,11 but no consensus has been reached yet. Overall the CMM metastatic process results from a series of sequential steps. The transition in gene expression occurs at different tumor thicknesses depending on the different genes, thus suggesting that the transition zone is crucial in the development of the metastatic phenotype.
A clonal analysis of human CMM tumors has revealed their heterogeneity in terms of metastatic potential. In addition, growth in specific organ areas appears to be selective. Although some of these features show a stochastic pattern,8 the basic aspect of CMM metastases reflects a combination of survival and growth of selected neoplastic cell subpopulations. Each metastasis has presumably a clonal origin, and different metastases possibly originate from an amplification proliferation in sets of active cells. The outcome of metastases depends on the interactions of metastatic CMM cells with various host factors. Organspecific CMM metastases can be identified occasionally in a given location within the organ. These findings suggest that systemic physiologic signals, including age and gender,12-14 are potentially recognized by CMM cells presumably through mechanisms similar to those of their regular cell counterparts.
A fundamental prerequisite for the initial steps of CMM metastatic process is the ability of CMM cells to dissociate from primary or already metastatic CMM, and to breach sequential structural and functional barriers. The progression from any primary CMM to its metastases is subject to various factors which influence their survival and proliferation. CMM metastases predominantly involve a variety of body sites where other cancer metastases are commonly found, namely skin, lymph nodes and lungs. A natural selection of CMM cell phenotypes occurs probably during the CMM invasion. The same mechanism is probably present in any cluster of metastatic CMM cells in different body sites.15 It should also be noted that some local immune mechanisms detect and destroy selected metastatic cells, whereas other metastatic cells with specific attributes survive. Just as the thick primary CMM is likely to become polyclonal, like bulky metastases.2
Cutaneous malignant melanoma stem cells and amplification growth rate
A major biologic characteristic of human CMM is the significant influence of the primary tumor thickness on the prognosis.1 The thicker (according to Breslow thickness) a primary CMM, the worse the prognosis will be. In general, a primary CMM thinner than 1 mm is associated with a high cure rate, whereas thicker CMM is associated with a different progression and outcome. However, at similar thickness, different groups of CMM patients may be identified on the basis of the degree of neoplastic progression in the primary neoplasms.5 The group of slowly progressing CMMs shows primary neoplasms that remain local for months or years. The group of rapidly progressing CMMs includes neoplasms that develop much faster in the matter of weeks. The group of orphan CMM metastases is formed by primary neoplasms in complete regression having, however, released overt metastases.
In histopathological terms, the global CMM growth rate largely depends on a positive balance between the overall proliferative activity and the apoptosis of its neoplastic cells.16 The mitotic rate,1,10,17,18 the S-phase index,19 and the growth fraction represented by the Ki-67 index5,9,20-23 are different, yet they are somehow correlated kinetic parameters of each neoplastic tumor. Globally, a high proliferative activity in the primary CMM, irrespective of its thickness, predicts early neoplastic involvement of the regional lymph nodes, which can occasionally be even more widespread.2,9,20 CMM cells fail to form bulky metastases unless they host the genotype and the phenotype which allow them to intravasate, extravasate, permeate the basal membranes, and ultimately proliferate in foreign tissue sites.11,24 These biologic characteristics are expressed by specific subpopulations of metastatically competent CMM cells which are likely to be already present in the primary neoplasm.
The putative impact of CMM stem cells is recognized in primary and metastatic CMM.25-27 CMM stem cells are slow-cycling tumor cells that do not express differentiated tumor-associated antigens. In general, the genomic and antigenic patterns found in primary CMM are heterogeneous.28,29 Different gene products are commonly involved in the malignant transformation of melanocytes, including melanoma stem/progenitor cells. These effects occur during CMM initiation and progression to locally advanced and metastatic disease stages.6,30 The frequent deregulated gene products contain the oncogenic B-RafV600E and N-RasQ61R mutants, different receptor tyrosine kinases and developmental pathways, such as epidermal growth factor receptor, stem cell-like factory (SCF) receptor KIT, hedgehog, Wnt/ -catenin, Notch, stroma cell-derived factor-1 (SDF-1)/CXC chemokine receptor-4 (CXCR4) and vascular endothelial growth factor (VEGF)/VEGFR receptor. These growth factors and some epithelial-mesenchymal transitionassociated molecules cooperate and activate various tumorigenic downstream signaling pathways.6,7,31
Stem cells have the potential for almost unlimited self-renewal, which is a typical function in neoplastic growth. Although selected anticancer therapies can target most malignant cells, they remain potentially inefficient against chemo-resistant cancer stem cells that in turn are ultimately responsible for recurrence and progression. Interestingly enough, CMM stem cells exhibit an intrinsic sluggish proliferative rate. Thus, they commonly appear at rest for extended periods of time before initiating the amplification of a proliferative pool of CMM cells leading to overt metastases.5,9
CMM cells undergo a possible self-destruction through programmed apoptosis. The molecular agents responsible for apoptosis include the balance between apoptotic and anti-apoptotic regulators. The former regulators include p53, Bid, Noxa, PUMA, Bax, TNF, TRAIL, Fas/FasL, PITSLRE, interferons, and c-KIT/SCF. The latter regulators include Bcl-2, Bcl-XL, Mcl-1, nuclear factor-kappa B (NF- B), survivin, livin and ML IAP.16,31-33 Other molecules, such as TRAF-2, c-Myc, endothelins, and integrins, exhibit either pro-apoptotic or anti-apoptotic effects.
The balance between cell proliferation and apoptosis is a key aspect in the determination of the CMM tumorigenic potential.9,16,31,34 The proliferative activity of CMM is in part regulated by its tumor vascularity.16,35-37 Furthermore, the influence of the extravascular extracellular matrix on CMM progression should not be ruled out.38
The impact of immune-surveillance on neoplastic proliferation is still controversial. In early phases of CMM progression, tumors rarely cause harm and are often either ignored by the immune system or induce specific tolerance. Slowly-developing CMM possibly eludes the immune system using various mechanisms. For instance, some CMM cell clones potentially compromise multiple immune functions, including immune cell recognition of the neoplasm and immune cell-mediated killing of CMM cells. In the presence of CD8+ T cells, the number of Ki67+ tumor cells is low, but it increases when CD8+ T cells are absent. Therefore the tumor immune-surveillance probably controls the growth of primary CMM, and keeps metastatic outgrowth under control.
As the expression of Ki67 is associated with cell proliferation and cell cycle progression, it can be definitely argued that, besides its cytotoxic effects, a cytostatic tumor silencing mode may prevent the disseminated tumor cells from forming metastases.
Cutaneous malignant melanoma progression and the host
CMM releasing metastases exhibit some attributes in relation to the host.39 The early spread of single neoplastic cells occurs during the initial steps of tumor development,23,40,41 and is eventually followed by a latency period before metastatic outgrowth. It should be noted that the initial stage of CMM evolution does not commonly led to any effective and complete immunologic rejection from the host. A possible exception occurs when an incipient CMM is readily destroyed and possibly remains undisclosed.
CMM cell genotypes and phenotypes are generally heterogeneous in a given neoplasm, although they apparently look alike when observed in a standard histopathologic examination.5 During CMM progression, any neoplastic cell deprived of its initial ability to survive and escape the host defense would be inexorably destroyed. This process affects a number of circulating intravascular CMM cells.42,43 It is responsible for partial CMM regression which is so common in superficial CMM. This regression phase occurs either spontaneously or as a consequence of immune-surveillance.44
Striking evidence suggests that single dormant tumor cells start to disseminate to distant sites during the initial steps of tumor development immediately after the clinical onset of the primary tumor (Figure 1). They seem to be of prognostic relevance, and some of them are responsible for metastases which appear at a later stage. Patients with these dormant cancer cells live with an increased risk of sudden metastases, which may occur more than a decade after the primary tumor surgery. CD8+ T cells inhibit the growth of disseminated tumor cells not only due to cytotoxic effects, but also through cytostatic effects. The host defense against CMM metastases apparently has not the same efficacy in every tissue. Therefore, selective survival of CMM cells is possible in some specific organs. In addition, the host defense against CMM is clearly altered by immune-suppression.44 A specific control mechanism relies on the nature of the stroma where metastatic CMM cells are enclosed. Indeed, the stroma adjacent to the primary CMM exhibits a peculiar configuration38 that can favor or be even required for the survival and growth of the neoplastic cells. When these conditions are not met at the ultimate location of the metastatic cells, these cells probably fail to develop overt metastases.
Figure 1.
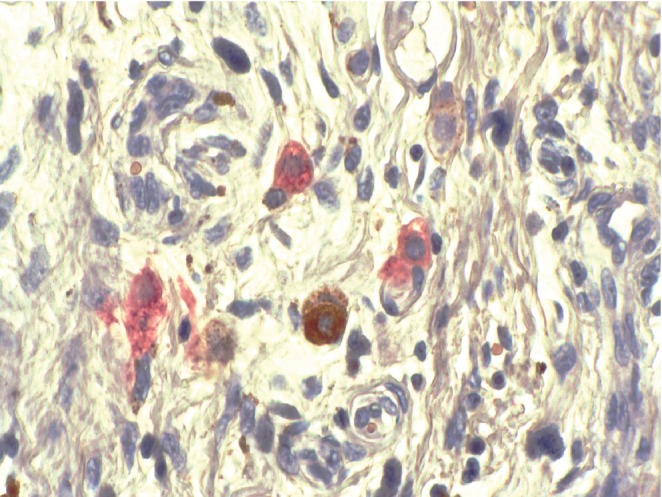
Single cell metastases corresponding to a growth-stunted malignant melanoma (CD63 immunohistochemistry).
Cutaneous malignant melanoma microsatellites
Some CMM melanocytes are keen to invade neighboring tissues, and metastasize to distant sites. Such a process is associated to a series of qualitative neoplastic deposits at the molecular and functional levels. The CMM neoplastic progression is characterized by increasingly uneven growth accompanied by a variety of cell abnormalities. In some cases, these events can be identified in specific areas inside the neoplasm. The combination of functional cell interactions through a crosstalk by soluble agents and close cell-cell contacts is probably active. The resulting CMM progression is related to a series of factors including: i) the renewal kinetics of CMM cells; ii) the antigenic characteristics of the molecular cell components; and iii) the release of growth factors acting on the peri-tumoral microenvironment, including its fibrovascular patterns. These events appear distinctly activated from the stage of neoplastic induction to full-blown bulky CMM metastases.
Each metastasis refers to a malignant cell cluster arising from a primary or metastatic malignancy, discontinuing any close contact with the sender neoplasm. In recent decades a significant progress has been made in the understanding of the mechanisms involved in CMM metastases. In particular, several migration paths for CMM micro-metastases were identified in the skin.24 In the early step of their evolution, most of the molecular traits of CMM micro-metastases are apparently already present in the primary tumor. Thin CMM tumors apparently lack competence for establishing overt metastases. Consequently, the required metastatic steps apparently depend on: i) the rate of cell renewal in the initial sender neoplasm; ii) the number of cells migrating outside the initial site; iii) the amount of malignant cells entering and surviving in both blood and lymph vascular pathways of neoplastic dissemination; iv) the density of cells coming from the vascular pathways; and v) the number of cells surviving and replicating at the terminal recipient metastatic site.
Any step in the metastatic pathway is ultimately related in part to the production rate of neoplastic cells in the primary CMM.9,19,20 As a whole, the induction of a local micro-metastatic process does not ineluctably lead to overt bulky metastases. Indeed, at the onset, micrometastases may be destroyed, or remain quiescent over a prolonged period of time, or occasionally grow in the presence of a positive proliferation-apoptosis-autophagy balance.45 Typically, the fate of metastatic CMM cells varies over time. Overt metastasis may develop even after a prolonged quiescence period44 or, on the contrary, enter a spontaneous regression phase.
Metastatic cells are found within some vessel lumina,19,46,47 embedded within the vessel walls,48 adjacent to the outer aspect of endothelial cells49 and scattered in the stroma.40 Flow cytometry used to analyze the DNA content in CMM metastases reveals the presence of a combination of diploid, tetraploid and various other polyploid cells.
Some CMM microsatellites are commonly followed by overt bulky locoregional metastases.50 Their presence represents a negative predictor for relapse-free survival, and they are reported to alter the overall survival.50 CMM microsatellites appear to be intimately tied to other specific markers of CMM severity. In this respect immunohistochemistry reveals small lesions down to discrete CMM micrometastases.2,3,40
By definition, cells at a distant metastatic site have full metastatic competence. Consequently, it seems likely that metastases from metastases represent a routine event in neoplastic biology. However, the role of the stroma hosting any CMM metastasis appears important, because it enables them to grow in a distant tissue. Indeed, the perimetastatic mesenchyme is significantly different from the regular stroma.
As the stroma at any distant site from the primary CMM is likely to be unprepared to host metastases, it requires a period of neoplastic adaptation prior to prominent neoplastic growth. Therefore metastases commonly remain restricted to a single organ or a few organs for a certain period of time. Such proliferation patterns probably reflect the presence of cells which may only grow in the mesenchyme of a given organ or tissue. Common restricted pathways include skin metastases.
In some instances, metastases appear in groups and grow simultaneously as if they were synchronized by a systemic control. In such instance, the sizes of the various metastases remain quite similar any time in their evolution. In other circumstances, metastases appear to grow independently of each other. In extreme cases, some grow, while others regress simultaneously. This pattern is defined CMM smoldering phenomenon.2,51
A potential absence of metastases could be explained by a subset of neoplastic cells which grow in the stroma at the primary site, but they are not capable of completing any step in a metastatic pathway, except for some motility and local micro-invasion. According to such hypothesis, invasiveness and metastatic development would not occur simultaneously, but as subsequent supplementary cell adaptations.
Cutaneous malignant melanoma persistent dormancy
Any unusual long latency period between the eradication of the primary CMM and the development of metastases corresponds to a clinically disease-free condition considered a CMM clinical dormancy.2,52 The relationship between such condition and the cause of CMM cell dormancy is complex and probably multifactorial.53 Dormant CMM cells live with an increased risk of unexpected late metastases, which may show up even after a decade from the primary tumor eradication. The process is not stable and commonly leads to relapse.
Tumor dormancy and autophagy are partially associated. Autophagy is a homeostatic and catabolic process that enables the sequestration and lysosomal degradation of cytoplasmic organelles and proteins.45 This process is important for the maintenance of genomic stability and cell survival in tissues. Autophagy represents a mechanism of stress tolerance maintaining partial cell viability and possibly leading to variable neoplastic dormancy, progression, and therapeutic resistance.54
Immune-mediated CMM dormancy may be involved in the absence of CMM cell killing. CMM dormancy appears related to IFN- and TNF-mediated signaling.52 In addition, T-cell immunity may also induce tumor dormancy. It involves a cytostatic mode of tumor silencing resulting from the suppression of both neoplastic cell proliferation and cell cycle progression. Therefore the suspension of tumor growth does not require discernable signs of CMM cell killing or apoptosis.
Any delay in the development of CMM metastases suggests a possible host defense mechanism, or a particular nature of non-actively proliferative CMM cells including CMM stem cells. CD4+ Th1 cells could induce a state of tumor dormancy by reducing tumor cell proliferation and cell cycle progression through strictly IFN- - and TNF-dependent signals.52 The suspension of tumor growth required neither detectable signs of tumor cell killing nor apoptosis. Therefore T-cell immunity can induce tumor dormancy with little or no signs of either tumor cell killing or apoptosis. These findings suggest that tumor immune-surveillance correlated with a substantial reduction in Ki67+ tumor cells, which is a key finding typical of the inhibition of cell proliferation and cell cycle progression. Tumor-specific T-cells prevent tumor growth in the absence of histological evidence of major tumor cell killing or apoptosis. Furthermore, both IFN and TNF are not only involved in killing and apoptosis, but they also exhibit pleiotropic effects on cell metabolism by controlling both the signaling cascades that regulate cell proliferation and cell cycle progression, and the signaling molecules that influence cell differentiation and transformation. Anti-angiogenic effects might also contribute to the dormancy phenomenon.52
Globally about 40% of CMM patients develop bulky metastases more than 5 years after the primary treatment.52 A typical example of persistent dormancy is provided by the appearance of metastases in the liver, a long time following the excision of uveal malignant melanoma (UMM).55 When UMM metastases develop, they nearly always appear first in the liver and often affect no other organ. The disease-free interval before the onset of such overt metastases may sometimes be as long as 25 years. The involvement of the liver early in the course of the disease remains unexplained. Also the reason for such a long interval from the primary UMM removal to the onset of metastases is still unclear. Since the eye has no lymphatic drainage, the neoplastic UMM cells must have already metastasized prior to the enucleation, and yet a long period of time commonly elapses before these metastases manifest themselves clinically in the liver. The growth rate of these overt UMM metastases may vary considerably, once they become clinically detectable. It is tempting to speculate that such metastases remain dormant for prolonged periods in response to some intrinsic control mechanisms of the neoplastic cells themselves, or a systemic control by the host.
Another example of persistent CMM dormancy concerns metastases developing from transplant organs in immunocompromised patients.44,56 Some silent micro-metastases that were present in the donor subject develop in an uncontrolled briskly way after transplantation of the invaded organ to an immunocompromised recipient. This unintentional transmission of CMM highlights the subtlety of tumor cell dissemination and the extent to which disseminated tumor cells remain dormant.
Cutaneous malignant melanoma smoldering dormancy
The relationship between the host and CMM cells is influenced by a large number of factors. These events occasionally result from a continuous fluctuating balance between two systems engaged in natural selection. They correspond to alternate fueling and silencing metastatic processes. Such changes in time of the combination of different cell processes involving CMM and its microenvironment lead to the smoldering CMM phenomenon.2
The concept of smoldering CMM was proposed to reflect the variable evolution of some CMMs with an established metastatic disease. The smoldering CMM phenomenon is a rare condition in which metastases appear and disappear on a small part of the body over a period of months or years.51 Smoldering metastases wax and wane in an apparently stochastic way, usually reaching at the most the size of a pea or a bean. The process appears to be difficult to control and differs from more common synchronized metastatic patterns. It is reported in particular after the excision of a primary CMM, when the metastatic disease remains concealed. Although common metastatic neoplasms may colonize a wide variety of tissues, the smoldering CMM phenomenon frequently and almost exclusively develops in the skin area extending between the primary CMM and the first group of drainage lymph nodes. Therefore, this phenomenon is controlled by some regional influences in the metastatic-colony formation. The metastatic process appears to be under great pressure.
The smoldering CMM highlights that metastatic progression is not inevitable.2 Indeed, the evolution of CMM metastasis early in tumor progression is characterized by a sort of balance between growth and regression. This is in part due to the prevalence of sequential apoptosis over proliferation, the multifaceted nature of antitumor immunity, and deficient stromal receptivity, including any angiogenesis defects. It should also be noted that the smoldering inflammation in the neoplasm microenvironment may promote the proliferation and survival of malignant cells, angiogenesis, metastasis, reversal of adaptive immunity, as well as a response to hormones and chemotherapeutic agents.52 Such a multifaceted process may occasionally lead to a programmed pathway of apoptosis and necrosis.
The smoldering CMM phenomenon probably involves a combination of distinct site-specific cell properties related to CMM cell growth, adhesion molecules, metastatic cell invasiveness, and regulation of metastatic cell growth and regression. Novel concepts regarding early seeding of metastases coupled with parallel progression, self-seeding of primary tumors by circulating neoplastic cells and the induction of premetastatic niches in distant sites by primary cancers still need to be thoroughly investigated.23
Highlights and perspectives
CMM development begins with the malignant transformation of regular human melanocytes. The development of CMM metastases may not derive from a single process, but it may be the result of many different factors related to the nature of the neoplastic cells and the reactivity of the host, which interfere in a complex pattern. CMM cells disseminate to distant sites early during the primary tumor development, but they do not cause a rapid onset of overt metastasis. They indeed rarely cause bulky metastases even after many years after they reached their distant site. In order to identify new therapeutic strategies, it is essential to understand in greater depth why these disseminated tumor cells fail to cause early metastasis and what are the events that cause tumor dormancy. More light remains to be shed on the ways in which the immune system controls single, disseminated tumor cells and micrometastasis, and the mechanisms that lead to tumor dormancy and awaken disseminated tumor cells making them grow into clinical metastases.
The gene expression profiling of primary, non-metastatic and metastatic CMM has led to identify a series of genes that are involved in the progression and the metastatic potential of CMM.7,57 Once CMM has metastasized, the overall patient survival drops. Smoldering CMM reflects the individual life of each metastasis confined to a restricted skin area. The lesions wax and wane in an apparently haphazard and uncoordinated way. Persistent CMM dormancy is a condition in which overt metastases appear after an extended period of time. It is possible to intervene with metastatic CMM stem cells or metastatic CMM cells with a blocked amplification cell cycle. The usual immunological concepts about CMM cell eradication by tumor cell killing should therefore be reviewed.
The two phenomena, i.e. smoldering process and persistent dormancy, might also occur at the primary site. The smoldering CMM phenomenon at the primary site could correspond to a fluctuating partial CMM regression, which is a frequent observation. The primary persistent CMM dormancy would correspond to a long period between the triggering of CMM and the early clinical stage of CMM development.
These two conditions should be further investigated, because the control of these processes might lead to new therapeutic progress. Also a clear understanding of the inhibition of cell proliferation and cell cycle progression may offer interesting results. In particular, tumor immune-surveillance and other mechanisms remain promising aspects which may lead to control tumor growth in the absence of histopathological evidence of major neoplastic cell killing or apoptosis.
Acknowledgements
the authors acknowledge Mrs. Ida Leclercq for her excellent support as assistant.
References
- 1.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27:6199-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piérard GE, Piérard-Franchimont C, Reginster MA, Quatresooz P. Smouldering malignant melanoma and metastatic dormancy: an update and review. Dermatol Res Pract 2012;2012:461–278.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu VH, Tetzlaff MT, Torres-Cabala CA, et al. Impact of the 2009 (7th edition) AJCC melanoma staging system in the classification of thin cutaneous melanomas. Biomed Res Int 2013;2013:898719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lian CG, Mihm MC, Piérard GE, Tommasino M. Skin cancer. Stewart BW, Wild CP, ed. World Cancer Report 2014. Geneva: World Health Organization Publ.; 2014. pp 495-502. [Google Scholar]
- 5.Piérard GE, Piérard-Franchimont C, Hermanns-Lê T, Delvenne P. Cutaneous malignant melanoma: one single disease? Rev Med Liege 2012;67:458-60. [PubMed] [Google Scholar]
- 6.Riker AI, Enkemann SA, Fodstad O, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics 2008;1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Peng Y, Tobin DJ. A new 12-gene diagnostic biomarker signature of melanoma revealed by integrated microarray analysis. Peer J 2013;1:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piérard GE, Piérard-Franchimont C. Stochastic relationship between the growth fraction and vascularity of thin malignant melanomas. Eur J Cancer 1997;33:1888-92. [DOI] [PubMed] [Google Scholar]
- 9.Piérard GE. Cell proliferation in cutaneous malignant melanoma: relationship with neoplastic progression. ISRN Dermatol 2012; 2012:828146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson JF, Soong SJ, Balch CM, et al. Prognostic significance of mitotic rate in localized primary cutaneous melanoma: an analysis of patients in the multi-institutional American Joint Committee on Cancer melanoma staging database. J Clin Oncol 2011;29:2199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van den Hurk K, Niessen HE, Veeck J, et al. Genetics and epigenetics of cutaneous malignant melanoma: a concert out of tune. Biochim Biophys Acta 2012;1826:89-102. [DOI] [PubMed] [Google Scholar]
- 12.Balch CM, Thompson JF, Gershenwald JE, et al. Age as a predictor of sentinel node metastasis among patients with localized melanoma: an inverse correlation of melanoma mortality and incidence of sentinel node metastasis among young and old patients. Ann Surg Oncol 2014;21:1075-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermanns-Lê T, Piérard S. Streamlining cutaneous melanomas in young women of the Belgian Mosan Region. Biomed Res Int 2014;2014:320767: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piérard GE, Hermanns-Lê T, Piérard S, et al. In vivo skin fluorescence imaging in young Caucasian adults with early malignant melanoma. Clin Cosmet Invest Dermatol 2014[In press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang E, Voiculescu S, Le Poole IC, et al. Clonal persistence and evolution during a decade of recurrent melanoma. J Invest Dermatol 2006;126:1372-7. [DOI] [PubMed] [Google Scholar]
- 16.McKenzie JA, Grossman D. Role of the apoptotic and mitotic regulator survivin in melanoma. Anticancer Res 2012;32:397-404. [PubMed] [Google Scholar]
- 17.Attis MG, Vollmer RT. Mitotic rate in melanoma: a reexamination. Am J Clin Pathol 2007;127:380-4. [DOI] [PubMed] [Google Scholar]
- 18.Vollmer RT. A probabilistic analysis of mitotic counts in melanoma. Am J Clin Pathol 2014;141:213-8. [DOI] [PubMed] [Google Scholar]
- 19.Piérard GE, Piérard-Franchimont C. The proliferative activity of cells of malignant melanomas. Am J Dermatopathol 1984;6:317-23. [PubMed] [Google Scholar]
- 20.Frahm SO, Schubert C, Parwaresch R, Rudolph P. High proliferative activity may predict early metastasis of thin melanomas. Hum Pathol 2001;32:1376-81. [DOI] [PubMed] [Google Scholar]
- 21.Plaza JA, Suster D, Perez-Montiel D. Expression of immunohisto-chemical markers in primary and metastatic malignant melanoma: a comparative study in 70 patients using a tissue microarray technique. Appl Immunohistochem Mol Morphol 2007;15:421-5. [DOI] [PubMed] [Google Scholar]
- 22.Muller-Hermelink N, Braumuller H, Pichler B, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008;13:507-18. [DOI] [PubMed] [Google Scholar]
- 23.Eyles J, Puaux AL, Wang X, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 2010;120:2030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sleeman JP, Nazarenko I, Thiele W. Do all roads lead to Rome? Routes to metastasis development. Int J Cancer 2011;128:2511-26. [DOI] [PubMed] [Google Scholar]
- 25.Klein WM, Wu BP, Zhao S, et al. Increased expression of stem cell markers in malignant melanoma. Mod Pathol 2007;20:102-7. [DOI] [PubMed] [Google Scholar]
- 26.Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res 2008;21:39-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shakhova O. Neural crest stem cells in melanoma development. Curr Opin Oncol 2014;26:215-21. [DOI] [PubMed] [Google Scholar]
- 28.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mimeault M, Batra SK. Novel biomarkers and therapeutic targets for optimizing the therapeutic management of melanomas. World J Clin Oncol 2012;3:32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussein MR, Haemel AK, Wood GS. Apoptosis and melanoma: molecular mechanisms. J Pathol 2003;199:275-88. [DOI] [PubMed] [Google Scholar]
- 32.Vucic D, Stennicke HR, Pisabarro MT, et al. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 2000;10:1359-66. [DOI] [PubMed] [Google Scholar]
- 33.Kasof GM, Gomes BC. Livin, a novel inhibitor of apoptosis protein family member. J Biol Chem 2001;276:3238-46. [DOI] [PubMed] [Google Scholar]
- 34.Ma S, Rubin BP. Apoptosis-associated tyrosine kinase 1 inhibits growth and migration and promotes apoptosis in melanoma. Lab Invest 2014;94:430-8. [DOI] [PubMed] [Google Scholar]
- 35.Piérard-Franchimont C, Henry F, Heymans O, Piérard GE. Vascular retardation in dormant growth-stunted malignant melanomas. Int J Mol Med 1999;4:403-6. [DOI] [PubMed] [Google Scholar]
- 36.Eelen G, Cruys B, Welti J, et al. Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab 2013;24:589-96. [DOI] [PubMed] [Google Scholar]
- 37.Quatresooz P, Piérard GE, Piérard-Franchimont C, et al. Spectral analysis of the microvasculature of primary cutaneous melanoma. Pathol Biol (Paris) 2012;60:149-53. [DOI] [PubMed] [Google Scholar]
- 38.Mikesh LM, Kumar M, Erdag G, et al. Evaluation of molecular markers of mesenchymal phenotype in melanoma. Melanoma Res 2010;20:485-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elder DE. Pathological staging of melanoma. Methods Mol Biol 2014;1102:325-51. [DOI] [PubMed] [Google Scholar]
- 40.Claessens N, Piérard GE, Piérard-Franchimont C, et al. Immunohistochemical detection of incipient melanoma micrometastases. Relationship with sentinel lymph node involvement. Melanoma Res 2005;15:107-10. [DOI] [PubMed] [Google Scholar]
- 41.Ulmer A, Fischer JR, Schanz S, et al. Detection of melanoma cells displaying multiple genomic changes in histopathologically negative sentinel lymph nodes. Clin Cancer Res 2005;11:5425-32. [DOI] [PubMed] [Google Scholar]
- 42.Juratli MA, Sarimollaoglu M, Nedosekin DA, et al. Dynamic fluctuation of circulating tumor cells during cancer progression. Cancers (Basel) 2014;6:128-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodic S, Mihalcioiu C, Saleh RR. Detection methods of circulating tumor cells in cutaneous melanoma: a systematic review. Crit Rev Oncol Hematol 2014;91:74-92. [DOI] [PubMed] [Google Scholar]
- 44.Strauss DC, Thomas JM. Transmission of donor melanoma by organ transplantation. Lancet Oncol 2010;11:790-6. [DOI] [PubMed] [Google Scholar]
- 45.Yang ZJ, Chee CE, Huang S, Sinicrope F. Autophagy modulation for cancer therapy. Cancer Biol Ther 2011;11:169-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mumford BS, Robertson GP. Circulating melanoma cells in the diagnosis and monitoring of melanoma: an appraisal of clinical potential. Mol Diagn Ther 2014;18:175-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu X, Gimotty PA, Guerry D, et al. Lymphatic invasion as a prognostic biomarker in primary cutaneous melanoma. Methods Mol Biol 2014;1102:275-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saluja A, Money N, Zivony DI, Solomon AR. Angiotropic malignant melanoma: a rare pattern of local metastases. J Am Acad Dermatol 2001;44:829-32. [DOI] [PubMed] [Google Scholar]
- 49.Lugassy C, Barnhill RL. Angiotropic melanoma and extravascular migratory metastasis: a review. Adv Anat Pathol 2007;14:195-201. [DOI] [PubMed] [Google Scholar]
- 50.Shaikh L, Sagebiel RW, Ferreira CM, et al. The role of microsatellites as a prognostic factor in primary malignant melanoma. Arch Dermatol 2005;141:739-42. [DOI] [PubMed] [Google Scholar]
- 51.Bodenham DC. A study of 650 observed malignant melanomas in the South-West region. Ann R Coll Surg Engl 1968;43:218-39. [PMC free article] [PubMed] [Google Scholar]
- 52.Rocken M. Early tumor dissemination, but late metastasis: insights into tumor dormancy. J Clin Invest 2010;120:1800-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev 2011;21:42-9. [DOI] [PubMed] [Google Scholar]
- 54.Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer 2010;10:871-7. [DOI] [PubMed] [Google Scholar]
- 55.Gaudi S, Messina JL. Molecular bases of cutaneous and uveal melanomas. Patholog Res Int 2011;2011:159421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med 2003;348:567-8. [DOI] [PubMed] [Google Scholar]
- 57.Soikkeli J, Lukk M, Nummela P, et al. Systematic search for the best gene expression markers for melanoma micrometastasis detection. J Pathol 2007;213:180-9. [DOI] [PubMed] [Google Scholar]