

Abstract
Repetitive DNA, including transposable elements (TEs), is found throughout eukaryotic genomes. Annotating and assembling the “repeatome” during genome-wide analysis often poses a challenge. To address this problem, we present dnaPipeTE—a new bioinformatics pipeline that uses a sample of raw genomic reads. It produces precise estimates of repeated DNA content and TE consensus sequences, as well as the relative ages of TE families. We shows that dnaPipeTE performs well using very low coverage sequencing in different genomes, losing accuracy only with old TE families. We applied this pipeline to the genome of the Asian tiger mosquito Aedes albopictus, an invasive species of human health interest, for which the genome size is estimated to be over 1 Gbp. Using dnaPipeTE, we showed that this species harbors a large (50% of the genome) and potentially active repeatome with an overall TE class and order composition similar to that of Aedes aegypti, the yellow fever mosquito. However, intraorder dynamics show clear distinctions between the two species, with differences at the TE family level. Our pipeline’s ability to manage the repeatome annotation problem will make it helpful for new or ongoing assembly projects, and our results will benefit future genomic studies of A. albopictus.
Keywords: transposable elements, repeated DNA, TE analysis, Aedes albopictus, Trinity, bioinformatic pipeline
Introduction
Repeated DNA, including transposable elements (TEs), is widespread within eukaryotic genomes. In such a “repeatome,” the spread of TEs, which might bear coding sequences and can reach thousands of base pairs in length, contributes substantially to genomic size and evolution. Because of their ability to insert within genes or regulatory regions and to cause ectopic recombination due to their repetitive nature, TEs are assumed to be frequently deleterious to their hosts (Goodier and Kazazian 2008; Beck et al. 2011; Vela et al. 2014). However, an increasing number of studies have shown that TE insertions can sometimes be adaptive and can be co-opted by their host genomes (Rebollo et al. 2010; Casacuberta and González 2013). Thus, understanding genomic evolution demands a comprehensive knowledge of TE composition within the genome, as well as of their dynamics and interactions with host genome. To this end, genome annotations that include TE annotation and quantification are crucial.
In the current era of short-read sequencing, the assembly of genomes bearing a significant amount of repeated sequence is a complex task. Reads overlapping a repeated element might correspond to several positions in the genome and thus can be misplaced and can produce chimeric assembly. Therefore, repeats produce a large number of short contigs that cannot be properly positioned or annotated within the assembly. Accordingly, the quality of the assembly for TEs is often poor and can result in underrepresented and/or incorrect annotation of their sequences (Modolo and Lerat 2014).
The Asian tiger mosquito Aedes albopictus (Diptera: Culicidae) presents a striking example of a genome that is difficult to assemble due to its repeatome. This species—a vector of Dengue and Chikungunya viruses that is often viewed as one of the most threatening invasive species in the world—still has not had its genome sequence released, even though several projects have been aimed at this task over the last few years (see Bonizzoni et al. 2013 for a review). Aedes aegypti, the closest species whose genome has been fully sequenced and annotated, possesses a similar genome size, and repeated DNA comprises more than 50% of its genome. Unlike A. albopictus, the whole genome of A. aegypti has been fully sequenced using Sanger technology, which produces longer reads than current Next-Generation Sequencing (NGS) methods and therefore allowed the construction of a large library of TEs and repeats (Nene et al. 2007). Moreover, intraspecies variation of the A. albopictus genome size—ranging from 0.62 to 1.66 pg—has been suggested (Rao and Rai 1987; Kumar and Rai 1990), supporting the hypothesis of a significant amount of TE activity, with more copies present in some populations than in others (McLain et al. 1987; Black et al. 1988). However, no study is currently aimed at finding and quantifying TEs in a comprehensive manner in this species.
Several bioinformatic solutions now enable the de novo assembly of TE sequences directly from NGS genomic data sets without the need for a reference genome. These methods assume that reads belonging to TEs or other repetitive DNAs are overrepresented among the sequenced reads. Current pipelines such as RepARK (Koch et al. 2014) and TEdna (Zytnicki et al. 2014) use whole NGS genomic data sets or only the unassembled reads left after a genome assembly. These two programs use overrepresented k-mers to assemble TE sequences: Velvet (Zerbino and Birney 2008) or CLC (CLCbio, http://www.clcbio.com/products/clc-assembly-cell/, last accessed April 13, 2015) are used in RepARK, and an implementation of a de Bruijn graph assembler is used in TEdna. Although these programs are dedicated to TE assembly, they do not allow repeat quantification or annotation. An alternative way to explore a genome’s repetitive content is to use low coverage sequencing. In such data sets, only TEs and other repetitive DNA sequences are expected to have a sufficient representation in the pool of reads to be assembled. For example, in average, for a sample with 0.1× coverage, only sequences that are present at least 10 times within the genome can be assembled. Based on this principle, the RepeatExplorer (RE) pipeline (Novák et al. 2010) was designed to cluster and then assemble similar reads from a small uniform genomic sample in order to retrieve repeats. In a uniform genomic sample, the proportion of reads assigned to a given cluster directly corresponds to the proportion of reads assigned to the relevant TE family in the genome. In addition to computing a direct quantification of each repeat family, RE can annotate repeat families using RepeatMasker (RM) and protein domain search (Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. 1996–2010, http://www.repeatmasker.org, last accessed April 13, 2015). However, although the RE pipeline can process NGS data sets, most of the tools it uses are not designed for this type of data, especially during the assembly step performed by CAP3 (Huang 1999)—a Bacterial Artificial Chromosome (BAC)-clone sequence type assembler.
Here, we present a new pipeline, dnaPipeTE (De Novo Assembly and Annotation Pipeline for Transposable Elements), that combines previous methods by allowing fast and accurate assembly of repeat sequences from a small genomic sample with dedicated NGS tools and by performing quantification and annotation of TEs and repeats for comparative analysis. The cornerstone of dnaPipeTE is the use of Trinity (Grabherr et al. 2011)—originally designed for RNAseq data assembly—to assemble repeats from low-coverage genomic data sets, which produce complete repeat sequences and enable the recovery of alternative consensuses within one TE family. Our pipeline also performs an automatic annotation of repeats using RM and the Repbase database (Jurka et al. 2005) and produces different data and figures for the quantification of repeats. We also implemented a computation of the TE age distribution for the most recent copies, using the divergence between reads and contigs.
With this pipeline and annotations from known TEs, we aimed to 1) estimate the number of repeated DNAs in A. albopictus, 2) annotate and quantify the diversity of TEs in its genome, and 3) compare this repeatome with that of A. aegypti, to infer the dynamics of TEs since the divergence of these two species.
Materials and Methods
dnaPipeTE: A Pipeline to Assemble, Annotate, and Quantify Repetitive Sequences from Small Unassembled NGS Data Sets
dnaPipeTE is a fully automated pipeline designed to assemble and quantify repeats from genomic NGS reads. It is freely available for download at https://lbbe.univ-lyon1.fr/-dnaPipeTE-.html (under the GPLv3). Figure 1 shows the main steps in the dnaPipeTE pipeline. Our pipeline takes as input a FASTQ (Cock et al. 2010) file containing quality filtered short reads. dnaPipeTE then performs uniform samplings of the reads to produce low coverage data sets used during analysis. The samples must represent less than 1× coverage to avoid the assembly of nonrepeated genome content; using a sample size of less than 0.25× of the genome is often sufficient to obtain a precise estimate of the repeated content (see supplementary fig. S1, Supplementary Material online, for examples with 0.1× and 0.25×). dnaPipeTE requires at least three samples of the original genomic data set: Two for the assembly step and an independent third used for the quantification steps. Our pipeline is currently designed to use only single-end reads because training analyses showed that using paired-end reads could produce chimeras during repeat assembly (data not shown). We developed dnaPipeTE using 100-bp reads, which are currently the most frequently generated NGS data sets, but our implementation would work with any read size.
Fig. 1.—

Overview of the dnaPipeTE pipeline. First, genomic reads in FASTQ format are sampled. Then, assembly of repeats is performed using two or more iterations of Trinity. For each iteration, the previously assembled reads are added to the next sample to improve the repeat assembly. In the next step, assembled contigs are annotated using RepeatMasker. Finally, reads from the “BLAST sample” are blasted against all the contigs to estimate the relative abundance of each assembled repeat and to compute the TE landscape. In a second BLAST, the same sample is successively blasted against the annotated contigs joined to the Repbase library, then with the unannotated contigs in order to retrieve copies that would not have been assembled and to obtain a more global repeat content estimation. See text for additional details.
Repeat Assembly with Trinity
After uniform sampling of the reads, dnaPipeTE builds contigs from the repeated sequences using Trinity. In an RNAseq experiment, a given gene can produce different transcripts, and the Trinity software is equipped to handle alternative transcripts with a hierarchical procedure: after identifying a “gene” (a subpart of the assembly graph), Trinity can produce different contigs that represent all the alternative transcripts of this gene. Similarly, TE copies from the same family, which may display an accumulation of mutations, deletions, insertions, or other structural changes, are treated by Trinity as alternative sequences of the same gene (TE family). Thus, with Trinity one can recover complete alternative consensus sequences from a given TE family. Retrieving good consensus increases the ability to perform an accurate estimation of TE abundance by improving read mapping to TEs. The rarest elements in the genome are predicted to generate few (or no) reads in the subset samples; thus, dnaPipeTE performs iterative runs of Trinity using new samples to decrease such risk. The first run uses a first sample; then, any reads mapping to k-mer contigs belonging to repeats (“inchworm” contigs; see Trinity manual) are added to a second independent sample, and Trinity is performed one more time. Each iteration enriches the number of reads associated with a repeat in the next sample and allows the recovery of more and larger contigs (some examples are given in supplementary Material, Supplementary Material online). In the case of A. albopictus sequences, our tuning experiments showed that two iterations performed on a data set with 0.1× coverage ensured the best assembly N50 and that supplementary iteration showed no significant improvement in the quality of the assembly (supplementary fig. S2, Supplementary Material online). In the latest versions of Trinity (≥r20140717), contigs are built from “clusters” that correspond to units of the de Bruijn graph made during the assembly. These clusters are divided into genes and finally “isoforms” that represent the alternative transcripts of a gene in RNAseq studies. Applied to low-coverage DNA data, one gene ideally represents one repeat family, in which isoforms are structural variant copies belonging to one family (copies with insertions or deletions for example) or to closely related families. An isoform present in Trinity.fasta output following all iterations of the Trinity program is referred to as a “dnaPipeTE contig.” During the assembly step in dnaPipeTE, Trinity (version r20140717) was used with default parameters for single-end reads, with the exception of the minimum coverage to join k-mer contigs set to 1 to retain contigs from low copy repeats (Haas B, personal communication).
Contig Annotation with RepeatMasker
After the assembly step, dnaPipeTE contigs are annotated using RM, for which a built-in or custom repeat library can be specified. Following the 80-80-80 rule proposed by Wicker et al. (2007), contigs with 80% query coverage on 80% of subjects (databases) were stored as “full-length,” and queries with 80% hits on fewer than 80% of subjects were stored as “partial” (fig. 2). Of the other contigs annotated by RM, only the order information (according to Wicker et al. 2007 classification)— Long Terminal Repeat (LTR), Long INterspersed Element (LINE), Short INterspersed Element (SINE), DNA, Miniature Inverted-repeat Transposable Elements (MITEs) (short TEs harboring terminal inverted repeats but without coding sequences), Ribosomal RNA, low complexity, and simple/tandem repeats—is retained. For our analysis, we used the Repbase libraries (version 2014-01-31 downloaded from http://www.girinst.org/, last accessed April 13, 2015) and the TEFam library (accessed at http://tefam.biochem.vt.edu/tefam/index.php, last accessed April 13, 2015). RM (version open-4.0.5) parameters were set to default values, slow-research mode with the NCBI BLAST program (RMBLASTN program, NCBI BLAST 2,2,23+), and only the best hit was kept following dnaPipeTE contig analysis, as determined by the highest Smith–Waterman score provided by RM.
Fig. 2.—
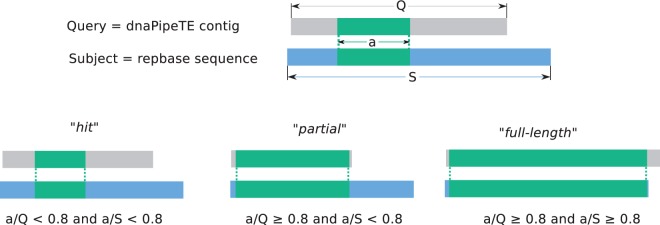
Classification procedure of RepeatMasker annotation for the dnaPipeTE contigs. According to the alignment overlap between the query (a/Q) and the subject (a/S), the dnaPipeTE contigs are annotated as one of the three categories. “Hit” is the weakest annotation, while partial and full-length indicate that the dnaPipeTE contig has annotated along more than 80% of its length.
Repeat Quantification
For quantifying the repeats, BLASTN software (Altschul et al. 1990) was found to perform better than classic short-read aligners such as Bowtie2 (Langmead and Salzberg 2012). Indeed, the divergence between a dnaPipeTE contig—that is, a consensus sequence for a repeat family—and its reads belonging to different copies can be higher than the divergence between a gene or a transcript and its reads, and requires a more sensitive approach. During the “BLAST 1” step (fig. 1), reads from the “BLAST” sample are matched against all the dnaPipeTE contigs to estimate the genome proportion of each assembled repeat. However, we cannot quantify the unassembled repeats during this step. Thus, to obtain an overall estimation of repeat content, the BLAST sample is first matched against a database composed of the annotated contigs of dnaPipeTE and the repeat library in order to recover reads associated with misassembled or missing repeats (“BLASTN 2,” fig. 1). Then, the unmapped reads are matched against the unannotated contigs supplied by dnapipeTE (“BLASTN 3,” fig. 1), and the remaining reads are assumed to belong to nonrepeated sequences. We use the BLAST sample for both estimations, and reads are mapped using discontinuous BLASTN (NCBI BLAST 2.2.29+), which keeps matches with 80% minimum identity and only the best hit per read. To speed-up computation, dnaPipeTE uses GNU Parallel (version 20 140 622) (Tange 2011) to parallelize BLASTN runs.
Finally, the divergence computed between one read and its contigs during the BLAST 1 step is used as a proxy of the divergence time between TE copies in a given family. This proxy is shown to be relevant compared with previous analyses of TE age distribution that used Kimura distances from a full-length TE copy and its consensus sequence in Repbase (“TE Landscapes,” http://www.repeatmasker.org/ (last accessed April 13, 2015); several examples are given in supplementary fig. S3, Supplementary Material online).
Efficiency of dnaPipeTE
Prior to A. albopictus genome analysis, we tested the efficiency of dnaPipeTE on well-annotated genomes that varied in size and TE content. We used available Illumina reads from the species Drosophila melanogaster (Diptera: Drosophilidae), Anopheles gambiae (Diptera: Culicidae), Caenorhabditis elegans (Rhabditida: Rhabditidae), Ciona intestinalis (Enterogona: Cionidae), Gasterosteus aculeatus (Gasterosteiformes: Gasterosteidae), and A. aegypti—the closest fully sequenced species to A. albopictus. We also tested the behavior of dnaPipeTE on older repeatomes, such as that of the human genome (Homo sapiens), in which copies of one TE family are highly divergent. All data management information and references are given in supplementary table S1, Supplementary Material online.
Analysis of the A. albopictus Repeatome and Comparison with A. aegypti
Genomic Data
The two mosquito genomes were sequenced with Illumina NGS technology (Illumina HiSeq2000). The A. albopictus strain originated from La Reunion Island, Indian Ocean. Genomic DNA was prepared from four female individuals of generation F5 bred in an insectarium. Sequencing generated 440.2 million 100-bp paired-end reads (ProfilXpert platform, Lyon, France). A total sample of 4,243,902 single-end reads was also generated (R1’s were used). Aedes aegypti female genomic reads (SRR871496; strain Liverpool; 213.4 million 100-bp paired-end reads; ∼16.4× coverage, Virginia Tech) were downloaded from the short-read archive collection (http://www.ncbi.nlm.nih.gov/sra, last accessed April 13, 2015); only the first read of each pair was used for analysis.
Read Preprocessing
According to quality statistics, all reads were trimmed to 82 bp, keeping the nucleotides 10 through 91 in both A. albopictus and A. aegypti species. Then, sequences were filtered using FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/, last accessed April 13, 2015) with a minimum 20 average Phred score on 90% of the reads. Finally, reads from mitochondrial DNA were removed from the data with Bowtie 2 software (version 2.1.0) under default parameters to map reads to the whole mitochondrial genome sequence for each Aedes species available through the NCBI website (http://www.ncbi.nlm.nih.gov/, last accessed April 13, 2015).
Aedes albopictus and A. aegypti Sampling
In the literature, the genome size of A. albopictus is reported to be variable, ranging from 0.6 to 1.6 Gbp. Flow cytometry performed on the heads of A. albopictus females estimated the genome size of our sequenced strain to be 1.16 Gbp (1.19 pg, unpublished data). The number of reads comprising the three independent samples used by dnaPipeTE was set to represent 0.1× of each genome. The subset sample of 4,243,902 reads (0.3×) was used to assemble TEs and repeats for A. albopictus, consisting of 2 samples of 0.1× genomic coverage for assembly and a third sample of 0.1× for the quantification step. This sample size was chosen after a preliminary analysis showed that 0.1× per Trinity run maximizes the assembly N50 for this genome (supplementary fig. S2, Supplementary Material online). We suggest that this will balance finding as many repeats as possible with limiting the assembly of nonrepeated DNA (noise). For A. aegypti, coverage was also set to 0.1×, using reads taken from the full sequencing experiment based on a genome size of 1.3 Gbp, according to the whole-genome assembly size and mean genome size estimations (Nene et al. 2007; Gregory, T.R. (2015); Animal Genome Size Database. http://www.genomesize.com, last accessed April 13, 2015).
TE Family Recovery and Quantification
To cluster dnaPipeTE contigs into TE families, we used the cd-hit-est program from the CD-HIT suite (version 4.6.1) (Li and Godzik 2006) with local alignment and the greedy algorithm. We set the clustering parameters to group pairs of sequences with at least 80% of the shortest sequence aligned, with a minimum of 80% identity in the longest sequence (parameters -aS 0,8 -c 0,8 -G 0 -g 1). This method results in better performance than grouping contigs per Trinity gene or by RM annotation. In the first case, contigs from one Trinity gene could be joined when they shared a conserved fragment (such as a protein domain), even if they did not actually belong to the same TE family. In the second case, RM annotations include only the closest sequences known, and one sequence could easily match to multiple TE families. This method allowed us to report the most abundant repeats (in relative genome proportion) and to estimate the number of TE copies for fully assembled repeats (dnaPipeTE contigs full-length, see above).
We then estimated the copy number of the fully assembled repeats (table 1) using the following formula:
where n is the number of read-matching contigs from a TE family (contigs from one CD-HIT cluster), N is the total number of reads in the BLAST sample, G is the genome size in bp, and L is the length of the representative sequence of the TE family (reference sequence of the CD-HIT cluster) in bp.
Table 1.
The Most Abundant Identified Repeat Families in Aedes albopictus
| Genome% | RM Annotation | RM Superfamily | dnaPipeTE Contig Size | Estimated Copy Number |
|---|---|---|---|---|
| 1.26% | Lian-Aa1 | LINE/LOA | 4,080 | 3586 |
| 1.25% | RTE Ele4 | LINE/RTE-BovB | 3,447 | 4203 |
| 1.16% | JAM1 | LINE/RTE-BovB | 2,356 | 5728 |
| 1.10% | R1_Ele1 | LINE/R1 | 5,797 | 2195 |
| 0.54% | RTE_Ele3 | LINE/RTE-BovB | 3,283 | 1911 |
| 0.41% | CACTA-3_AA | DNA/CMC-EnSpm | 1,626 | |
| 0.37% | TF001239_mTA_Ele24Aedes | MITE | 638 | |
| 0.33% | Chapaev3-2_AA | DNA/CMC-Chapaev-3 | 1,611 | |
| 0.29% | Loner_Ele2 | LINE/I | 6,335 | 526 |
| 0.28% | TF001239_mTA_Ele24_Aedes | MITE | 469 | |
| 0.28% | Loner Ele1 | LINE/I | 6,329 | 513 |
| 0.23% | Lian-Aa1 | LINE/LOA | 934 | |
| 0.23% | FEILAI_AA | S1NE/tRNA | 324 | 8215 |
| 0.22% | TF001248_mTA_E1e33_Aedes | MITE | 2,407 | 1071 |
| 0.18% | MSAT-1_AAe Satellite | 2,133 | ||
| 0.17% | RTE Ele5 | LINE/RTE-BovB | 2,642 | |
| 0.17% | Lian-Aa1 | LINE/LOA | 1,865 | 1053 |
| 0.17% | LSU-rRNADme | rRNA | 4,681 | |
| 0.16% | R1_Ele1 | LINE/R1 | 3,362 | |
| 0.16% | JAM1B_AAe | LINE/RTE-BovB | 793 | |
| 0.16% | LOA_Ele5 | LINE/LOA | 3,724 | 500 |
| 0.16% | TF001244_mTA_Ele29Aedes | MITE | 578 | |
| 0.15% | MSAT-2_AAe | Satellite | 1,301 | |
| 0.15% | TF001312_m8bp_Ele20_Aedes | MITE | 1,532 | |
| 0.15% | TF000681_m4bp_Ele5_Aedes | MITE | 674 | 2548 |
| 0.14% | CR1-50_AAe | LINE/CR1 | 678 | |
| 0.14% | Sola2-4_AAe | DNA/Sola | 1,232 | |
| 0.14% | TF001310_m8bp_E1e19_Aedes | MITE | 1,840 | |
| 0.14% | TF001280_otherMITEs_Ele7Aedes | MITE | 252 | |
| 0.13% | JAM1B_AAe | LINE/RTE-BovB | 424 | |
| 0.13% | MSAT-1_AAe | Satellite | 663 | |
| 0.13% | MSAT-2_AAe | Satellite | 575 | |
| 0.13% | Gecko | SINE/tRNA-I | 249 | 5967 |
| 0.13% | TF001295_mTA_Ele38c_Aedes | MITE | 1,377 | |
| 0.12% | MSAT-1AAe | Satellite | 204 | |
| 0.12% | TF001257_m4bp_E1e16_Aedes | MITE | 887 | |
| 0.12% | TF001280_otherMITEs_Ele7Aedes | MITE | 1,379 | |
| 0.12% | TF001313_otherMITEs_Ele27Aedes | MITE | 2,209 | |
| 0.12% | MSAT-1_AAe | Satellite | 852 | |
| 0.12% | TF000746_mTA_Ele22_Aedes | MITE | 557 | 2439 |
| 0.11% | LOA_Ele2B_AAe | LINE/LOA | 2,484 | |
| 0.11% | Sola1-3_AA | DNA/Sola | 349 | |
| 0.11% | otherMITEs_Ele11 | DNA/hAT-hATm | 421 | |
| 0.11% | TF001251_m3bp_Ele8a_Aedes | MITE | 900 |
Note.—An estimation of copy number was made only for TEs identified as full-length elements and was based on the size of the dnaPipeTE reference contig after TE family clustering. RM annotation, repeat family hit found by RepeatMasker; RM superfamily, repeat superfamily name in Repbase.
TE Transcriptional Activity
To identify transcriptionally active TEs among the discovered repeats in A. albopictus, we mapped the A. albopictus transcriptome assembly (adult, embryo, and oocyte transcriptome merged reference assembly downloaded from http://www.albopictusexpression.org/, last accessed April 13, 2015) onto the dnaPipeTE contigs using BLAT. We filtered the results of the BLAT analysis such that only TE consensus sequences matching 80% of a transcriptome contig (minimum alignment 80 bp) with 80% minimum identity were retained.
Comparison between A. albopictus and A. aegypti
To avoid annotation bias due to the abundance of reference sequences from A. aegypti in Repbase, we performed a second analysis with dnaPipeTE on A. albopictus and A. aegypti using a TE library devoid of reference sequences from A. aegypti. Then, we used BLAT to match cd-hit-clustered dnaPipeTE contigs between species in order to identify shared TE families. We filtered the results of the BLAT analysis such that alignments with at least 80 bp and 75% identity and only one reference contig per species were retained. Finally, for each species we summed the total number of reads in the cluster for which the references belonged. Thus, we obtained pairs of counts for putatively shared TE families.
dnaPipeTE Comparison with RepeatExplorer
Compared with dnaPipeTE, RE requires only one sample for assembly and annotation. We thus ran it using the “BLAT” sample generated by dnaPipeTE for the A. albopictus data set, on which an estimation of repeated content and a quantification of the main repeat families is performed. Computations were performed online with the “clustering” tool of the RE Galaxy server (http://repeatexplorer.umbr.cas.cz/, last accessed April 13, 2015) with the following parameters: 44 bp (55% of the read length) minimum overlap for clustering, 0.01% cluster threshold for detailed analysis, 40 bp minimal overlap for read assembly and RepeatMasking against the “all” database. Computation time, contig number, N50, proportion of repeats in the sample, and percentage of annotation of the repeated content were calculated for comparison.
Results
Efficiency of dnaPipeTE
We report here the results obtained for D. melanogaster (fig. 3). Details and results from other species are presented in supplementary figures S1 and S3, Supplementary Material online. In D. melanogaster, as well as the other fully annotated genome tested, dnaPipeTE estimations for the different families of TEs are accurate when only a small subset sample of NGS sequencing reads was used as input (three samples of 0.25× coverage). The relative proportion of each TE order is respected in dnaPipeTE estimations. In D. melanogaster, however, the whole repeat content is underestimated (17.78% vs. 28.21%). For this species, our results indicate that dnaPipeTE seems to have underestimated the simple and tandem repeat content of the genome. For A. aegypti (supplementary fig. S1, Supplementary Material online), we estimate the TE content to be 45.6%, which is very close to the estimation of 47% made by Nene et al. from the assembled genome. Using genomes variable in size and TE content as benchmark, we also noticed that the more the genome is filled with repeated DNA, the less the number of Trinity iteration is needed, as well as the coverage provided as input.
Fig. 3.—

Relative genome proportions of the main repeat classes (pie charts) and TE landscapes (bar plots) from RepeatMasker on assembled genome (left) and dnaPipeTE (right, BLASTN with 0.25× genome coverage) for Drosophila melanogaster strain w1118. RepeatMasker analysis data were downloaded from http://repeatmasker.org and retranscribed according to the name used for annotation in dnaPipeTE.
Comparisons of TE age distributions obtained with dnaPipeTE (fig. 3 and supplementary fig. S3, Supplementary Material online) and those made from fully assembled genomes available on the RM website (TE landscapes) (http://repeatmasker.org/genomicDatasets/ RMGenomicDatasets.html, last accessed April 13, 2015) were performed. These comparisons showed that dnaPipeTE provides a good estimate of the recent TE age distribution. As with other de novo TE assemblers, dnaPipeTE is limited in its ability to detect old TE families with degraded and divergent copies. For example, in D. melanogaster or H. sapiens, TEs with more than 30% divergence between reads and the consensus sequence are not identified (fig. 3 and supplementary fig. S4, Supplementary Material online). Our tuning tests show that dnaPipeTE performs well in the estimation of TE proportion and dynamics, with consensus-read divergence ranging from 0% to 15%, which is sufficient to compare closely related species and is close to the definition of a TE family as per the 80-80-80 rule (Wicker et al. 2007).
Aedes albopictus Repeatome Analysis
Repeat Assembly with dnaPipeTE
Assembly of the repeats produced 8,102 contigs with an N50 of 677 bp. Although no reference genome for A. albopictus exists at this point in time, dnaPipeTE was able to annotate 5,141 contigs including 949 “partial TEs” and 30 full-length elements. Among these, some full-length annotated dnaPipeTE contigs were found to represent different variants of the same family, including some internal deletions. Taking this into account, a total of 24 annotated families with full-length consensus sequences were quoted for A. albopictus.
Repeated DNA Content of A. albopictus
dnaPipeTE reported that the repeatome of A. albopictus comprises 49.73% of the genome. Annotation of this repeated DNA showed that TEs occupy 33.58% of the genome. Tandem repeats (satellites and microsatellites) occupy 8% (fig. 4), while unannotated repeats represent 7.23%. The most abundant repeats were Class II (DNA) transposons and LINE (Class I non-LTR) retrotransposons, followed by LTR retrotransposons and SINEs. Details regarding the most abundant repeat families are reported in table 1. The most abundant TE family in terms of genome percentage is a “Lian-like” LINE element (similar to Lian-a1 in A. aegypti), which occupies 1.267% of the genome with 3,586 estimated copies (table 1). The most highly represented families in terms of copy number among the full-length elements annotated by dnaPipeTE are two LINE elements from the “Loner” superfamily, with more than 6,000 estimated copies each. Thirteen other LINE families represent more than 0.10% of the genome each. Fourteen MITEs (non-autonomous Class II) also appear among the most repeated TE families.
Fig. 4.—

Relative genome proportions of the main repeat classes found in Aedes albopictus using dnaPipeTE, from a nucleotide BLAST of 1,414,634 reads (0.1×) against the repeat assemblies performed with a total of 2,829,268 reads (0.2×).
In addition, we found using BLAT that 7,005 of the 8,102 dnaPipeTE contigs have significant hits with a sequence from the A. albopictus transcriptome assembly reported for adult, embryo, and oocyte (Poelchau 2011; http://www.albopictusexpression.org/ [last accessed April 13, 2015]; supplementary table S2, Supplementary Material online).
Comparison of TE Dynamics between A. albopictus and A. aegypti
Aedes albopictus TE age distribution was compared with that of the yellow fever mosquito, A. aegypti (the only available assembled genome for the Aedes genus). We showed that in both species, most of the reads are highly similar to their respective dnaPipeTE contigs (fig. 5). This indicates that most of the detected TE families are recent and possess a high degree of similarity between their copies. This similarity is particularly strong for the detected LTR retrotransposons and, to a lesser extent, for the LINEs that are the most represented TEs in these distributions. Class II DNA transposons are less represented than expected in these comparisons, as their detection suffered from the removal of A. aegypti reference sequences from the library for comparison (fig. 4 for the full analysis in A. albopictus vs. fig. 5 for the interspecies comparison). Between species, the most striking result is that the genomic proportion of LINE/Jockey reads in A. aegypti is high and is composed of mostly recent but also some older TEs, while this family is much less abundant in A. albopictus, with less divergence between reads and contigs. In addition, the distribution of the read divergence of LINE/R1 elements is strongly concentrated at the left of the graphic (representing recent TE copies) in A. aegypti, while in A. albopictus the proportion of reads in superfamilies of higher divergence decreases more slowly (representing older TE copies).
Fig. 5.—

TE age distribution comparisons between Aedes albopictus (left) and Aedes aegypti (right). For each species, the nucleotide divergence from BLASTN is reported between a repeat read and the contig, where it matches the dnaPipeTE assembly.
The weak positive correlation between A. aegypti and A. albopictus in the genomic abundance of the shared families (fig. 6, r2 = 0.186, P < 0.01 on the log10 scale) is mostly due to the less abundant families (<0.1% of the genome). Some families display very high differences, such as the Juan-A (LINE/Jockey retrotransposon) family which represents almost 3% of the genome proportion in A. aegypti but only 0.08% in A. albopictus, or Copia_Ele122 which displays a 5-fold change between the two species, while R1-Ele1 and RTE-3 are good examples of the mirror case. Globally, very few shared families have the same genomic proportion, with the exception of CACTA-3 (DNA transposon) and, less markedly, Jam-1 or Lian-Aa1 (LINEs), which contrast the general trend.
Fig. 6.—
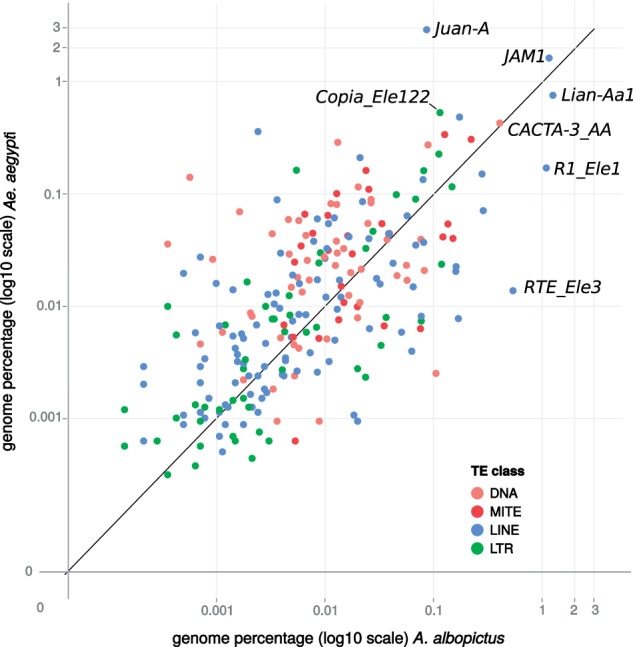
Comparison of the relative genome proportions of shared TE families between Aedes albopictus and Aedes aegypti in terms of genome percentage (log10 scale). Each dot represents a shared TE family, defined by a more similar BLAT hit between the TE family reference contig of each species. Names on the graphs correspond to the main TE annotation (from A. aegypti) discussed in the text.
Comparison between dnaPipeTE and RepeatExplorer
Our pipeline dnaPipeTE operates on the same principles as RE to estimate, assemble, and annotate the repeatome of a species from a sample of reads. Therefore, it was expected that similar estimates of global repeated content in A. albopictus would be obtained by RE and dnaPipeTE (table 2). However, dnaPipeTE, in addition to being much faster, was also able to annotate a larger fraction of TEs and to compute larger contigs. However, RE seems to more sensitively estimate the proportion of low complexity and tandem repeat sequences (data not shown).
Table 2.
Performance Comparison between dnaPipeTE and RepeatExplorer Using Aedes Albopictus and Drosophila melanogaster Samples
| Computing Time | Contig Number | Assembly N50 (bp) | Repeat Content Estimation | Repeat Annotation | ||
|---|---|---|---|---|---|---|
| A. albopictus | dnaPipeTE | 3 h 07 min (8 CPUs/40 Go RAM) | 8102 | 677 | 49.13% | 85.3% |
| RepeatExplorer | 2 days 5 h 12 min (8 CPUs/16 Go RAM) | 14615 | 198 | 51.0% | 25.5% | |
| D. melanogaster | dnaPipeTE | 0 h 40 min (8 CPUs/15 Go RAM) | 2054 | 2,590 | 18% | 98.8% |
| RepeatExplorer | 6 h 05 min (8 CPUs/16 Go RAM) | 1352 | 287 | 16.5% | 86.1% |
Note.—Repeat annotation percentage was computed by counting the number of genomic reads receiving an annotation for each method.
Discussion
The A. albopictus repeatome
We report the first description of the A. albopictus repeatome using dnaPipeTE, a new bioinformatic pipeline for the de novo estimation, annotation, and assembly of repeatomes from raw genomic reads. We found that the total amount of repeated DNA reached 49.13% of the genome that includes at least 33.58% TEs. Taking into account that this method will underestimate low copy number TEs as well as older copies that were unable to be assembled due to mutation accumulation, our estimation should be viewed as a lower bound for the TE content of A. albopictus. As 7.23% of the genome is still unannotated repeats, it is possible that the TE content of A. albopictus ranks the largest among mosquitoes (fig. 7; Holt et al. 2002; Nene et al. 2007; Arensburger et al. 2011; Marinotti et al. 2013; Zhou et al. 2014). The large repeatome of A. albopictus contributes to half of its genome size, which is consistent with the observed relation between genome size and TE content (Biémont and Vieira 2004; Chénais et al. 2012). This relation exists between published genome sizes and TE content of other mosquitoes (fig. 7, r2 = 0.82, P < 0.01).
Fig. 7.—
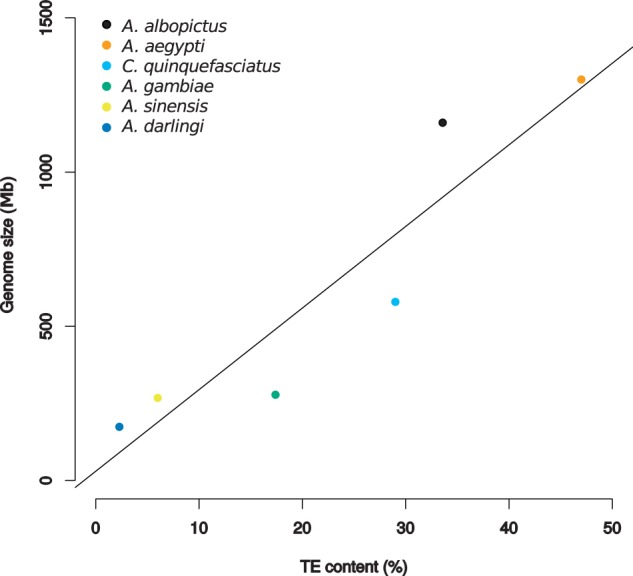
Linear regression of genome size over TE content in mosquitoes. Except for Aedes albopictus, data come from complete sequenced genomes cited in the text. (r2 = 0.827, P < 0.01).
TE families can be extremely different from each other and are classified into several subfamilies. In a given genome, some TE families are present in few copies, while others can reach hundreds of thousands of copies. In A. albopictus, the largest TE families in terms of genome proportion and copy numbers are LINE (non-LTR) retroelements, which harbor thousands of copies per family and represent 12.09% of the genome. These LINEs represent several well-known superfamilies that have been described in mosquitoes, such as I (Lian, R1, Loa, and Loner families) and RTE (Tu et al. 1998; Biedler and Tu 2003; Boulesteix and Biémont 2005). LINEs are also found in high copy number in A. aegypti, where they represent 14% of the genome (Nene et al. 2007). At the class level, the most abundant class of TE is the Class II, with a majority of DNA transposons and MITEs. This feature is shared by the A. aegypti genome, in which Class II elements are also the most abundant repeats, comprising 20% of genome proportion, including 16% of MITEs.
TE Dynamics and Comparison with Aedes aegypti
Comparison of the two related Aedes species highlighted a convergence in TE landscapes at the superfamily level. Both species display a similar distribution of sequenced TE reads against their contig sequences for the three TEs studied (LTR, LINEs [Class I], and Class II). In these species, Class I elements (RNA-mediated transposition) showed a right-skewed distribution, meaning that copies of each TE family share a high identity. This is typical of recent or active TE families, in which the copy number increases faster than the accumulation of mutations within the copies (Lerat et al. 2011; Staton et al. 2012). This pattern can be seen in species such as D. melanogaster or An. gambiae, in which Class I elements showed recent amplifications (Biedler and Tu 2003; Kapitonov and Jurka 2003; see also the genome analysis available online at http://repeatmasker.org/genomicDatasets/RMGenomicDatasets.html, last accessed April 13, 2015).
In both mosquito species, DNA-based transposons (Class II) are poorly represented compared with their relative genome proportion. However, this result might be explained by the removal of A. aegypti TE references from the library to avoid any bias toward this species in the annotation, which might have removed elements specific to the Aedes genus. Another explanation is that DNA transposons could belong to families with very few copies and/or result from an old invasion of the genome. Thus, our methodology, which is weaker beyond 15% divergence and for elements with few copies, could have missed old Class II elements. Ultimately, this could mean either that members of Class II are the first TEs to have invaded Aedes genomes or that Class I TEs are undergoing a new expansion wave.
Despite these similarities in the TE age distributions, the LINE/Jockey superfamily is different between these two species. Indeed, these elements are rare (0.04% of the blasted reads) in A. albopictus, where only recent copies are found. However, in A. aegypti, they represent half of the LINEs, and the LINE/Juan-A is the most abundant TE, representing 3% of the genome (Nene et al. 2007). Conversely, A. albopictus harbors more LINE/I elements than A. aegypti, and their distribution indicates a higher number of divergent copies, which suggests that their amplification in the A. albopictus genome could have begun earlier than in A. aegypti following the divergence of these two species.
The distinction between A. albopictus and A. aegypti is even more striking when observing the abundance of the TE families they share. Indeed, the abundance of TEs copies is very different from one genome to another. This indicates that while both species share similar trends in TE class dynamics, a TE expansion occurred independently in each species. This observation could be interpreted in the ecological framework of TE dynamics and evolution (Venner et al. 2009; Linquist et al. 2013). Indeed, “ecological” factors affecting the genome, such as GC content or genome size, have been shown to be linked to TE abundance and distribution in related species (Jurka et al. 2011). Thus, inheritance of a common genome and ecosystem from an ancestor could have constrained superfamily dynamics in both species, considering either the possible interaction between TEs (identical to interspecific competition) or between TEs and the genome architecture (Venner et al. 2009; Linquist et al. 2013). However, at the family level, the spread of one TE family instead of another is not subject to ecological constraint (Jurka et al. 2011). For instance, the general pattern of a recent invasion of LTRs and LINEs in the Aedes species studied here can still be observed, while the specific TE families amplified in each species differ. In addition, both A. albopictus and A. aegypti are examples of species with numerous subdivided populations in their native areas (Hawley 1988; Mousson et al. 2005; Brown et al. 2014) and a relatively limited natural dispersion capability (Reiter 1996; Bellini et al. 2010; Medley et al. 2015), which increases the probability of differential TE fixation in isolated subpopulations (Jurka et al. 2011). Therefore, the sequenced individuals are only representative of the subpopulations to which they belong, and it would be interesting to compare TE family diversity at the subpopulation level with regard to intraspecific genome size variation imparted by TEs in A. albopictus (McLain et al. 1987; Black and Rai 1988).
dnaPipeTE: A Novel Tool for TE Comparative Studies
Preliminary work on the A. albopictus repeatome led us to develop our own pipeline in order to address specific unmet needs. As the A. albopictus genome is especially large, we were interested in solutions using low coverage sequencing to find and quantify TEs and interspersed repeats. The most advanced software for this task previously available was RE (Novák et al. 2010), which allows the simultaneous location, quantification, and annotation of repeats from unassembled sequencing reads. However, we felt that some points could be improved by using NGS-specific tools. By using Trinity as a TE assembler on small genomic data sets, dnaPipeTE can recover larger TE contigs and can improve this step by performing multiple iterations with additional independent samples. dnaPipeTE can annotate and quantify TE families with its contigs and the number of mapped reads, while RE annotation is given only for sampled reads. Our method allowed the identification of more repeats in A. albopictus than RE, with a substantial decrease in computational time. As with other library-based tools, this automatic annotation should be considered with caution when working on species with very few reference libraries, where the similarities between hits might be weak and could lead to annotation errors. However, tests on model species showed that dnaPipeTE performed well in the estimation of the TE content and the proportions of the main TE families. Although it was not designed for de novo identification of new TE families, dnaPipeTE can produce full-length contigs of TEs that could be manually annotated at a later point. dnaPipeTE also provides a large amount of usable output (summary tables, graphs, sorted data sets). Finally, dnaPipeTE is the first method capable of generating a representation of TE age distribution without prior genome assembly. This analysis of course has some limitations. First, the BLAST method allows the detection of variation only from 0% to 15% divergence. Second, considering two divergent copies in a TE family, the accumulation of mutations will not be evenly distributed along the sequence; reads from a conserved protein domain will be more similar to the contig than nonfunctional regions due to selective constraints, biasing the TE age distribution toward recent divergence. In the future, the effects of these drawbacks will be reduced by the use of longer reads, which dnaPipeTE is already equipped to handle. In conclusion, this new bioinformatic pipeline, available for download at https://lbbe.univ-lyon1.fr/-dnaPipeTE-.html, allowed us to perform a fast and comprehensive analysis of TEs and repeat elements in a newly sequenced genome using NGS raw data with only 0.3× genome coverage. It allows the design of “low sequencing experiments” that reduce sequencing cost and facilitate an increase in the number of samples compared. The consistency and the robustness of dnaPipeTE also allow for comparative studies such as the one presented in this article.
Our study showed that the repeatome of A. albopictus is huge, encompassing 50% of the genome, and that it shares notable similarities with A. aegypti at the main TE order level. The intrafamily dynamics of TEs show high variation between species. Since the divergence of A. albopictus and A. aegypti 10 million years ago (Pashley and Rai 1983), TE families seemed to have evolved independently from ancestral TE ecology. These pictures of the two Aedes species’ repeatomes could explain the large genome size variation due to repetitive DNA reported at the intraspecific level (McLain et al. 1987; Black and Rai 1988).
Supplementary Material
Supplementary figures S1–S4, tables S1 and S2, and Material are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank the members of the Transposable Elements, Evolution, and Population team for testing dnaPipeTE and for providing suggestions and constructive discussion about this article. We also thank Petr Novák for his availability to provide help and information about the RepeatExplorer pipeline and Murray Patterson for English revisions. This work was performed using the computing facilities of the CC LBBE/PRABI. This work was supported by the Agence Nationale de la Recherche (ANR Genemobile), the Centre National de La Recherche Scientifique, the Institut Universitaire de France, and the Federation de Recherche 41 “Bio-Environnement et Santé.” C.G. received a grant from the French Ministry of Superior Education. This study was also supported by The A. albopictus genome sequencing project which is partly funded by the Agence Nationale de la Recherche (ANR Immunsymbart).
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arensburger P, Hice RH, Wright JA, Craig NL, Atkinson PW. The mosquito Aedes aegypti has a large genome size and high transposable element load but contains a low proportion of transposon-specific piRNAs. BMC Genomics. 2011;12:606. doi: 10.1186/1471-2164-12-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini R, et al. Dispersal and survival of Aedes albopictus (Diptera: Culicidae) males in Italian urban areas and significance for sterile insect technique application. J Med Entomol. 2010;47:1082–1091. doi: 10.1603/me09154. [DOI] [PubMed] [Google Scholar]
- Biedler J, Tu Z. Non-LTR retrotransposons in the African malaria mosquito, Anopheles gambiae: unprecedented diversity and evidence of recent activity. Mol Biol Evol. 2003;20:1811–1825. doi: 10.1093/molbev/msg189. [DOI] [PubMed] [Google Scholar]
- Biémont C, Vieira C. [The influence of transposable elements on genome size] J Soc Biol. 2004;198:413–417. [PubMed] [Google Scholar]
- Black WC, Ferrari JA, Sprengert D. Breeding structure of a colonising species: Aedes albopictus (Skuse) in the United States. Heredity (Edinb) 1988;60(Pt 2):173–181. doi: 10.1038/hdy.1988.29. [DOI] [PubMed] [Google Scholar]
- Black WC, Rai KS. Genome evolution in mosquitoes: intraspecific and interspecific variation in repetitive DNA amounts and organization. Genet Res. 1988;51:185–196. doi: 10.1017/s0016672300024289. [DOI] [PubMed] [Google Scholar]
- Bonizzoni M, Gasperi G, Chen X, James AA. The invasive mosquito species Aedes albopictus: current knowledge and future perspectives. Trends Parasitol. 2013;29:460–468. doi: 10.1016/j.pt.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulesteix M, Biémont C. Transposable elements in mosquitoes. Cytogenet Genome Res. 2005;110:500–509. doi: 10.1159/000084983. [DOI] [PubMed] [Google Scholar]
- Brown JE, et al. Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito. Evolution. 2014;68:514–525. doi: 10.1111/evo.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta E, González J. The impact of transposable elements in environmental adaptation. Mol Ecol. 2013;22:1503–1517. doi: 10.1111/mec.12170. [DOI] [PubMed] [Google Scholar]
- Chénais B, Caruso A, Hiard S, Casse N. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene. 2012;509:7–15. doi: 10.1016/j.gene.2012.07.042. [DOI] [PubMed] [Google Scholar]
- Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley WA. The biology of Aedes albopictus. J Am Mosq Control Assoc Suppl. 1988;1:1–39. [PubMed] [Google Scholar]
- Holt RA, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- Huang X. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Bao W, Kojima KK. Families of transposable elements, population structure and the origin of species. Biol Direct. 2011;6:44. doi: 10.1186/1745-6150-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci U S A. 2003;100:6569–6574. doi: 10.1073/pnas.0732024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P, Platzer M, Downie BR. RepARK—de novo creation of repeat libraries from whole-genome NGS reads. Nucleic Acids Res. 2014;42:e80. doi: 10.1093/nar/gku210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Rai KS. Intraspecific variation in nuclear DNA content among world populations of a mosquito, Aedes albopictus (Skuse) Theor Appl Genet. 1990;79:748–752. doi: 10.1007/BF00224239. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat E, Burlet N, Biémont C, Vieira C. Comparative analysis of transposable elements in the melanogaster subgroup sequenced genomes. Gene. 2011;473:100–109. doi: 10.1016/j.gene.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Linquist S, et al. Distinguishing ecological from evolutionary approaches to transposable elements. Biol Rev Camb Philos Soc. 2013;88:573–584. doi: 10.1111/brv.12017. [DOI] [PubMed] [Google Scholar]
- Marinotti O, et al. The genome of Anopheles darlingi, the main neotropical malaria vector. Nucleic Acids Res. 2013;41:7387–7400. doi: 10.1093/nar/gkt484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLain DK, Rai KS, Fraser MJ. Intraspecific and interspecific variation in the sequence and abundance of highly repeated DNA among mosquitoes of the Aedes albopictus subgroup. Heredity (Edinb) 1987;58:373–381. doi: 10.1038/hdy.1987.65. [DOI] [PubMed] [Google Scholar]
- Medley KA, Jenkins DG, Hoffman EA. Human-aided and natural dispersal drive gene flow across the range of an invasive mosquito. Mol Ecol. 2015;24:284–295. doi: 10.1111/mec.12925. [DOI] [PubMed] [Google Scholar]
- Modolo L, Lerat E. Identification and analysis of transposable elements in genomic sequences. In: Poptsova MS, editor. Genome analysis: current procedures and application. Norfolk (UK): Caister Academic Press; 2014. pp. 165–181. [Google Scholar]
- Mousson L, et al. Phylogeography of Aedes (Stegomyia) aegypti (L.) and Aedes (Stegomyia) albopictus (Skuse) (Diptera: Culicidae) based on mitochondrial DNA variations. Genet Res. 2005;86:1–11. doi: 10.1017/S0016672305007627. [DOI] [PubMed] [Google Scholar]
- Nene V, et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316:1718–1723. doi: 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Macas J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 2010;11:378. doi: 10.1186/1471-2105-11-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashley DP, Rai KS. Comparison of allozyme and morphological relationships in some Aedes (Stegomyia) mosquitoes (Diptera: Culicidae) Ann Entomol Soc Am. 1983;76:388–394. [Google Scholar]
- Rao PN, Rai KS. Inter and intraspecific variation in nuclear DNA content in Aedes mosquitoes. Heredity (Edinb) 1987;59:253–258. doi: 10.1038/hdy.1987.120. [DOI] [PubMed] [Google Scholar]
- Rebollo R, Horard B, Hubert B, Vieira C. Jumping genes and epigenetics: towards new species. Gene. 2010;454:1–7. doi: 10.1016/j.gene.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Reiter P. [Oviposition and dispersion of Aedes aegypti in an urban environment] Bull Soc Pathol Exot. 1996;89:120–122. [PubMed] [Google Scholar]
- Staton SE, et al. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012;72:142–153. doi: 10.1111/j.1365-313X.2012.05072.x. [DOI] [PubMed] [Google Scholar]
- Tange O. GNU parallel: the command-line power tool ;login USENIX Mag. 2011;3:42–47. [Google Scholar]
- Tu Z, Isoe J, Guzova JA. Structural, genomic, and phylogenetic analysis of Lian, a novel family of non-LTR retrotransposons in the yellow fever mosquito, Aedes aegypti. Mol Biol Evol. 1998;15:837–853. doi: 10.1093/oxfordjournals.molbev.a025989. [DOI] [PubMed] [Google Scholar]
- Vela D, Fontdevila A, Vieira C, García Guerreiro MP. A genome-wide survey of genetic instability by transposition in Drosophila hybrids. PLoS One. 2014;9:e88992. doi: 10.1371/journal.pone.0088992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venner S, Feschotte C, Biémont C. Dynamics of transposable elements: towards a community ecology of the genome. Trends Genet. 2009;25:317–323. doi: 10.1016/j.tig.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, et al. Genome sequence of Anopheles sinensis provides insight into genetics basis of mosquito competence for malaria parasites. BMC Genomics. 2014;15:42. doi: 10.1186/1471-2164-15-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zytnicki M, Akhunov E, Quesneville H. Tedna: a transposable element de novo assembler. Bioinformatics. 2014;30:2656–2658. doi: 10.1093/bioinformatics/btu365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.