

Abstract
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disorder caused by GAA triplet expansions or point mutations in the FXN gene on chromosome 9q13. The gene product called frataxin, a mitochondrial protein that is severely reduced in FRDA patients, leads to mitochondrial iron accumulation, FeS cluster deficiency and oxidative damage. The tissue specificity of this mitochondrial disease is complex and poorly understood. While frataxin is ubiquitously expressed, the cellular phenotype is most severe in neurons and cardiomyocytes. Here, we conducted comprehensive proteomic, metabolic and functional studies to determine whether subclinical abnormalities exist in mitochondria of blood cells from FRDA patients. Frataxin protein levels were significantly decreased in platelets and peripheral blood mononuclear cells from FRDA patients. Furthermore, the most significant differences associated with frataxin deficiency in FRDA blood cell mitochondria were the decrease of two mitochondrial heat shock proteins. We did not observe profound changes in frataxin-targeted mitochondrial proteins or mitochondrial functions or an increase of apoptosis in peripheral blood cells, suggesting that functional defects in these mitochondria are not readily apparent under resting conditions in these cells.
Keywords: Friedreich ataxia, Frataxin, Blood cells, Mitochondria, Biomarkers
1. Introduction
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disorder caused by a GAA triplet expansion or point mutations in the frataxin gene (FXN) on chromosome 9q13 (Campuzano et al., 1996). Its gene product (FXN) is a ubiquitously expressed, nuclear-encoded mitochondrial protein that is severely reduced in FRDA patients. Deficits of FXN are associated with mitochondrial iron accumulation, deficient Fe-S cluster biogenesis, heme protein deficiency, increased sensitivity to oxidative stress, deficits of respiratory chain complex activities and in vivo impairment of tissue energy metabolism (Babcock et al., 1997; Foury and Cazzalini, 1997; Wilson and Roof, 1997; Rotig et al., 1997; Chantrel-Groussard et al., 2001; Pandolfo, 2002). Clinically, FRDA mostly impacts neural and cardiac tissue, even though frataxin is ubiquitously expressed in all tissues. Muscle and islet cells are also affected in these patients to a lesser extent. Thus, the tissue specificity of this mitochondrial disease is complex and poorly understood. Platelets and mononuclear cells are a non-invasive, readily accessible material that can be purified easily and are the main source of mitochondria in blood. We hypothesized that sub-clinical abnormalities exist in the mitochondria of blood cells from FRDA patients, because (i) frataxin is also expressed in blood cell mitochondria, (ii) iron homeostasis and heme synthesis are essential for these cells, (iii) and several antioxidant enzymes as well as glutathione homeostasis in blood cells of FRDA patients are impaired while serum iron and ferritin concentrations are normal. If mitochondria from blood cells are abnormal, such cells would provide a powerful, readily accessible means to analyze the impact of frataxin deficiency on mitochondrial metabolism of FRDA patients by proteomic and functional analyses. They could also provide tissue for use in identifying clinically useful biomarkers for diagnoses and evaluating progression of therapies.
In the present study, we have developed proteomic, metabolic and functional approaches to analyze the changes in mitochondria from FRDA peripheral blood cells from FRDA patients. We have demonstrated that, while the frataxin protein level is decreased in blood cells of FRDA patients, this deficiency is associated only with a decrease in two mitochondrial heat shock proteins. However, we did not observe any profound change in target mitochondrial proteins or mitochondrial functions or an increase of apoptosis in peripheral blood cells.
2. Materials and methods
2.1. Patient characterization and samples process
Blood samples of 25 patients with FRDA and 15 normal healthy volunteers (NHV) as controls were obtained from subjects seen in the Department of Neurology, University of Pennsylvania School of Medicine. All protocols were approved by the IRB at the University of Pennsylvania and written informed consent was obtained from each subject.
Peripheral blood mononuclear cells (PBMCs) and platelets were purified by differential centrifugation and Ficoll gradients. The purity of the cell preparations was assessed by Wright–Giemsa and FACS analysis. We obtained 1–5 109 platelets and 50–100 106 peripheral blood mononuclear cells isolated from 30 mL of citrated peripheral blood draw of normal healthy volunteers and FRDA patients. We routinely observed purity greater than 85 and 96% for mononuclear cells and platelets, respectively, using FACS analysis and Giemsa–Wright Hema3 staining. Apoptosis was assayed by flow cytometry using 7-AAD and Annexin-V-APC as recommended by the manufacturer.
2.2. Lateral flow immunoassay (dipstick assay)
MitoSciences dipstick (MSF31) assays were used to measure frataxin levels in PBMCs and platelets. Briefly, 10 μg of PBMC protein or 5 μg of platelet protein in 25 μL of extraction buffer was mixed with 25 μL blocking buffer and added to individual wells on a 96-well plate with gold-conjugated mAb at the bottom of each well. After samples were allowed to equilibrate with the antibodies, dipsticks were inserted into the well and sample was allowed to wick up the membrane, where frataxin was immunocaptured onto designated capture zones on the dipstick. Capture zones on developed dipsticks were quantified with a Hamamatsu immunochromato reader (MS1000 Dipstick reader). A standard curve using a range (1–50 μg) of total protein extracted from PBMCs and platelets was run to determine an appropriate concentration of sample to use within the working range of the assay. Raw milliabsorbance units (mABS) were corrected for protein concentration and normalized to the control goat anti-mouse IgG band (internal positive control), and the data were expressed as percentages of the average controls run on the same assay.
2.3. SDS-PAGE and western blot analysis
40 μg of PBMC and platelet proteins was solubilized in SDS-PAGE sample loading buffer containing 80 mM dithiothreitol. Samples were denatured by heating to 95 °C for 5 min prior to electrophoresis on a 13% Laemmli-type gel. Proteins were electrophoretically transferred to Protran BA83 nitrocellulose membranes (Schleicher & Schuell) and briefly stained with a solution of amido black to position the protein markers. Membranes were incubated in the blocking solution (5% dry milk in TBST buffer (0.1 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween 20)) followed by incubations with appropriate primary antibodies, washing steps, and horseradish peroxidase-conjugated donkey anti-rabbit IgGs as secondary antibodies (Amersham Biosciences). The signal was detected by exposing the membrane to an x-ray film after incubating with luminol and p-coumaric acid as ECL substrates. The primary antibodies used were as follows: rabbit polyclonal antibodies against the GAPDH, cytochrome c, ACO1 and lipoic acid were purchased from Calbiochem. Anti-VDAC1, SOD1, SOD2, SDHC and FXN antibodies were mouse monoclonal antibodies obtained from Molecular Probes/Invitrogen and MitoSciences.
2.4. 2-D gel electrophoresis
Mitochondrial lysates were processed for 2-DGE as described previously (Sarry et al., 2006). Proteins in the lysates were precipitated by the addition of 10% (w/v) trichloroacetic acid dissolved in ice-cold acetone and centrifuged at 16,000 g for 20 min. After two washes with ice-cold acetone containing 0.05% (w/v) dithiothreitol, protein pellets were resuspended in isoelectric focusing loading buffer containing 2 M thiourea, 6 M urea, 4% (w/v) Chaps and 50 mM dithiothreitol, incubated with stirring at room temperature for 1 h, and centrifuged at 16,000 g for 15 min. Aliquots (300 μL) of the isoelectric focusing buffer containing 200 μg protein and supplemented with 0.2% (v/v) pH 3–10 ampholines were loaded onto the IPG strips (pH 3–10 NL; 11 cm; Bio-Rad) by passive rehydration for 12 h and active rehydration at 50 V for 8 h. Isoelectric focusing was performed for 120 kVh in a Protean IEF cell (Bio-Rad) at 20 °C. Voltage was increased linearly from 50 to 200 V for the first 3 h, held at 200 V for another 3 h, before being increased linearly to 1000 V over the next 3 h and held at that voltage for another 3 h. In the final two periods of 6 h each, the voltage was increased linearly to 5000 V and held at that value. Upon completion of the isoelectric focusing separations, the isoelectric focusing strips were equilibrated with 50 mM Tris/HCl (pH 8.8) containing 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS and 2% (w/v) dithiothreitol for 20 min with gentle stirring before the addition of 2.5% (w/v) iodoacetamide, and alkylation of the samples for 20 min. Electrophoresis in the second dimension was performed on Criterion 8.7×13.3 cm precast 8–16% (w/v) polyacryl-amide gradient gels (Bio-Rad) at 50 V for the first hour and at 200 V for the second hour in a Protean Cell Criterion apparatus (Bio-Rad).
For analyses of the protein profiles, the gels were stained with colloidal Coomassie Brilliant Blue G-250 (GE Healthcare), scanned with an Epson flatbed scanner, and the TIFF files generated using Adobe Photoshop 7.0 were subjected to spot detection, matching and quantification using the PDQuest software package (v. 7.2, Bio-Rad). Three-dimensional Gaussian images of the spots were generated and their volumes were estimated after background subtraction. The intensity of each spot was normalized to the sum of the total spots on the gel such that the integrated and normalized intensity for each spot was expressed as a percentage of the sum of the intensities of all the spots in the Gaussian image.
2.5. Protein identification by mass spectrometry
The Coomassie Brilliant Blue-stained protein spots on the gels were excised manually and transferred to 96-well microtiter plates for fully automated washing, destaining, tryptic digestion, elution, and preparation for LC-QIT-MS/MS in a Multiprobe II MassPREP Station (Waters/Micromass, Inc., Milford, MA). Routinely, the excised gel plugs were washed several times for 15 min, first with aqueous 50 mM ammonium bicarbonate (pH 7.8) and then with 50% (v/v) acetonitrile containing 50 mM ammonium bicarbonate before dehydration for 5 min in acetonitrile. The gel plugs were digested at 37 °C in 25 μL 50 mM ammonium bicarbonate (pH 7.8) containing 6 μg mL−1 trypsin (sequencing grade, modified, Promega) for 7 h. The resulting peptide digests were successively extracted in 20 μL 0.1% (v/v) formic acid in 2% (v/v) acetonitrile, and with 0.1% (v/v) formic acid in 50% (v/v) acetonitrile.
The LC-QIT-MS/MS analyses were performed on an LCQ Ion Trap MS (Deca XP Plus, Thermo-Finnigan, San Jose, CA). The LC-QIT-MS/MS was interfaced to a Famos autosampler (LC-Packings, Sunnyvale, CA) which was used to sample the tryptic digests after their resuspension in 5% (v/v) acetonitrile, 0.1% (v/v) formic acid. The aqueous phase (A) (0.1% v/v formic acid in 5 : 95 acetonitrile/water) and organic phase (B) (0.1% v/v formic acid in 80 : 20 acetonitrile/water) were delivered using an Ultimate HPLC (LC-Packings). After first injecting the samples onto a μ-Precolumn (300 μm i.d. ×5 mm, 5 μm particle size, 100 Å pore diameter, PepMap C18; LC-Packings) which was washed with solvent A at a flow rate of 0.25 μL·min−1 for 4 min, a Switchos microcolumn switching module (LC-Packings) was used to direct the flow from waste to the analytical column (75 μm i.d. ×150 μm, 3 μm particle size, 100 Å pore diameter, PepMap C18; LC-Packings). The analytical column was developed with 5% B for the first 8 min, 5–50% B from 8 to 38 min, 50–95% B from 38 to 39 min, 95% B from 39 to 49 min, 95–5% B from 49 to 50 min, and 5% B from 50 to 75 min. The solvents delivered by the pump were split at a flow ratio of 1: 1250 with the postsplitter flow rate set at 250 nL min−1. An 8 μm aperture fused silica needle (New Objective, Woburn, MA) was used to introduce samples. ESI was accomplished by applying a voltage difference of ±1.3 kV across the fused silica needle. The aperture of the needle was positioned 1–2 mm from the opening of the ion transfer capillary and slightly off axis. Most mass spectra were acquired using the repetitive ‘triple-play’ sequence as recommended by the manufacturer, which consisted of a full scan event for ions with m/z ratios of 400–1200, a zoom scan event acquired within an m/z window of 10 units centered on the chosen ion, and an MS/MS scan event. Ions were selected for the zoom scan and for the MS/MS scan automatically in a data-dependent manner, whereby ions of sequentially decreasing abundance were chosen and two scan events were allowed for any given ion in a 3-s time window. For complex samples, the zoom scan event was omitted. The tolerance for the selection of the precursor ions ranged from 1.5 to 3.0m/z (low to high m/z).
The measured MS/MS spectra were matched with tryptic peptide amino acid sequences from a human HPI database. Raw MS/MS data files that had a minimum total ion current of 105 and contained 15 or more fragment ions were selected. The tolerance window for the grouping of raw MS/MS data files into input files for the Finnigan Sequest/Turbo Sequest software (version 2.0; ThermoQuest) was set to 1.4 amu. The Sequest algorithm was used to identify and retrieve peptide sequences from the database possessing at least one tryptic end with a theoretical mass within 1.25 amu of that measured for the precursor ion and a theoretical y- and b-ion profile bearing a high degree of similarity to the experimental MS/MS spectrum. The similarity between an experimental and theoretical MS/MS spectrum, reported as the cross-correlation factor (Xcorr) and the difference between the unit-normalized Xcorr values of the first- and second-ranked hits (ΔCorr) provided criteria for the preliminary assignment of amino acid sequences based on the experimental MS/MS spectra. Sequences were reported if the Xcorr values were equal to or greater than 1.50, 2.25 and 3.00 for singly, doubly or triply charged precursor ions, respectively, and if the ΔCorr value exceeded 0.1 against the database background.
2.6. Mitochondrial enzyme assays
Enzyme assays were performed in a total reaction volume of 1 mL using standard methods (Trounce et al., 1996). Absorbance changes were continuously monitored using the dual-beam mode of an OLIS-converted DW2a spectrophotometer. NADH dehydrogenase was measured as NADH:ferricyanide oxidoreductase at 410 nm minus 480 nm (ε=1 mM−1 cm−1) in the presence of 2 μM rotenone, 2 μM antimycin A and 5 mM NaN3. Complex II was measured as TTFA-sensitive succinate:DCIP oxidoreductase at 600 nm minus 750 nm (ε=19.0 mM−1 cm−1). Complex IV (cytochrome c oxidase) was measured at 550 nm minus 540 nm (ε=21 mM−1 cm−1) as azide-sensitive ferrocytochrome c oxidase. Citrate synthase was measured at 412 nm minus 360 nm (ε=13.6 mM−1 cm−1) using 5,5-dithio-bis (2-nitrobenzoic acid) to detect free sulfhydryl groups in coenzyme A. SOD activity was assayed spectrophotometrically at 550 nm minus 540 nm by the capacity to lower the rate of superoxide-mediated reduction of acetylated-ferricytochrome c generated by xanthine oxidase (Azzi et al., 1975; Kuthan et al., 1986). All SOD assays were performed at pH 7.8 to allow for balanced measurement of both Mn and Cu/Zn activities. Each sample was assayed in duplicate or triplicate using different protein concentrations. Freeze-thawed mitochondria were assayed in the presence of n-dodecyl-β-D-maltoside (0.1 mM) to allow full access of electron donors/acceptors to the matrix. Specific activities of electron transport chain complexes and citrate synthase are reported as nmol/min/mg protein. One unit of SOD was defined as the amount required to inhibit the rate of reduction of cytochrome c by 50% ([SOD]0.5). The concentration of SOD in the sample was defined by [SOD]=[(v0/v1)−1], with final activity expressed as Units/mg protein. Protein concentration was determined by the BCA assay using BSA as standard.
2.7. Oxygen consumption measurements
Oxygen consumption was measured polarographically using a Strathkelvin oxygen electrode in a magnetically stirred and thermostatically regulated chamber (37 °C). Aliquots of cells were suspended in a total volume of 0.15 mL of air-saturated medium and rates are expressed as nanomoles O2 consumed/ min/ 106 cells for PBMCs and 109 cells for platelets.
2.8. Low temperature optical absorbance difference spectra to assay cytochrome abundance
Low temperature difference spectra were recorded using the split-beam mode of an OLIS-converted DW2a spectrophotometer equipped with a low-temperature module according to the modified method of Wilson (1967). Sonicated mitochondria were oxidized with 5 uM ClCCP and reduced by addition of 10 mM sodium hydrosulfite (dithionite). Following 2 min incubation at ambient temperature, mitochondrial samples were diluted 1:1 with 2 M sucrose and transferred to the appropriate chambers of the cuvette and frozen by complete emersion of the cuvette for 3 min in liquid nitrogen. The cuvette light path was 2 mm and the bandwidth of the measuring light was set at 2 nm. Intensification factors for lymphocyte cytochromes c, b, and a in 1 M sucrose, calculated from the ratio of the observed absorbance at 77°K to the expected absorbance at 22 °C, were found to be 25, 43 and 18, respectively. Baselines for low temperature-spectra were established by employing apparent isobestic points at 540 and 582 nm and the known isobestic point at 615 nm. Cytochrome content was normalized to protein.
3. Results
3.1. Subjects
Age, gender, GAA repeat length, antioxidants and treatments used by patients were recorded (Table 1). The average age was 35 years for FRDA patients (9 females/25 males) while the average age for NHV controls was 32 years (8 females/15 males). The GAA repeat lengths of FRDA patients ranged from 42 to 790 repeats and two patients carried point mutations (R165C and a splice site mutation).
Table 1.
Clinical features of FRDA patient subjects.
| Study
|
Gender | Age | GAA repeat
|
Drug use or treatment |
|---|---|---|---|---|
| # | Length | |||
| FA0 | F | 40 | 760, 860 | Toprol x1 12 mg>1 yr |
| FA02 | F | 30 | 590, 990 | Vit E 400>1 yr; Co Q10 800 > 1 yr; NAC 600 > 1 yr; Buspar 10 mg>1 yr; Flexaril>1 yr; Vit C 500 6–12 mos; Aspirin 81 6–12 mos; Vit B 100 6–12 mos; Albeutrol pm 6–12 mos; Singulair 10 6–12 mos |
| FA03 | M | 22 | 550, 848 | Vit E 400 In>1 yr; Co Q10 200>1 yr |
| FA04 | M | 23 | 750, 920 | Vit E 400>1 yr; Co Q10 600 > 1 yr |
| FA05 | M | 36 | 500, 570 | Asacol>1 yr |
| FA06 | F | 21 | 790, 1120 | Keppra 1–2 day>1 yr; Birth control 6–12 mos; Celexa 20 mg>1 yr |
| FA07 | M | 40 | 500, 660 | Vit E 400 mg>1 yr; Co Q10 100 mg > 1 yr |
| FA08 | F | 28 | 400, 450 | Vit E 1200>1 yr; Co Q10 800 > 1 yr; Idebenone 345>1 yr; NAC 1500>1 yr; Selenium 600 > 1 yr; Mutli Vit>1 yr; Silymarin 600>1 yr; Alpha Lipoic Acid 800 > 1 yr; Vit A 25000 > 1 yr; Beta Carotene 50,000 > 1 yr; Amino Acid 2 tsps>1 yr |
| FA09 | F | 54 | 42, 696 | Vit E 200 mg>1 yr; Co Q10 800 mg > 1 yr; Idebenone 900 mg > 1 yr; Baclofen 60 mg>1 yr; Enablex>1 yr; Celebrex 200 mg 6–12 mos |
| FA10 | M | 22 | 750, 850 | Co Q10 100 mg |
| FA11 | M | 28 | 690, 1120 | None |
| FA12 | M | 30 | 750, 1000 | EPO for the past three months |
| FA13 | M | 20 | 690, 1120 | None |
| FA14 | F | 24 | 796, 1129 | Vit E 1000 IU, Lyrica 150 mg, Zoloft 100 mg, Requip 0.5 M, Ambien 5 mg, Tylenol Arthris 400 mg, YAZ Birth control once dai ly, Senekot SII once daily, Ultram once daily |
| FA15 | M | 75 | 114, 1047 | 1200 mg CoQ10, 1200 mg NAC, Omeprazole 20 mg, Citracal, Vit C 1000 mg |
| FA16 | M | 21 | 620, 1350 | 400 mg CoQ10 |
| FA17 | F | 41 | 359, 359 | Trazadone, Buspar |
| FA18 | M | 20 | pt mutation | 400 mg Vit E, 400 mg Q10, 20 mg baclofen, 600 mg neurtontin, 0.088 synthoid, B12 injection 1 X monthly (all>1 yr) |
| FA19 | F | 46 | 500, 670 | 80 mg Baclofen, 20 mg Prilosec, 4 mg Detrol LA, Bactrim, 2 mg Tinzanidine, 40 mg Nexium, Vitamin D, Calcium citrate |
| FA20 | M | 54 | pt mutation | 1 aspirin in past 7 days, 2.5 mg Detrol LA, Loporamide prn, 4 mg Aciphex, 4 mg Amaryl, 1000 mg Metformin, 45 mg Actose, Insulin |
| FA21 | F | 24 | 603, 1001 | Birth control |
| FA22 | M | 65 | 90, 1025 | CoQ10 300 mg, Neurotin 600 mg, Protonix 40 mg, ENALAPRIL 20 mg, DYNACIRC CR 10 mg, PENTASA 3000 mg, Remicade every 8 weeks by IV, Ambien CR 10 mg, Wellbutrin 150 mg |
| FA25 | M | 65 | 760, 860 | None |
| FA26 | M | 18 | 566, 866 | Lomactil 200 mg, Abilli fy 15 mg, Zoloft 100 mg, Minicycl ine (acne) 200 mg |
| FA27 | M | 30 | 430, 430 | Vit E 200 IU, CoQ10 200 mg, Magnesuim 500 mg, CLONAZEPAM 0.5 mg, TOPROL XL 25 mg Vit C 100 mg, IC KLOR-CON M10 40 meq |
3.2. Proteomic analysis of frataxin and its mitochondrial protein targets level
Mononuclear cells and platelets, the most abundant source of mitochondria in peripheral blood (Fig. 1), were purified to assess the level of frataxin protein. Using Western blots, frataxin was clearly detected in both platelets and PBMCs of normal healthy donors, whereas markedly decreased levels were found in mitochondria of both cell types from FDRA patients (Fig. 2A). Frataxin was detected as a single 15 kDa band as was previously observed in peripheral blood lymphocytes (Condò et al., 2007). This result was confirmed by lateral flow (“dipstick”) immunoassay (Fig. 2B). In both blood cell types, the average level of frataxin was decreased by 3-fold in patient mitochondria compared to normal healthy donor mitochondria. When normalized per mg of protein, PBMCs contained a three-fold higher level of frataxin. However, as whole blood contains approximately 30 times more platelet mitochondria than PBMC mitochondria (Fig. 1), the level of frataxin in whole blood is more likely to reflect the platelet level than the PBMC level. There was an overlap in the absolute levels of FXN between some normal healthy donors and some FRDA patients as assessed by immunoassay.
Fig. 1.
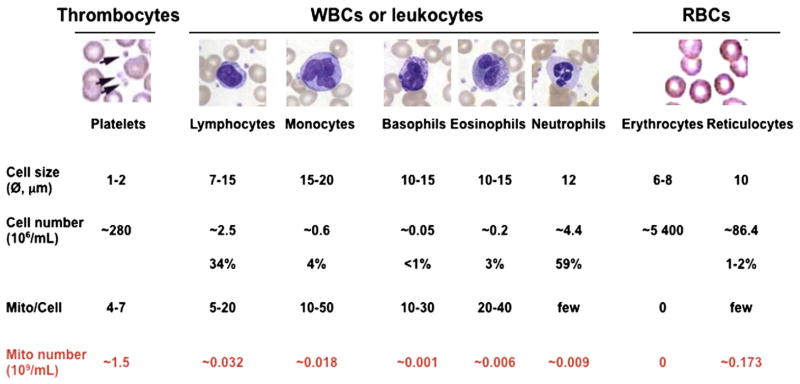
Assessment of the total number of mitochondria per mL of blood in hematopoietic cell types. Adapted from Hoffman et al. (2000). Hematology: basic principles and practice; and Pon and Schon (2001). Methods in Cell Biol. Vol. 65. Mitochondria.
Fig. 2.

Analysis of the frataxin (FXN) protein expression in isolated resting platelets (PLTs) and peripheral blood mononuclear cells (PBMCs) by western blot (A) and dipstick immunoassay (B). This figure shows that frataxin is clearly detected in mitochondria of both platelets and PBMCs of normal healthy donors. We also observed a dramatic decrease of this protein in mitochondria of both cell types from patients with both methods. Cytc, cytochrome c and GAPDH, glyceraldehyde 3-phosphate dehydrogenase, were used as a loading controls. 40 μg of protein were loaded in each lane and western blot analyses were performed in pair-wise manner (each FRDA sample is always processed with its matched NHV samples). The number below the patient identification represents the GAA repeat length of the corresponding patient as well as the treatment received at the time of the blood draw.
We also performed Western blot analysis to assess the protein level of several known mitochondrial targets of frataxin and observed only a decrease in cytochrome c (Fig. 2A). The level of cytochrome c was decreased in 4 out of 6 FRDA platelet and 2 out of 6 FRDA patient PBMC samples, respectively. Additionally, we examined the expression levels of several other mitochondrial proteins relevant to FRDA, including succinate dehydrogenase (SDHA, SDHB), superoxide dismutase (SOD1, SOD2), aconitase (ACO2), ferritin (FTH1), pyruvate dehydrogenase E2 subunit (DLAT), α-ketoglutarate dehydrogenase E2 (DLST), ATP synthase subunit alpha (ATP5A1) and ETC complex IV subunit Va (CoxVa) by Western blotting. None of these mitochondrial proteins were affected in patient platelets or PBMCs of patient blood samples (data not shown).
In parallel, we performed an unbiased comparative analysis of the PBMC mitochondrial proteome of FRDA patients with those of age-matched normal volunteers. First, we refined the method for purification of mitochondria from mononuclear cells and assessed sample purity by Western blot (Fig. 3A). All of the mitochondrial proteins (DLAT, CoxVa, SOD2) are enriched in mitochondrial pellet extracts (MP) of mononuclear cells, while cytosolic proteins (LDH, GAPDH) were decreased or absent in these extracts as expected. Using this material, we performed mitochondrial protein separation by 2D-gel electrophoresis (200 μg; IPG strip 3–10NL, 11 cm; 11% SDS-PAGE; Colloidal Coomassie blue staining) and identified 21 mitochondrial proteins by mass spectrometry using LC-QIT-MS/MS. The abundance of these proteins was quantified by PDQuest v7.0 imaging analysis (Fig. 3B and Table 2) and we found that two of these proteins were significantly decreased (p<0.05; two-tailed independent two homoscedastic samples Student’s t-test) in PBMC mitochondrial lysates of FRDA patients (Table 2). These proteins corresponded to the mitochondrial HSP60 (HSPD1) and HSP70 (HSPA9B).
Fig. 3.

A. Purification of the mitochondria from peripheral blood mononuclear cells and assessment of their purity by western blot. Almost all of the mitochondrial proteins (DLAT, CoxVa, SOD2) are enriched in the mitochondrial pellet extract (MP) of mononuclear cells, while cytosolic proteins (LDH, GAPDH) are markedly decreased or absent in this extract. B. A representative 2-DE gel (IPG strip 3–10NL, 11 cm; 11% SDS-PAGE; Colloidal Coomassie blue staining) of mitochondrial proteins (200 ug) from PBMCs. Protein identification was performed using LC-QIT-MS/MS.
Table 2.
Identities of 2-DGE-separated mitochondrial proteins from PBMCs mitochondrial lysates. Ratio=[averaged spot abundance in FRDA’s gels]/[averaged spot abundance in NHV’s gels].
| Gene | Protein name | Access # | Theoretical
|
Normal PBMCs
|
FRDA PBMCs
|
Ratio | p-value | |||
|---|---|---|---|---|---|---|---|---|---|---|
| p/ | Mr | Average (n=) | SD | Average (n=) | SD | |||||
| HSPD1 | Heat shock protein 60 kDa | P10809 | 5.7 | 61.1 | 1.278 | 0.179 | 0.549 | 0.139 | 0.4 | 0.01 |
| HSPA9B | Heat shock protein 70 kDa | P38646 | 5.9 | 73.7 | 0.514 | 0.020 | 0.196 | 0.098 | 0.4 | 0.01 |
| ATP5A1 | ATP synthase subunit alpha | P25705 | 9.2 | 60.0 | 0.874 | 0.556 | 0.366 | 0.004 | 0.4 | 0.19 |
| ALDH2 | Aldehyde dehydrogenase | P05091 | 6.6 | 56.4 | 0.061 | 0.016 | 0.035 | 0.009 | 0.6 | 0.07 |
| ATP5A1 | ATP synthase subunit alpha | P25705 | 9.2 | 60.0 | 0.197 | 0.049 | 0.118 | 0.049 | 0.6 | 0.12 |
| GLUD1 | Glutamate dehydrogenase 1 | P00367 | 7.7 | 61.4 | 0.197 | 0.049 | 0.118 | 0.049 | 0.6 | 0.12 |
| FH | Fumarase | P07954 | 6.9 | 50.2 | 0.040 | 0.014 | 0.029 | 0.022 | 0.7 | 0.50 |
| ATP5B | ATP synthase subunit beta | P06576 | 5.3 | 56.6 | 1.663 | 0.150 | 1.315 | 0.697 | 0.8 | 0.45 |
| PRDX3 | Peroxiredoxin 3 | P30048 | 7.7 | 27.7 | 0.278 | 0.106 | 0.221 | 0.108 | 0.8 | 0.55 |
| PHB | Prohibitin | P35232 | 5.6 | 29.8 | 0.217 | 0.094 | 0.189 | 0.056 | 0.9 | 0.69 |
| HSD17B | HSD17B10 Hydroxysteroid (17-beta) dehydrogenase | Q99714 | 7.7 | 26.9 | 0.110 | 0.004 | 0.105 | 0.020 | 1.0 | 0.67 |
| PRDX3 | Peroxiredoxin 3 | P30048 | 7.7 | 27.7 | 0.088 | 0.046 | 0.092 | 0.080 | 1.1 | 0.94 |
| C1QBP | Complement component 1 Q subcompo | Q07021 | 4.7 | 31.4 | 0.146 | 0.063 | 0.177 | 0.092 | 1.2 | 0.65 |
| SOD2 | Superoxide dismutase [Mn] | P04179 | 8.4 | 24.7 | 0.359 | 0.010 | 0.447 | 0.207 | 1.2 | 0.50 |
| ACO2 | Aconitase | Q99798 | 7.4 | 85.4 | 0.526 | 0.009 | 0.793 | 0.512 | 1.5 | 0.42 |
| ATP5B | ATP synthase subunit beta | P06576 | 5.3 | 56.6 | 0.068 | 0.013 | 0.124 | 0.092 | 1.8 | 0.36 |
| ATP5B | ATP synthase subunit beta | P06576 | 5.3 | 56.6 | 0.499 | 0.172 | 0.918 | 0.288 | 1.8 | 0.10 |
| ATP5B | ATP synthase subunit beta | P06576 | 5.3 | 56.6 | 0.525 | 0.234 | 1.016 | 0.471 | 1.9 | 0.18 |
| MRPL55 | Mitochondrial ribosomal protein L55 isof | Q7Z7F7 | 11.0 | 18.9 | 0.073 | 0.036 | 0.152 | 0.161 | 2.1 | 0.45 |
| NDUFA6 | NADH dehydrogenase (ubiquinone) 1 | P56556 | 10.0 | 15.1 | 0.038 | 0.011 | 0.152 | 0.087 | 4.0 | 0.09 |
3.3. Analysis of metabolic and functional changes in FRDA mitochondria
Next, we analyzed several mitochondrial enzymatic activities and oxygen consumption to determine if there are differences in FRDA patient blood cells compared to normal cells. Spectrophotometric enzyme analyses showed that the specific activities of cytochrome c oxidase (ETCIV), citrate synthase (CS), succinate dehydrogenase (ETCII) and total superoxide dismutase (SOD) were not affected in either platelets or PBMCs of FRDA patients (Table 3).
Table 3.
Mitochondrial enzymatic activities and basal oxygen consumption of platelets and PBMCs isolated from peripheral blood of normal healthy volunteers and patients with Friedreich ataxia.
| Mithochondrial activities | PBMCs
|
Platelets
|
||
|---|---|---|---|---|
| FRDA | NHV | FRDA | NHV | |
| NADH dehydrogenase (nmol/min/mg prot) | 558±101 | 487±73 | 1830±689 | 1931±404 |
| Complex II (nmol/min/mg prot) | 42.4±15.0 | 50.3±6.9 | 28.3±11.2 | 26.1±11.2 |
| Complex IV (nmol/min/mg prot) | 20±13 | 13±4 | 30±11 | 62±12 |
| Citrade (nmol/min/mg prot) | 45±16 | 46±9 | 60±21 | 62±12 |
| Total SOD (nmol/min/mg prot) | 11.1±3.3 | 10.4±3.1 | 35.8±10.7 | 41.1±10.7 |
| Basal (nmol/min/10+6 or 10+9) | 0.08±0.03 | 0.12±0.05 | 2.8±0.9 | 3.2±1.0 |
Mitochondrial oxygen consumption of platelets and PBMCs measured using a oxygen electrode also was not altered in FRDA patients (Table 3). Furthermore, no significant differences in the basal rate of oxygen consumption were observed, regardless of patient treatment or disease severity phenotype. Finally, we also assessed apoptosis of patient mononuclear cells and lymphocytes by flow cytometry using Annexin V and 7-AAD and observed no differences in the level of apoptosis in patient cells as compared to normal volunteers (Fig. 4).
Fig. 4.

Flow cytometric analyses of apoptosis level of normal (n=6; black bar) and patient (n=8; gray bar) PBMCs and gated lymphocytes using Annexin V assay.
4. Discussion
We performed a comprehensive characterization of blood cell mitochondria from patients with FRDA by assessing the consequences of frataxin deficiency on heme biosynthesis (cytochrome c), heme-dependent proteins (ETC complex IV), non-heme iron-sulfur cluster proteins and iron-binding proteins (aconitase, SDH), oxidative stress (SOD), mitochondrial proteome, apoptosis, and overall mitochondrial function (mitochondrial enzyme activities, oxygen consumption) in mononuclear cells and platelets from FRDA patient blood. The most significant difference observed in this study was the decrease of FRDA protein level in both platelets and PBMCs of FRDA patients. All patient samples tested showed a dramatic decrease in the amount of frataxin observed of less than 30% of the normal level, consistent with observations in whole blood and buccal cells (Deutsch et al., 2010). We observed a partial overlap of frataxin levels between some patients and controls that, over time, this unaffected tissue may select for cells with the minimum defect and producing most frataxin in patients. Most frataxin-deficient blood cells are cleared by the hematopoiesis. This might also attribute to late-onset and less severely affected FRDA patients, or reflect induced level of frataxin as observed with certain treatments. Interestingly, while the FXN level was 2.5-fold higher in PBMCs compared to platelets, the platelet FXN level reflects better the whole blood level, indicating the need for PBMC fractionation to study FXN in the hematopoietic system as well as to monitor any therapeutic response in clinical trials for this disease. However, with this relatively small cohort size, it was not possible to establish a correlation between the level of frataxin in platelets and PBMCs with any clinical data (age, gender, GAA repeats length, treatments) using Western blot and lateral-flow immunoassay (data not shown) as opposed to what has been demonstrated in buccal cells (Deutsch et al., 2010).
From the proteomic, metabolic and functional screen of FRDA blood mitochondria, we also observed a decrease in two mitochondrial HSPs (Hsp60/70). Frataxin directly interacts with numerous proteins associated with iron homeostasis and iron-sulfur cluster biogenesis, including aconitase, ferrochelatase, ISD11 protein of the NFS1/ISCU complex and multiple mitochondrial chaperones (Goldberg et al., 2008; Shan et al., 2007; Sun et al., 2003). Both mitochondrial Hsp60/70 are mostly involved into the mitochondrial protein transport and have important connections to Fe-S cluster assembly. Hsp60 has been implicated in folding and Fe-S cluster insertion into aconitase (Horwich Cell). Hsp 70 has been associated with the Fe-S cluster assembly as part of the machinery that releases Fe-S clusters from the scaffolds. Our proteomic results from FRDA mitochondria differ from those of the two other proteomic studies published to date using whole lysates of frataxin KO mouse heart and total lysates of yhf1Δ; mutant yeast (Sutak et al., 2008; Irazusta et al., 2006). These investigators observed both a marked rearrangement of proteins associated with energy metabolism (presumptively to compensate for the decreased expression of several electron transport proteins) and an up-regulation of enzymes involved in cellular stress response and defenses. The most striking difference between these studies is the absence of marked up-regulation of MnSOD in frataxin-deficient mouse heart (consistent with our measurements of total cellular SOD levels in patient PBMC mitochondria) compared to frataxin-deficient yeast. Both of these models are characterized by complete frataxin deficiency, which contrasts with the more limited reductions noted in FRDA patients.
Only a few studies have shown significant differences in markers from serum, blood or peripheral blood cells from FRDA patients. A recent publication reported a significant decrease of electron transport chain complex I (ETCI) activity and ATP content in FRDA patient lymphocytes (Heidari et al., 2009). Interestingly, they also detected a large deletion in mtDNA in 75% of FRDA patients, which affected several mitochondrial ETC genes. However, ETCI activity was assessed in this study as NADH:ferricyanide oxidoreductase which measures only the NADH dehydrogenase portion of complex I and does not measure flux through the entire complex as does rotenone-sensitive NADH:ubiquinone oxidoreductase. A reduction of free glutathione levels in serum, a significant increase of glutathione bound to hemoglobin in erythrocytes and increase of oxidative stress markers (urinary 8-hydroxydeoxyguanosine and serum peroxides) have also reported (Piemonte et al., 2006; Boesch et al., 2008). More recently, Haugen et al. (2010) have observed down-regulation of genes involved in immune response, oxidative phosphorylation and protein synthesis by transcription profiling and increase of DNA damage in blood DNA from patients.
With the exception of these reports, other major investigations have observed normal protein levels and activities in serum, blood, and platelets of patients with Friedreich ataxia, including serum iron and ferritin concentrations, lactate and pyruvate concentrations, cholesterol, apolipoproteinemia, Vitamin E, very long chain fat acid concentrations, arylsulfatase, hexosaminidase activities and pyruvate dehydrogenase complex activities (Stumpf and Parks, 1981; Constantopoulos et al., 1980). Blood counts (Hgb, wbc, differential wbc count, platelet count) in FRDA’s are also normal. Similar to almost all observations published to date, our data suggest that frataxin deficiency in blood cells does not lead to severe pathogenic phenotype observed in affected neurons and cardiomyoctes of Friedreich ataxia patients. Several explanations may account for this observation: i) the specific role of frataxin in blood and/ or the residual amount of frataxin may be sufficient to maintain iron homeostasis, mitochondrial metabolism and ATP generation by oxidative phosphorylation; ii) compensatory mechanisms specific to blood cells, such as a significant expression of mitochondrial ferritin, may offset frataxin deficiency; iii) ATP synthesis by glycolysis may provide adequate energy levels for quiescent platelets and PBMCs under resting (i.e. non-stimulated) conditions, whereas very metabolically active tissues such as cardiac myocytes and neurons are particularly vulnerable to defects which negatively impact aerobic metabolism and mitochondrial ATP synthesis. Mitochondrial defects are well established to be preferentially manifest in tissues that are most reliant on mitochondrial respiratory capability. It is thus highly relevant that several studies have observed mitochondrial defects in cultured lymphoblasts, which are transformed, metabolically active and proliferating cells derived from patient PBMCs (Napoli et al., 2006). This suggests that the ‘parent’ cells from which such transformed lymphoblast lines are derived may harbor a sub-threshold mitochondrial defect resulting from frataxin deficiency but its manifestation is too subtle to be readily detectable in the resting state in these cells.
Given the need to assess new therapeutic agents in development and those being used in clinical trials, comprehensive and clinically useful blood biomarkers would be highly beneficial to monitor drug activity in FRDA patients. Performing a metabolite profiling and functional screening of platelets from blood samples obtained from children with FRDA would be a useful approach to achieve this objective. In addition, treatments with ROS-generating agents and mitochondrial inhibitors, which induce oxidative/metabolic stress, would be expected to amplify and unmask sub-threshold mitochondrial defects in these terminally differentiated and resting blood cells from patients. Understanding the function of frataxin in blood cell mitochondria represents a biochemical dilemma, which clearly merits further investigation. It remains an open question why the frataxin deficiency in these cells (as outlined by this work) does not induce the same pathogenic phenotype observed in other tissues such brain or heart, as otherwise these patients would be anemic and exhibit major hematological deficiencies. The answer to this question will be important for understanding the tissue specificity of FRDA, to providing useful and relevant blood biomarkers for monitoring the drug responses, and to proposing therapies using hematopoietic stem cells. Finally, while the tissue specificity of FRDA and the use of blood as biomarker in this disease are still questioning, this study might be quite relevant to blood diseases associated with mitochondrial dysfunction (e.g. Pearson’s large mtDNA deletion with sideroblastic anemia, Bart’s cardiolipin deficiency associated with neutropenia…).
Acknowledgments
We thank the patients and health donors who participated in this study as well as Lisa Friedman, Prof. Robert Wilson, Dr. Jennifer Farmer, Dr. Mary Kearney and Marianne Kerdougli. This work was supported by a grant from NIH GM38237 (F.D.), EurAtaxia and Association Francaise Ataxie de Friedreich (J-E.S.), MDA and Friedreich Ataxia Research Association (D.L.).
Abbreviations
- 2-DGE
two-dimensional gel electrophoresis
- ETCI
electron transport chain complex I
- FRDA
Friedreich ataxia
- FXN
frataxin
- FXN
frataxin gene
- HSP
heat shock protein
- NHV
normal healthy donor
- PBMC
peripheral blood mononuclear cell
References
- Azzi A, Montecucco C, Ricter C. The use of acetylated ferricytochrome c for the detection of superoxide radical produced in biological membranes. Biochem Biophys Res Commun. 1975;65:597–603. doi: 10.1016/s0006-291x(75)80188-4. [DOI] [PubMed] [Google Scholar]
- Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of Frataxin. Science. 1997;276:1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- Boesch S, Sturm B, Hering S, Scheiber-Mojdehkar B, Steinkellner H, Goldenberg H, Poewe W. Neurological effects of recombinant human erythropoietin in Friedreich’s ataxia: a clinical pilot trial. Mov Disord. 2008;23:1940–1944. doi: 10.1002/mds.22294. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani S, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Chantrel-Groussard K, Geromel V, Puccio H, Koenig M, Munnich A, Rotig A, Rustin P. Disabled early recruitment of antioxidant defenses in Friedreich ataxia. Hum Mol Gen. 2001;10 (19):2061–2067. doi: 10.1093/hmg/10.19.2061. [DOI] [PubMed] [Google Scholar]
- Condò I, Ventura N, Malisan F, Rufini A, Tomassini B, Testi R. In vivo maturation of human frataxin. Hum Mol Genet. 2007;16 (13):1534–1540. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- Constantopoulos G, Chang CS, Barranger JA. Normal pyruvate dehydrogenase complex activity in patients with Friedreich’s ataxia. Ann Neurol. 1980;8:636–639. doi: 10.1002/ana.410080618. [DOI] [PubMed] [Google Scholar]
- Deutsch EC, Santani AB, Perlman SL, Farmer JM, Stolle CA, Marusich MF, Lynch DR. A rapid, noninvasive immunoassay for frataxin: utility in assessment of Friedreich ataxia. Mol Genet Metab. 2010;101:238–245. doi: 10.1016/j.ymgme.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foury F, Cazzalini O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997;411:373–377. doi: 10.1016/s0014-5793(97)00734-5. [DOI] [PubMed] [Google Scholar]
- Goldberg AV, Molik S, Tsaousis AD, Neumann K, Kuhnke G, Delbac F, Vivares CP, Hirt RP, Lill R, Embley TM. Localization and functionality of microsporidian iron-sulphur cluster assembly proteins. Nature. 2008;452 (7187):624–628. doi: 10.1038/nature06606. [DOI] [PubMed] [Google Scholar]
- Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN, Halweg C, Collins JB, Durr A, Fischbeck K, Van Bouten B. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PLoS Genet. 2010;15 (61):e1000812. doi: 10.1371/journal.pgen.1000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari MM, Houshmand M, Hosseinkhani S, Nafissi S, Khatami M. Complex I and ATP content deficiency in lymphocytes from Friedreich’s ataxia. Can J Neurol Sci. 2009;36:26–31. doi: 10.1017/s0317167100006260. [DOI] [PubMed] [Google Scholar]
- Hoffman R, Benz E, Shattil S, Furi B, Cohen H. Hematology: basic principles and practice. 3. Elsevier; Philadelphia: 2000. [Google Scholar]
- Irazusta V, Cabiscol E, Reverter-Branchat G, Ros J, Tamarit J. Manganese is the link between frataxin and iron-sulfur deficiency in the yeast model of Friedreich ataxia. J Biol Chem. 2006;281 (18):12227–12232. doi: 10.1074/jbc.M511649200. [DOI] [PubMed] [Google Scholar]
- Kuthan H, Hausman HJ, Weringloer J. A spectrophotometric assay for superoxide dismutase activities in crude tissue fractions. Biochem J. 1986;237:175–180. doi: 10.1042/bj2370175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli E, Taroni F, Cortopassi GA. Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid Redox Signal. 2006;8:506–516. doi: 10.1089/ars.2006.8.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfo M. Iron metabolism and mitochondrial abnormalities in Friedreich ataxia. Blood Cells Mol Dis. 2002;29 (3):536–552. doi: 10.1006/bcmd.2002.0591. [DOI] [PubMed] [Google Scholar]
- Piemonte F, Pastore A, Tozzi G, Tagliacozzi D, Santorelli FM, Carrozzo R, Casali C, Damiano M, Federici G, Bertini E. Glutathione in blood of patients with Friedreich’s ataxia. Eur J Clin Invest. 2006;31:1007–1011. doi: 10.1046/j.1365-2362.2001.00922.x. [DOI] [PubMed] [Google Scholar]
- Pon LA, Schon EA. Mitochondria. Methods in Cell Biol. 2001;65:809–812. doi: 10.1016/s0091-679x(01)65010-6. [DOI] [PubMed] [Google Scholar]
- Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17 (2):215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Sarry JE, Kuhn L, Le Lay P, Garin J, Bourguignon J. Dynamics of Arabidopsis thaliana soluble proteome in response to different nutrient culture conditions. Electrophoresis. 2006;27:495–507. doi: 10.1002/elps.200500561. [DOI] [PubMed] [Google Scholar]
- Shan Y, Napoli E, Cortopassi G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum Mol Genet. 2007;16 (8):929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- Stumpf DA, Parks JK. Human mitochondrial electron transport chain: assay of succinate: cytochrome c reductase in leukocytes, platelets and cultured fibroblasts. Biochem Med. 1981;25:234–238. doi: 10.1016/0006-2944(81)90080-6. [DOI] [PubMed] [Google Scholar]
- Sun G, Gargus JJ, Ta DT, Vickery LE. Identification of a novel candidate gene in the iron-sulfur pathway implicated in ataxia-susceptibility: human gene encoding HscB, a J-type co-chaperone. J Hum Genet. 2003;48 (8):415–419. doi: 10.1007/s10038-003-0048-9. [DOI] [PubMed] [Google Scholar]
- Sutak R, Xu X, Whitnall M, Kashem MA, Vyoral D, Richardson DR. Proteomic analysis of hearts from frataxin knockout mice: marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics. 2008;8 (8):1731–1741. doi: 10.1002/pmic.200701049. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts and transmitochondrial cell lines. Meth Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- Wilson DF. Effect of temperature on the spectral properties of some ferrocytochromes. Arch Biochem Biophys. 1967;121:757–768. doi: 10.1016/0003-9861(67)90065-3. [DOI] [PubMed] [Google Scholar]
- Wilson RB, Roof DM. Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat Genet. 1997;16:352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]