

Abstract
Epigenetic information regulates gene function and has important effects on development in eukaryotic organisms. DNA methylation, one such form of epigenetic information, has been implicated in the regulation of gene function in diverse metazoan taxa. In insects, DNA methylation has been shown to play a role in the regulation of gene expression and splicing. However, the functional basis for this role remains relatively poorly understood, and other epigenetic systems likely interact with DNA methylation to affect gene expression. We investigated associations between DNA methylation and histone modifications in the genome of the ant Camponotus floridanus in order to provide insight into how different epigenetic systems interact to affect gene function. We found that many histone modifications are strongly predictive of DNA methylation levels in genes, and that these epigenetic signals are more predictive of gene expression when considered together than when considered independently. We also found that peaks of DNA methylation are associated with the spatial organization of chromatin within active genes. Finally, we compared patterns of differential histone modification enrichment to patterns of differential DNA methylation to reveal that several histone modifications significantly covary with DNA methylation between C. floridanus phenotypes. As the first genomic comparison of DNA methylation to histone modifications within a single insect taxon, our investigation provides new insight into the regulatory significance of DNA methylation.
Keywords: DNA methylation, epigenetics, gene expression, gene regulation, histone modification, Camponotus floridanus
Introduction
Most organisms are capable of developing different phenotypes in response to distinct environmental conditions. The molecular information regulating such developmental plasticity is often heritable through cell divisions, yet is not directly encoded by the genome. Transmission of such information is known as epigenetic inheritance (Berger et al. 2009).
One of the most important forms of epigenetic information is the methylation of DNA. DNA methylation is present in all three domains of life (Klose and Bird 2006; Suzuki and Bird 2008; Glastad et al. 2011), and has been linked to variation in gene regulation in mammals (Maunakea et al. 2010; Shukla et al. 2011), plants (Ecker and Davis 1986; Zilberman et al. 2008; Zemach et al. 2010), and insects (Kucharski et al. 2008; Lyko et al. 2010; Li-Byarlay et al. 2013). In mammals, DNA methylation has traditionally been associated with gene repression, particularly when localized to promoter regions (Bird and Wolffe 1999; Weber et al. 2007; Suzuki and Bird 2008). However, in mammals, plants, and even insects, methylation of DNA within gene bodies (exons + introns) is associated with actively expressed genes (Lyko et al. 2010; Maunakea et al. 2010; Zemach et al. 2010; Glastad et al. 2011; Shukla et al. 2011). Notably, DNA methylation in insects is present at considerably lower levels than in plants or mammals, and is confined almost exclusively to gene bodies in holometabolous insects (Glastad et al. 2011; Hunt et al. 2013a). Despite this, DNA methylation has been linked to the regulation of alternative developmental outcomes in social insects (Kucharski et al. 2008), potentially through its association with alternative splicing (Lyko et al. 2010; Shukla et al. 2011; Flores et al. 2012; Herb et al. 2012; Li-Byarlay et al. 2013).
DNA methylation acts in concert with other types of epigenetic information. For example, histone protein posttranslational modifications (hPTMs) also affect gene regulation and organismal development. Like DNA methylation, hPTMs have been found to mediate the binding affinities of protein complexes, such as those related to transcriptional and splicing machinery (Kolasinska-Zwierz et al. 2009; Luco et al. 2010, 2011; Negre et al. 2011), as well as to control the local accessibility of chromatin (Henikoff 2008; Venkatesh et al. 2012; Zentner and Henikoff 2013).
Until recently, genomic profiles of DNA methylation and hPTMs were not both available for a single insect species, making it difficult to gain insight into the integration of DNA methylation in the greater chromatin landscape. Nevertheless, comparative epigenomic studies revealed that patterns of DNA methylation grossly mirror patterns of several hPTMs across insect orders (Nanty et al. 2011; Hunt et al. 2013b). These investigations suggest that DNA methylation acts in concert with hPTMs to affect gene regulation in insects, but the precise relationship between DNA methylation and hPTMs has yet to be explored. With the advent of genome-wide profiles of DNA methylation (Bonasio et al. 2012) and hPTMs (Simola et al. 2013) for distinct castes of the Florida carpenter ant Camponotus floridanus, it is now possible to investigate how these two important classes of epigenetic modifications relate to one another at a fine spatial scale. Here, we interrogate the relationship between hPTMs and DNA methylation genome-wide in C. floridanus in order to better understand DNA methylation and its epigenomic context.
We find that hPTMs are highly predictive of DNA methylation in C. floridanus. In particular, a strong spatial relationship exists between highly methylated regions (HMRs) and patterns of hPTM enrichment within actively expressed genes. This relationship is further supported by an observed association, as assessed between social insect phenotypes, between differential DNA methylation and differential hPTM enrichment. Overall, these findings expand our understanding of the function of gene body methylation and how it interacts with other epigenetic information, such as that encoded by modifications to histone proteins.
Materials and Methods
Analysis of DNA Methylation
DNA Methylation Level of Genomic Features
Genome-wide, processed DNA methylation data for C. floridanus were obtained from the Gene Expression Omnibus (GEO series: GSE31576, Bonasio et al. 2012) for males, minor works and major workers (castes with associated ChIP-sequencing [ChIP-seq] data). DNA methylation in animals is predominantly targeted to CpG dinucleotides (Yi and Goodisman 2009). Thus, fractional methylation levels were calculated as mCG/CG for each CpG, defined as the number of reads with methylated cytosines divided by the total number of reads mapped to the given CpG. False discovery rate (FDR)-corrected binomial P-values provided along with the CpG read data (Bonasio et al. 2012 supplementary files deposited in GEO series: GSE31576) were used to assign a status of “methylated” or “unmethylated” to each CpG (FDR < 0.01). Only CpG sites with ≥4 reads were considered in analyses. Fractional methylation was calculated for specific genomic features (e.g., exons, introns) as the mean fractional methylation value of all CpGs within that feature. A feature was called as methylated if at least three CpGs within the feature were called as methylated according to the binomial test.
Determination of Highly Methylated Regions (HMRs) of the Genome
We sought to detect HMRs of the genome, which we define as areas of high DNA methylation relative to much more lowly methylated regions directly up- and downstream of the HMR. HMRs were detected by identifying sharp transitions in DNA methylation levels using a sliding window method (length = 250, step = 50 bp), wherein focal window DNA methylation level was compared with all windows within 500 bp upstream (background). We determined that a focal window belonged to an HMR boundary if the focal window was greater than the background mean by a fractional DNA methylation level of at least 0.3, and if the difference between the focal window and the background mean exceeded 65% of the DNA methylation value of the focal window. Once established, an HMR boundary was extended to include all adjacent windows that exhibited a fractional methylation level greater than 50% of the level of the initial boundary window. This analysis was performed in both directions (5′ to 3′ and 3′ to 5′), and resulting HMR boundaries were connected to form contiguous regions of high methylation, provided all windows either 1) met the criteria for inclusion in both directional HMR boundaries or 2) possessed a fractional methylation level ≥ 50% of the mean of both boundaries. Unpaired HMR boundaries were themselves called as HMRs provided they did not fall within 500 bp of another HMR and possessed at least four methylated CpGs (according to the binomial test). Orientation was established by finding the closest gene (up to 2 kb) to a given HMR and assigning that HMR its strandedness (Glastad et al. 2011)—HMRs not falling within 2 kb of a gene were not assigned a strand.
HMRs in the genome were then compared with gene annotations (Cflo_OGSv3.3) and assigned a status of “exon,” “intron,” “5′-upstream,” or “NA” (not overlapping a genic future), as well as being called as “5′-proximal” (≤1,500 bp from start codon) or “non-5′-proximal” (any other genomic region).
Determination of Differentially Methylated Regions of the Genome between Castes
We identified differentially methylated regions (DMRs) of the genome between the male and worker castes by examining 200 bp windows (step = 100 bp; due to the very low number of DMRs [12] identified between minor and major worker castes, we only considered comparisons between males and workers). We modeled methylation levels for each genic feature as a function of two categorical variables: “caste” and “CpG position” using generalized linear models (GLMs) of the binomial family, implemented in the R statistical computing environment (R Development Core Team 2011). If caste contributed significantly (χ2 test of GLM terms, P-value < 0.01) to the methylation status of a window (after adjustment for multiple testing using the method of Benjamini and Hochberg [1995]), the window was considered differentially methylated between castes (Lyko et al. 2010). Only CpG sites that were significantly methylated (after multiple test correction) in one or both castes and covered by ≥4 reads in both libraries were used in these comparisons. Moreover, only features with ≥3 CpG sites were considered in these analyses. Once regions were assigned as DMRs, each DMR was then called as “elevated” in the caste with higher fractional methylation level. Overlapping windows of the same differential methylation status (Caste1 > Caste2, Caste2 > Caste1, or not differentially methylated) were then combined.
Analysis of Histone Modifications
ChIP-Seq Read Alignment and Signal Estimation
ChIP-seq data are the product of preferential enrichment of gDNA bound to a specific chromatin protein. For each hPTM, raw sequencing reads are processed followed by alignment to the reference genome of the organism in question. Once aligned, reads reflect quantitative levels of ChIP signal that can then be further normalized to a no antibody (input) control to produce a base-wise measure of the enrichment of ChIP signal reads over the control library—reflective of protein binding or prevalence (Park 2009).
We analyzed the prevalence of hPTMs H3K4me1, H3K4me3, H3K9ac, H3K9me3, H3K27ac, H3K27me3, H3K36me3, as well as the protein RNA polymerase (pol) II, in males, minor workers and major workers (Simola et al. 2013). After quality and adaptor trimming (trimmomatic: [Bolger et al. 2014]), raw sequencing reads (accession: SRX144014-SRX144044) were mapped to the C. floridanus genome (v3.0) with bowtie2 (Langmead et al. 2009) using the options “—sensitive -k 1 -N 0”. MACS2 (Zhang et al. 2008) was then used to estimate the read enrichment relative to an input control (as well as bulk histone H3 profiles for histone modifications to histone H3) for each ChIP library after removal of any duplicate reads using samtools (Li et al. 2009). Unless otherwise noted, all general comparisons between DNA methylation and hPTMs employed DNA methylation and hPTM enrichment averaged across all three castes.
Determination of Peaks of ChIP-Enrichment
Regions of significant ChIP signal enrichment (ChIP enrichment “peaks”) in the genome were established using MACS2 (FDR < 0.01), which identifies regions significantly enriched with a given ChIP signal relative to control libraries. Such peaks indicate regions that are likely to be strongly bound by a given chromatin protein. We considered a feature (e.g., exon, intron) to be significantly bound with a given protein if greater than 10% of its length was overlapped by a region of significant enrichment for that mark.
Determination of Regions of Differential ChIP Enrichment between Castes
Differentially bound regions (DBRs) were established using the program MAnorm (Shao et al. 2012), which uses common peaks between two libraries (as called by MACS2) to rescale and normalize ChIP data between two treatments, then estimate significance, direction and magnitude of differential ChIP enrichment for all confident ChIP enrichment peaks. Candidate DBRs with an FDR corrected P-value of < 0.01 were called as differentially enriched between castes, and the direction of differential binding enrichment was determined from the MAnorm-produced normalized between-comparison ChIP enrichment M-value (log2 ratio).
Analysis of Gene Expression
We determined levels of expression for given genes by analyzing RNA-seq data from the three castes which also have DNA methylation and ChIP-seq data (male, minor worker, major worker; Bonasio et al. 2010). Raw RNA-seq reads (GSM563074, GSM921123, and GSM921122) were filtered and aligned to the C. floridanus genome (v3.3; Bonasio et al. 2010) using Tophat (Trapnell et al. 2009), with the options “-r 50 –mate-std-dev 11(/20) -i 60 –no-discordant –read-realign-edit-dist 0 –coverage-search –b2-sensitive” specified. Cufflinks (Roberts et al. 2011) was run with multiread and fragment bias correction (“-u” and “-b” respectively), and upper quartile normalization was used. Assemblies across castes were merged using cuffmerge (“-s”). FPKM (fragments per kilobase of exon per million fragments mapped) produced by Cuffdiff was used to quantify expression levels at the level of the gene.
Combined Analysis of DNA Methylation, ChIP Analysis, and Gene Expression
We investigated if the patterns of DNA methylation were correlated with the presence of chromatin proteins in C. floridanus. In order to do so, we used measures of mean fractional DNA methylation level and average normalized ChIP enrichment for each coding sequence (CDS) to perform linear regressions and Spearman’s rank correlations between epigenetic marks with the JMP statistical software package (SAS Institute Inc.). For each hPTM we determined the correlation coefficients derived from its correlation with DNA methylation among all CpGs (allCpG), as well as among only those CpGs determined to have at least some significant DNA methylation (mCGs).
We next determined patterns of ChIP-seq enrichment relative to HMRs. ChIP-seq enrichment was calculated for each HMR, as well as for 0.5 kb regions up- and downstream of each HMR in order to identify relationships between levels of DNA methylation and the presence of hPTMs. For analyses of ChIP enrichment profiles relative to HMR boundaries, continuous ChIP enrichment signal was averaged at each base up to 1 kb up- and down-stream of HMR boundaries. Within HMRs, length-proportional bins were used to average between HMRs—allowing for differing HMR lengths.
We next investigated if there were relationships between DMRs and DBRs between C. floridanus castes. We first compared DMRs to DBRs genome-wide, in order to test whether DMRs are preferentially associated with DBRs. We tested for enrichment of DBRs among DMRs, relative to non-DMRs, using a Fisher’s exact test. We then tested if the directionality of a DMR showed any significant association with the direction of differential ChIP enrichment at that locus. For each caste pair we assigned each DMR and DBR the caste which showed the highest pairwise DNA methylation or ChIP enrichment levels, respectively, and then determined if hypermethylation in a specific caste was associated with consistent increases or decreases in that caste’s ChIP enrichment at the same locus.
Finally, we were interested in understanding if epigenetic factors, including hPTMs and DNA methylation, were jointly predictive of patterns of gene expression. In order to evaluate the contributions of DNA methylation to gene expression level, we performed multiple regression analyses between the epigenetic marks (methylation + hPTMs) and gene expression. We first performed regressions between gene expression and each mark independently. We then performed regression using all epigenetic marks in a multiple regression model. For single-term tests, each factor was regressed against gene expression (log2(FPKM+0.01)) and bias independently, then for the full test as a component of an additive model including all factors. This enabled a comparison of DNA methylation’s contribution to gene expression when controlling for hPTM enrichment and vice versa. All variables were standardized (0-centered after normalization) before model fitting.
Results and Discussion
DNA Methylation Is Strongly Associated with Active Histone Modifications
Recent studies in plants (Zilberman et al. 2008; Zemach et al. 2010; Coleman-Derr and Zilberman 2012) and animals (Ooi et al. 2007; Cedar and Bergman 2009; Shukla et al. 2011) have demonstrated that epigenetic information encoded by DNA methylation and hPTMs may interact to affect gene function. We thus sought to evaluate the relationships between DNA methylation and hPTM enrichment in the C. floridanus genome, and thereby improve our understanding of insect gene regulation.
Each hPTM we investigated was significantly over- or underrepresented among methylated genes (fig. 1 and supplementary table S1, Supplementary Material online). Consistent with previous comparative results (Nanty et al. 2011; Hunt et al. 2013b), the hPTMs that are generally most strongly associated with actively expressed genes (H3K4me3, H3K27ac, and H3K36me3; Kharchenko et al. 2011) were highly overrepresented among methylated genes. H3K4me3, H3K27ac, and H3K36me3 were present among over 79% of methylated genes, with 95% of methylated genes featuring at least one of these hPTMs (supplementary table S1, Supplementary Material online). Conversely, repressive hPTMs (H3K27me3 and H3K9me3; Kharchenko et al. 2011), which are generally associated with much less broadly expressed genes, were significantly and strongly underrepresented among methylated genes, with less than 2% of methylated genes significantly enriched for either modification (fig. 1 and supplementary table S1, Supplementary Material online).
Fig. 1.—

Histone modification enrichment as a function of DNA methylation levels for methylated (red) and unmethylated (blue) genes. Linear fits for all genes (black line) and methylated genes only (red line) are provided, along with their relevant R2 values and Spearman’s correlations, ρ. Bars represent the number of genes belonging to each class: those with significant histone posttranslational modification (hPTM) enrichment (+) and those without (−).
Similarly, when examining correlations between CDS DNA methylation levels and hPTM enrichment we found that the level of gene methylation was strongly positively associated with the quantitative level of ChIP enrichment for the active hPTMs H3K4me3, H3K27ac, H3K36me3, and H3K4me1, as well as for RNA polymerase II (RNA pol II) (mean ρ: 0.53; fig. 1 and supplementary table S2, Supplementary Material online). Conversely, the repressive hPTM H3K9me3 was strongly negatively correlated with CDS DNA methylation levels (ρ = −0.62; fig. 1 and supplementary table S2, Supplementary Material online). Thus, within insect genomes DNA methylation shows strong preferential targeting relative to most well-studied hPTMs, and is strongly biased to genes exhibiting active hPTMs. Consistent with this finding, hPTM levels explained 65% of the variance in CDS DNA methylation as inferred by the R2 value generated by multiple regression (supplementary fig. S1, Supplementary Material online).
We observed that many of the correlations between overall CDS methylation level and hPTM enrichment largely result from the fact that genes featuring any DNA methylation were also those most likely to exhibit significant regions of enrichment or depletion of hPTMs (i.e., binary associations; fig. 1). Consequently, when limiting our analysis to only genes displaying significant levels of DNA methylation, we found that many correlations between DNA methylation and hPTM enrichment were substantially weakened (fig. 1). hPTMs associated with actively expressed gene TSSs (namely, H3K4me3 and H3K27ac) and RNA pol II, however, maintained relatively strong relationships with DNA methylation level among significantly methylated genes (fig. 1). Interestingly, despite being considered an “activating” mark and being significantly colocalized to methylated genes, the hPTM H3K9ac exhibited a considerable negative correlation with DNA methylation in this methylation-limited analysis. This may be due to DNA methylation’s tendency to be most highly targeted to genes of intermediate expression, while H3K9ac is known to target very highly expressed genes. Moreover, a previous analysis found H3K9ac to be strongly preferentially targeted to high-CpG regions within promoters (supplementary fig. S9 of Simola et al. 2013), which are also the most consistently depleted of methylation.
Finally, though we observed strong relationships between DNA methylation and hPTMs at the gene level, we sought to evaluate the presence of direct spatial overlap between epigenetic marks within genes. We found that the observed relationships between DNA methylation and specific hPTMs remained largely intact when considering DNA methylation enrichment within regions of significant hPTM enrichment (supplementary fig. S2, Supplementary Material online) or within spatially restricted windows downstream of the TSS (supplementary fig. S3, Supplementary Material online).
Overall, active hPTMs seem to be highly predictive of genic DNA methylation levels. That is, active hPTMs are 1) targeted to the same loci as DNA methylation, 2) positively correlated with DNA methylation levels at these loci, and 3) spatially enriched for DNA methylation within hPTM-marked regions. The hPTM most consistently and strongly associated with DNA methylation in our analyses was H3K4me3 (fig. 1 and supplementary tables S1 and S2, Supplementary Material online).
DNA Methylation and Histone Modifications Bear Similar, but Nonredundant, Associations with Gene Expression
We next sought to evaluate how DNA methylation and hPTMs were related to patterns of gene expression in the broader context of the other epigenetic information studied here. We compared gene expression levels between genes possessing at least one region significantly enriched for a given histone modification and/or DNA methylation in order to evaluate the redundancy of DNA methylation to individual hPTMs in explaining gene expression levels. We found that, among genes possessing at least one region significantly enriched for a given histone modification, those with DNA methylation exhibited consistently higher expression levels and consistently lower expression bias than those with the same modifications but no DNA methylation (fig. 2 and supplementary fig. S4, Supplementary Material online).
Fig. 2.—

Methylated genes are more highly expressed, independent of hPTM status. Expression levels (log2(FPKM)) of genes associated (marked) or not associated (not marked) with particular histone modifications. Methylated genes exhibit consistently higher expression relative to unmethylated genes, regardless of their associations with particular histone posttranslational modifications.
We sought to further evaluate how epigenetic factors and their interactions related to gene expression in a combined framework using multiple regression analysis. We investigated if hPTMs and DNA methylation were predictive of gene expression level and gene expression bias among castes, as measured by RNA-seq. We first performed regressions between each epigenetic mark and gene expression separately. Not surprisingly, DNA methylation showed a significant positive association with gene expression when regressed singly (table 1). Moreover, when incorporated into a full regression involving all epigenetic marks, DNA methylation still contributed significantly to the modeling of gene expression. This indicates that, even after accounting for the contribution of hPTMs, DNA methylation remains independently associated with gene expression (table 1). Thus, though DNA methylation is highly correlated with active hPTMs, methylated genes were more highly and broadly expressed than unmethylated genes, even when controlling for hPTM status.
Table 1.
Regression Analysis for Estimating Gene Expression Level and Bias from Epigenetic Marks
| Gene Expression Level |
Gene Expression Bias |
|||||
|---|---|---|---|---|---|---|
| Effect | R2 (single term) | Coefficient (single term) | Coefficient (full model) | R2 (single term) | Coefficient (single term) | Coefficient (full model) |
| DNA-methylation | 0.279 | 1.875**** | 0.424**** | 0.165 | −0.401**** | −0.170**** |
| H3K4me3 | 0.273 | 1.869**** | −0.128** | 0.151 | −0.476**** | 0.162**** |
| H3K4me1 | 0.222 | 1.684**** | 0.238**** | 0.086 | −0.504**** | −0.067*** |
| H3K27me3 | 0.081 | 1.020**** | −0.537**** | 0.002 | 0.021**** | 0.161**** |
| H3K27ac | 0.343 | 2.096**** | 0.891**** | 0.207 | −0.567**** | −0.272**** |
| H3K36me3 | 0.344 | 2.097**** | 1.382**** | 0.205 | −0.618**** | −0.373**** |
| H3K9me3 | 0.307 | −1.983**** | −0.610**** | 0.279 | 0.723**** | 0.233**** |
| H3K9ac | 0.082 | 1.022**** | −0.119** | 0.084 | −0.390**** | −0.255**** |
| PolII | 0.124 | 0.558**** | −0.256**** | 1.22E-05 | −0.010*** | 0.164**** |
| R2 adj. (full model) | 0.5086 | 0.4126 | ||||
Note.—Coefficients for both single-term tests and full model are provided. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. N = 15,165.
Histone Modifications Are Strongly Spatially Organized Relative to Regions of DNA Methylation in Insect Genomes
Up to this point, we have described associations between DNA methylation and hPTMs as summarized at the level of genes. These analyses provide important insight into the coassociation of DNA methylation and hPTMs as it relates to patterns and levels of gene expression. However, such analyses are unable to provide insight into the precise localization of DNA methylation and hPTMs, let alone their interplay. Thus, we sought to evaluate levels and patterns of hPTM enrichment at a fine spatial scale relative to HMRs. This facilitates an evaluation of hPTM enrichment within the spatial context of DNA methylation, but independent of other genomic annotations (gene features, etc). To accomplish this aim, we first developed an algorithm to establish regions of high fractional DNA methylation bordered by regions of much lower DNA methylation (see Materials and Methods). This produced a set of 7,382 highly methylated regions, which were subsequently analyzed for hPTM enrichment.
HMRs represent loci displaying relatively high levels of methylation in the otherwise-sparsely methylated C. floridanus genome, with an average fractional methylation level of 0.63, and almost 70% of individual highly methylated CpGs (CpGs with >0.5 fractional DNA methylation) falling within an HMR. Despite this, HMRs were only an average of 650.3 bp (SD: 335.6 bp) long, and while over 85% of genes with significant DNA methylation featured at least one HMR (4,922/5,785 methylated genes), HMRs only covered about 33% of the area of these genes. Thus, even within methylated genes, regions of high methylation are often limited to only a portion of the gene, most frequently at the 5′-end of these genes (Bonasio et al. 2012; Hunt et al. 2013a). As expected, out of 7,382 HMRs, the great majority (6,927; 93.8%) were located within or near genes, and only 22/7,382 of such peaks did not fall within 2 kb of a gene annotation or RNA-seq-based cufflinks annotation (supplementary table S3, Supplementary Material online). Of these 22, only 14 showed no RNA-sequencing coverage from the samples analyzed here. Thus, the overwhelming majority of HMRs are associated with expressed genes.
Studies of hPTMs in C. floridanus and other insects have revealed that many hPTMs, particularly those associated with actively transcribed genes, exhibit a strong spatial organization relative to the TSS of genes (Kharchenko et al. 2011; Simola et al. 2013). TSSs and surrounding proximal regions of active genes are marked with highly accessible chromatin and enriched with the hPTM H3K4me3. In contrast, further-3′ regions of the same transcribed genes are marked with the hPTM H3K36me3, indicative of less-accessible regions of chromatin characterized by transcriptionally elongating RNA pol II (Bannister and Kouzarides 2011; Kharchenko et al. 2011). Recent investigations have revealed that DNA methylation in C. floridanus and other holometabolous insects is preferentially targeted to the 5′-region of genes, immediately downstream of the TSS (Bonasio et al. 2012; Hunt et al. 2013a). The common spatial organization of active hPTMs and DNA methylation relative to gene starts suggests a functional interdependence between DNA methylation and hPTMs within actively expressed insect genes.
Consistent with this idea, we found that HMRs exhibited significantly different levels of enrichment for most active hPTMs relative to regions directly up- and downstream of HMRs (fig. 3 and supplementary fig. S5, Supplementary Material online). More specifically, HMRs tend to lie between distinctive promoter- and gene body-associated hPTMs: TSS-associated active hPTMs, including H3K9ac, H3K4me3, H3K27ac, as well as RNA pol II, were enriched upstream of HMRs, while H3K36me3 was depleted upstream and enriched downstream of HMRs (fig. 3b and c and supplementary fig. S5, Supplementary Material online). For these active hPTMs, we also found that the level of HMR methylation correlated positively with quantitative levels of ChIP enrichment within or nearby HMRs (fig. 4), indicating a strong quantitative link between hPTM enrichment and DNA methylation at a local level. Notably, we found that active TSS-associated hPTMs were most strongly correlated with HMR methylation level directly upstream of the HMR and not within the HMR itself (fig. 4).
Fig. 3.—

Active histone modification enrichment significantly differs between HMRs and non-HMRs. (a) Example genome browser track showing stark spatial contrast between DNA methylation (HMRs) and promoter-proximal active chromatin (highlighted in red boxes). (b) Spatial relationship between DNA methylation and select histone posttranslational modifications or polymerases. (c) ChIP enrichment at HMRs separated by whether they fall within 1,200 bp of a gene start (“5′-proximal”) or not (“non-5′-proximal”). Significance values represent results of Kruskal–Wallis tests comparing HMRs and 500 bp regions in each direction of HMR boundaries. Repressive hPTMs showed little organization relative to HMRs, and were thus excluded.
Fig. 4.—
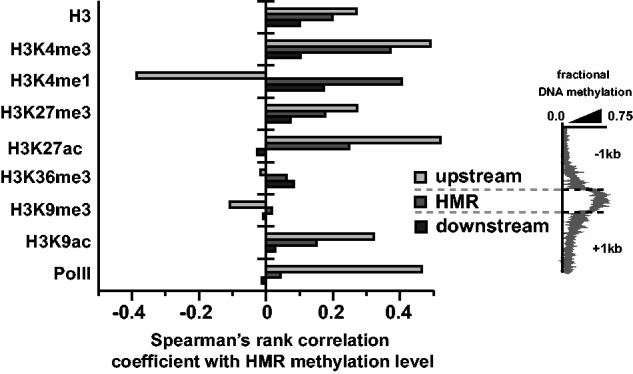
Distinct associations between DNA methylation and hPTMs upstream and downstream of HMRs. Spearman’s rank correlation coefficients between methylation level for HMRs and histone posttranslational modification enrichment within HMRs and 1 kb up- and downstream of HMRs.
The TSS-proximal boundary between H3K4me3 and H3K36me3 represents a boundary between two distinct, transcriptionally relevant chromatin states across the bodies of actively transcribed genes. These states are established (or maintained), at least in part, due to the fact that the histone methyltransferase responsible for establishing H3K4me3 binds preferentially to initiating RNA pol II associated with transcriptional start sites, while that responsible for H3K36me3 deposition binds the form of RNA pol II associated with transcriptional elongation (Bannister and Kouzarides 2011).
We found that RNA pol II exhibited significantly lower levels of enrichment at HMRs relative to up- and downstream regions, independent of the genomic context or length of the HMR (exon/intron, 5′-/3′-proximal localization; fig. 3b and c and supplementary fig. S6, Supplementary Material online), and was the only ChIP feature examined to exhibit considerable negative log-fold enrichment (indicative of depletion) at HMRs. This finding is particularly striking given that RNA pol II exhibits a signal of enrichment both directly up- and downstream of HMRs. It is possible this RNA pol II depletion at HMRs is related to an alteration of RNA pol II kinetics within or surrounding highly methylated DNA, a phenomenon observed in previous studies (Lorincz et al. 2004; Zilberman et al. 2007; Maunakea et al. 2013). Because of the strong tendency for H3K4me3 to be highly enriched upstream of HMRs, and H3K36me3 to be highly enriched downstream of HMRs, it is tempting to speculate that, through the alteration of RNA pol II dynamics, intragenic DNA methylation plays a role in the formation of a chromatin boundary that differentiates states of transcriptional initiation and elongation within actively expressed genes. Indeed, prior studies suggest that the conversion of TSS-proximal initiating RNA pol II into the elongating form plays an important role in the establishment of the distinct chromatin state associated with gene bodies (Brookes and Pombo 2009; Badeaux and Shi 2013). Thus, our finding that RNA pol II enrichment was lowest at HMRs relative to up- and downstream regions (fig. 3 and supplementary fig. S6, Supplementary Material online) suggests the possibility that the strong associations seen here between DNA methylation and hPTM enrichment may result from DNA methylation’s alteration of RNA pol II kinetics within and surrounding methylated DNA (Lorincz et al. 2004; Zilberman et al. 2007; Maunakea et al. 2013).
Of all chromatin marks we investigated, only H3K4me1 consistently showed its highest levels of enrichment within HMRs relative to up- and downstream regions (where it was consistently depleted; fig. 3c and supplementary fig. S5, Supplementary Material online). Interestingly, while positively correlated with HMR methylation level within the HMR, we found that H3K4me1 enrichment within 1 kb upstream of HMRs was negatively correlated with the level of HMR methylation (ρ: −0.39 vs. 0.37 for 1 kb upstream and within HMRs respectively; fig. 4). Thus, as the DNA methylation level of HMRs increases, the enrichment of H3K4me1 within those regions also increases; however, within the region directly upstream of HMRs, H3K4me1 is more depleted with increasing DNA methylation (fig. 4 and supplementary fig. S7, Supplementary Material online). At least one recent report has noted that, within active gene bodies, H3K4me1 is important to limiting domains of H3K4me3-marked open chromatin to promoter-proximal regions (Cheng et al. 2014). Indeed, H3K4me1 is often seen flanking TSS-proximal enriched regions of H3K4me3 within active gene bodies (Kharchenko et al. 2011).
It is possible that the patterning of hPTMs around HMRs is linked to H3K4me3 exclusion, either through DNA methylation informing or being targeted to this boundary. However, we are unable to determine whether DNA methylation plays a causal role in chromatin boundary formation in insects with the current data. Nevertheless, the fact that abrupt differences in RNA pol II, H3K4 methylation, and H3K36me3 exist within and around HMRs suggests that the hypothesis that DNA methylation may alter or maintain local chromatin states warrants testing in future investigations. Notably, both the patterning of hPTMs around active gene TSSs and the alternative splicing of exons involve differences in H3K4me1 and RNA pol II (Luco et al. 2010, 2011; Cheng et al. 2014; Stasevich et al. 2014), thus highlighting the possibility that the regulation of genic chromatin domains may help to explain DNA methylation’s link with alternative splicing in insects (Lyko et al. 2010; Bonasio et al. 2012; Herb et al. 2012).
Differential DNA Methylation Is Associated with Differential Histone Modification Enrichment
We next sought to examine whether regions exhibiting significant differences in levels of DNA methylation between C. floridanus castes also exhibited significant differences in hPTM enrichment. Thus, we compared DMRs to a set of regions exhibiting significantly different hPTM enrichment (differentially bound regions: DBRs) between males and female workers.
We found that DMRs were significantly enriched for several DBRs (hPTMs H3K27ac, H3K4me1, H3K4me3, and H3K9me3) relative to methylated regions not displaying significant differences between males and workers (table 2). Thus, even at the coarse resolution provided by whole body samples, DMRs exhibit significantly more DBRs than non-DMR genes.
Table 2.
Association Tests between a Genomic Region’s Differential Methylation Status and Differential ChIP Enrichment, as Assessed between Castes
| hPTM | Methylation Status | Non-DBR | DBR | Fold Enrichment | P-Value |
|---|---|---|---|---|---|
| H3K27ac | Non-DMR | 5,559 | 1,754 | 1.10 | 0.0002 |
| DMR | 2,754 | 980 | |||
| H3K27me3 | Non-DMR | 10 | 82 | −1.23 | 0.0184 |
| DMR | 11 | 29 | |||
| H3K36me3 | Non-DMR | 1,878 | 2,782 | −1.02 | NS |
| DMR | 1,148 | 1,607 | |||
| H3K4me1 | Non-DMR | 1,158 | 480 | 1.24 | 0.0006 |
| DMR | 466 | 267 | |||
| H3K4me3 | Non-DMR | 4,640 | 3,502 | 1.39 | <0.0001 |
| DMR | 1,950 | 2,912 | |||
| H3K9ac | Non-DMR | 4,460 | 857 | 1.00 | NS |
| DMR | 3,349 | 641 | |||
| H3K9me3 | Non-DMR | 166 | 104 | 1.20 | 0.0386 |
| DMR | 172 | 147 | |||
| RNA Pol II | Non-DMR | 1,266 | 426 | −1.02 | NS |
| DMR | 647 | 213 |
Note.—The numbers of genomic regions falling into each pairwise category for the different hPTMs are provided along with fold enrichment of DMRs coinciding with DBRs relative to regions not differentially associated by either epigenetic signal (negative fold enrichment represents hPTM for which DMRs are underrepresented among DBRs). P-values derived from a Fisher’s exact test (P < 0.05 in bold).
Moreover, we found that DNA methylation biased to either males or workers was significantly associated with hPTM enrichment in the opposite phenotype for H3K4me3, and RNA pol II (fig. 5 and supplementary table S5, Supplementary Material online). This is again consistent with a hypothesized functional link between DNA methylation and the patterning of genic chromatin, wherein DNA methylation exhibits spatial antagonism with RNA pol II and H3K4me3.
Fig. 5.—
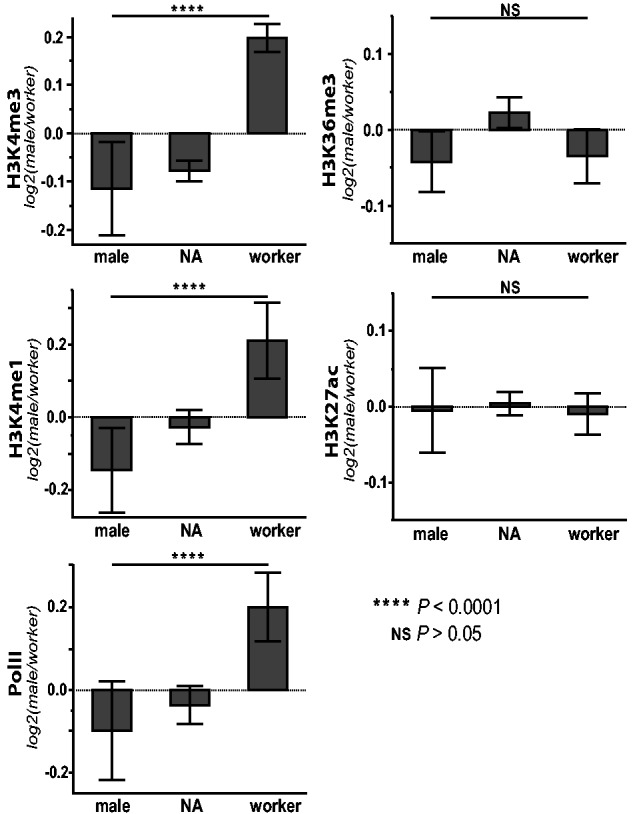
DMRs show significantly different, directional hPTM enrichment between male and worker phenotypes. Active histone posttranslational modification (hPTM; and RNA pol II) log2 fold differences between males andworkers as they relate to regions of significant, directional differential methylation (positive values on y axes indicate male-biased ChIP enrichment, whereas negative values indicate worker bias; x axes: “male,” male hypermethylated; “worker,” worker hypermethylated; NA, not differentially methylated between phenotypes).
In Arabidopsis thaliana (Zilberman et al. 2008; Coleman-Derr and Zilberman 2012), and likely vertebrates (Zemach et al. 2010), DNA methylation is known to play a role in altering chromatin within and directly surrounding methylated regions. Specifically, methylation acts as a boundary to H2A.Z, an important TSS-associated histone variant that is linked to chromatin activation (Zilberman et al. 2008; Zemach et al. 2010; Coleman-Derr and Zilberman 2012). Because H2A.Z is a highly conserved component of the epigenome of active genes, and has been shown to strongly correlate with DNA methylation and promoter-proximal active gene hPTMs (Zilberman et al. 2008), it is possible many of our observations are reflective of the conserved mechanism of H2A.Z exclusion by DNA methylation operating in insects. However, because this histone variant was not tested directly in our study, additional research will be required to test this hypothesis.
Conclusion
Our results provide several important insights into insect DNA methylation. By assessing, for the first time, the relationship between DNA methylation and hPTMs within a single insect taxon, we provide a foundation for understanding the greater epigenome in insects. In particular, our results suggest that the function of intragenic DNA methylation is linked to the function of key, active histone modifications, with over 90% of methylated genes also featuring the hPTMs H3K4me3 or H3K36me3. As additional support to this claim, we provide evidence that DNA methylation and active hPTM enrichment covary between distinct phenotypes in C. floridanus, suggesting that changes to DNA methylation are coupled with changes in chromatin modifications. Despite the striking concordance between DNA methylation and hPTMs, however, our results suggest the function of DNA methylation is not entirely redundant to hPTMs—DNA methylation retains explanatory power for gene expression levels when controlling for numerous hPTMs.
Studies in plants and animals have shown that variation in gene body DNA methylation affects gene regulation by altering local chromatin and the rate of elongation of RNA pol II (Zilberman et al. 2007; Maunakea et al. 2013). Likewise, our findings are consistent with a functional link between DNA methylation and the organization of chromatin. Our spatial analysis of DNA methylation and hPTMs reveals a strong patterning of multiple, functionally distinct hPTMs and RNA pol II relative to methylated regions. Most notably, RNA pol II is depleted, and H3K4me1 enriched, within HMRs. We hypothesize that intragenic DNA methylation contributes to changes in chromatin and chromatin boundaries within active insect genes, particularly those that differentiate states of transcriptional initiation and elongation, occurring near the transcription start site. This hypothesis may help to explain why DNA methylation is preferentially targeted to 5′-regions of genes in most investigated insects (Bonasio et al. 2012; Hunt et al. 2013a). Furthermore, as both alternative splicing and TSS-proximal chromatin organization have been linked to the dynamics of RNA pol II and H3K4me1 (among other hPTMs) (Luco et al. 2010, 2011; Cheng et al. 2014), it is possible that the previously observed link between DNA methylation and alternative splicing in insects (Lyko et al. 2010; Bonasio et al. 2012; Herb et al. 2012) is influenced by hPTMs.
As we look to the future, it is clear that studies seeking to establish the epigenetic basis for developmental regulation in insects, as with environmental caste determination (Kucharski et al. 2008), will benefit from investigating both DNA methylation and hPTMs. In doing so, a meaningful exploration of the causal links between epigenetic modifications, chromatin boundary formation, gene regulation, and developmental fate will require extensive advancement of reverse genetic approaches to the perturbation of enzymatic mediators of epigenetic modifications in previously nonmodel insects.
Supplementary Material
Supplementary tables S1–S5 and figures S1–S7 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the U.S. National Science Foundation (DEB-0640690) and the Georgia Tech-Elizabeth Smithgall Watts endowment.
Literature Cited
- Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14:211–224. doi: 10.1038/nrm3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329:1068–1071. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, et al. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr Biol. 2012;22:1755–1764. doi: 10.1016/j.cub.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, Pombo A. Modifications of RNA polymerase II are pivotal in regulating gene expression states. EMBO Rep. 2009;10:1213–1219. doi: 10.1038/embor.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Cheng J, et al. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell. 2014;53:979–992. doi: 10.1016/j.molcel.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman-Derr D, Zilberman D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 2012;8:e1002988. doi: 10.1371/journal.pgen.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker JR, Davis RW. Inhibition of gene expression in plant cells by expression of antisense RNA. Proc Natl Acad Sci U S A. 1986;83:5372–5376. doi: 10.1073/pnas.83.15.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores K, et al. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genomics. 2012;13:480. doi: 10.1186/1471-2164-13-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastad KM, Hunt BG, Yi SV, Goodisman MAD. DNA methylation in insects: on the brink of the epigenomic era. Insect Mol Biol. 2011;20:553–565. doi: 10.1111/j.1365-2583.2011.01092.x. [DOI] [PubMed] [Google Scholar]
- Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- Herb BR, et al. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat Neurosci. 2012;15:1371–1373. doi: 10.1038/nn.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Glastad KM, Yi SV, Goodisman MAD. The function of intragenic DNA methylation: insights from insect epigenomes. Integr Comp Biol. 2013a;53:319–328. doi: 10.1093/icb/ict003. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Glastad KM, Yi SV, Goodisman MAD. Patterning and regulatory associations of DNA methylation are mirrored by histone modifications in insects. Genome Biol Evol. 2013b;5:591–598. doi: 10.1093/gbe/evt030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, et al. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharski R, Maleszka J, Foret S, Maleszka R. Nutritional control of reproductive status in honeybees via DNA methylation. Science. 2008;319:1827–1830. doi: 10.1126/science.1153069. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Byarlay H, et al. RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc Natl Acad Sci U S A. 2013;110:12750–12755. doi: 10.1073/pnas.1310735110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative Pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, et al. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8:e1000506. doi: 10.1371/journal.pbio.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23:1256–1269. doi: 10.1038/cr.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanty L, et al. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 2011;21:1841–1850. doi: 10.1101/gr.121640.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SKT, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan G-C, Orkin S, Waxman D. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012;13:1–17. doi: 10.1186/gb-2012-13-3-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simola DF, et al. A chromatin link to caste identity in the carpenter ant Camponotus floridanus. Genome Res. 2013;23:486–496. doi: 10.1101/gr.148361.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasevich TJ, et al. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature. 2014;516:272–275. doi: 10.1038/nature13714. [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, et al. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455. doi: 10.1038/nature11326. [DOI] [PubMed] [Google Scholar]
- Weber M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- Yi SV, Goodisman MAD. Computational approaches for understanding the evolution of DNA methylation in animals. Epigenetics. 2009;4:551–556. doi: 10.4161/epi.4.8.10345. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456:125–129. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.