

The gut microbiota is known to have a considerable impact on health; some types of organisms, including bifidobacteria and lactobacilli, are considered to be ‘good’ colonizing bacteria, while other species are potentially pathogenic. The initial colonization of a newborn infant affects his/her health in the future. Recently, research has indicated that the fetus may actually be colonized with bacteria during gestation. Accordingly, this study was conducted to assess the similarities in the bacterial species detected in the infant’s first feces with samples obtained from the mother’s placenta and vagina.
Keywords: Bacterial community, Neonates’ feces, Placenta, Vagina
Abstract
BACKGROUND:
The gut microbiota plays an important role in human health. It is essential to understand how the composition of the gut microbiota in neonates is established.
OBJECTIVES:
To investigate the nature of the microbial community in the first feces of newborn infants compared with the mothers’ placentae and vaginas.
METHODS:
One infant who was delivered via Cesarean section was compared with an infant who was delivered vaginally. Bar-coded pyro-sequencing of 16S ribosomal RNA genes was used to investigate the bacterial community composition and structure of each site.
RESULTS:
Neonatal feces of both infants had similar bacterial communities, and they were similar to the mother’s placenta regardless of the method of delivery. The vaginal bacterial community differed between the two mothers, but not different sites within the vagina. The bacteria in the neonatal feces and the mothers’ placentae demonstrated considerably higher diversity compared with the vaginas. The family Lactobacillaceae dominated in the vaginal samples, while the most abundant family in the fecal and placental samples was Micrococcineae.
CONCLUSIONS:
These results may provide new directions for the study of infant gut microbial formation.
Abstract
HISTORIQUE :
Le microbiote intestinal joue un rôle important dans la santé humaine. Il est essentiel de comprendre comment il s’établit chez les nouveau-nés.
OBJECTIFS :
Examiner la structure et la composition de la communauté microbienne des premières fèces des nouveau-nés par rapport à celles du placenta et du vagin de la mère.
MÉTHODOLOGIE :
Les chercheurs ont comparé un nourrisson né par césarienne à un nourrisson né par voie vaginale. Ils ont utilisé le pyroséquençage à code-barres des gènes d’ARN ribosomique 16S pour examiner la composition et la structure de la communauté bactérienne de chaque foyer.
RÉSULTATS :
Les fèces des nouveau-nés contenaient des communautés bactériennes similaires, qui étaient également similaires à celles du placenta de la mère, quel que soit le mode d’accouchement. La communauté bactérienne vaginale n’était pas la même chez les deux mères, mais étaient similaires dans les différents foyers du vagin. Les bactéries contenues dans les fèces néonatales et le placenta de la mère ont démontré une beaucoup plus grande diversité que celles des vagins. La famille de Lactobacillaceae dominait dans les échantillons vaginaux, tandis que la famille des Micrococcineae était plus abondante dans les échantillons fécaux et placentaires.
CONCLUSIONS :
Ces résultats fournissent de nouvelles voies pour étudier la formation de la flore microbienne intestinale du nourrisson.
The gut microbiota harbours a vast array of microbes and provides important metabolic capabilities (1–4). Thus, it is important to understand how the composition of the gut microbiota in neonates is established. Bifidobacteria and lactobacilli are considered to be the most important health-beneficial bacteria for the human host, whereas bacteria such as staphylococci and clostridia are potential pathogens (5,6).
For a long time, the healthy human fetus was believed to develop in a bacteria-free environment, and the presence of bacteria was considered to be a serious threat to the growing fetus; however, this is no longer considered to be correct. The presence of bacteria may lead to inflammation, but there are some bacteria whose presence does not appear to harm the developing fetus (7). Bacterial infection is a frequent cause of preterm labour, and neonatal sepsis was believed to be caused by infections that that originate during pregnancy or perinatally (8,9). However, it has now become apparent that bacteria are quite often present in the placenta in normal pregnancies. In one study that examined 195 placentas, intracellular bacteria were detected in the placental basal plate in 27%; bacteria were present in placentas from preterm as well as from full-term gestations (10). Neonates are exposed to a wide variety of microbes, which are mostly provided by the mother during and after the passage through the birth canal (11). Today, many human infants are not exposed to vaginal microbes at birth because they are delivered by Cesarean section. In previous studies, researchers focused on the effects of these delivery methods. Gronlund et al (12) demonstrated that the establishment of primary gut flora in infants born by Cesarean section was delayed. Furthermore, as supported by other studies, the infant gut microbiota largely reflects the delivery modes (5,11,13). In another study, Dominguez-Bello et al (11) found that neonates harboured bacterial communities that were undifferentiated across multiple body habitats, regardless of the method of delivery; these results also showed that the mother’s vaginal and skin surface microbiota affected neonatal gut bacterial communities. At the same time, Trosvik et al (4) described the infant gut ecological dynamic using a combination of nonlinear data modelling and simulations of the early infant gut colonization processes, and confirmed that different delivery modes did not affect the phylum composition in the infant gut. These studies focused on the bacterial communities of the mother’s vagina or skin.
The aim of the present study was to investigate whether the microbial communities of the first feces of neonates were similar to the mothers’ vaginal or placental communities. Samples were taken from two sites in the mothers’ vaginas, as well as from the placentae and the first feces of neonates. The composition and structure of the bacterial communities were analyzed using bar-coded pyrosequencing by amplifying the V3–V6 regions of the bacterial 16S ribosomal RNA (rRNA) gene.
METHODS
Sampling
Specimens were collected one day before childbirth by wiping the posterior and side wall of the vagina with sterile cotton swabs. The placentas were collected after childbirth in delivery room or operating room to ensure sterility. The placenta samples were collected using sterile centrifuge tubes immediately after delivery of the placenta. To avoid the placenta being contaminated by the vagina, samples were collected from the inner surface of the placenta. The first feces of the neonates were also collected in the delivery room using sterile cotton swabs. All samples were immediately stored at −20°C. Sterile cotton swabs in the same environment were also analyzed as negative controls. Both mothers were 29 years of age and neither had experienced bacterial vaginosis during pregnancy. Both mothers and neonates did not receive any antibiotic treatment. Mother 1 delivered vaginally and mother 2 via Cesarean section. Ethics approval was granted by the First People Hospital of Yunnan Province, Yunnan, China.
DNA extraction and purification
Samples were stored at −20°C before DNA extraction. The cotton swabs were used for DNA extraction. Total DNA was extracted from 1 g samples of placenta or feces, or one cotton swab as previously described (14), with minor modifications. Briefly, samples were simultaneously treated with lysozyme (1 mg/mL) and lyticase (0.16 mg/mL). Subsequently, samples were treated with sodium dodecyl sulfate (1%) and cetrimonium bromide (1%). Three liquid nitrogen freeze/thaw cycles were also performed to ensure the homogeneity of lysed cell samples. The concentration of extracted DNA was determined using a spectrophotometer (NanoDrop ND 1000, Thermo Fisher, USA).
Polymerase chain reaction amplification and pyrosequencing
In all polymerase chain reaction (PCR) amplifications, reactions were performed with rTaq MasterMix (TaKaRa, China) with a total volume of 50 μL and approximately 50 ng DNA template. A modified primer set (338F and 907R) was generated according to a metagenomic database (15). All PCR programs consisted of an initial 5 min denaturation at 95°C, followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 53°C for 1 min, extension at 72°C for 1.5 min and a final elongation step of 10 min at 72°C. To obtain similar numbers of sequences from each sample, an equivalent amount of each purified PCR product was mixed for sequencing using the Genome Sequencer FLX System (Roche, Switzerland). Sequences were preprocessed to assess the overall quality, and classified using bar codes and primer sequences. All pyrosequencing samples were extracted using the specified bar code for each sample. Sequences containing ‘N’ or those <350 bp were excluded.
Classification and phylogenetic analysis
Classification information of all 16S rRNA gene sequences was determined with the Silva v108 database using the mothur program v.1.25.1 (www.mothur.org/wiki/Main_Page). The operational taxonomic units (OTUs) of the 16S rRNA gene sequences were determined using a 3% cut-off. To determine the phylogenetic position of the 16S rRNA genes, sequences were compared with available database sequences via a BLAST search; related sequences were obtained from the National Center for Biotechnology Information nonredundant database. The taxonomic information was further confirmed by the online analysis tool EzTaxon (16). Phylogenetic trees were constructed via MEGA version 4.0 (17) using the neighbour-joining method (18).
Diversity index analysis
Richness and diversity statistics were calculated using the mothur program v.1.25.1, including the abundance-based coverage estimator (ACE) (19,20) and the bias-corrected Chao1 (21). The estimated coverage of the 16S rRNA gene sequences was calculated as:
in which n1 is the number of singleton sequences, and N is the total number of sequences (22).
Community analysis
Microbial community similarity analyses were conducted using the weighted pair group method with arithmetic averages (WPGMA) clustering and principal component analysis (PCA) using the online UNIFRAC program (23), which measures the molecular evolutionary distances of the sequences and is able to compare the relationships among microbial communities.
Nucleotide sequences accession numbers
The sequences were available in the short read archive of the National Center for Biotechnology Information, using the accession number SRA060045.
RESULTS
Analysis of OTU richness
In the eight samples obtained from the two mothers/neonates, a total of 4967 (mean [± SD] sequence numbers 621±268) pyrosequencing samples were used for analysis. The estimated OTU numbers according to ACE and Chao1 were generated to allow for 3% sequence dissimilarities of 16S rRNA gene (Table 1). Based on a 97% cutoff, the numbers of the estimated OTUs ranged from 10 to 87, with an average of 43. The higher OTU numbers appeared in both placental samples. The OTU numbers in four samples from mother 2 were higher than those in mother 1. In all samples, the estimated coverage was >90%. The ACE and Chao1 estimates were calculated to compare the diversities and richness of the bacterial communities. Those indexes showed higher bacterial genotype diversities in fecal and placental samples compared with those recovered in vaginal samples in the present study.
TABLE 1.
Analysis of operational taxonomic units (OTUs) in eight samples
| Sample | Sequences, n | Average sequence length, bp | OTUs, n, 97% (99%) | Singletons | Coverage, % | ACE, n, 97% (99%) | Chao1, n, 97% (99%) |
|---|---|---|---|---|---|---|---|
| Mother 1* | |||||||
| Vagina side | 405 | 558 | 12 (55) | 6 | 99 | 20 (1673) | 16 (619) |
| Vagina posterior | 557 | 551 | 10 (75) | 6 | 99 | 37 (700) | 18 (336) |
| Placenta | 1001 | 533 | 87 (269) | 57 | 94 | 395 (2287) | 187 (1040) |
| Infant’s first feces | 296 | 532 | 34 (91) | 25 | 92 | 103(554) | 77 (383) |
| Mother 2† | |||||||
| Vagina side | 668 | 558 | 39 (130) | 24 | 96 | 141 (1038) | 74 (511) |
| Vagina posterior | 1035 | 565 | 49 (185) | 32 | 97 | 263 (1578) | 120 (773) |
| Placenta | 498 | 532 | 71 (179) | 42 | 92 | 211 (1227) | 133 (584) |
| Infant’s first feces | 507 | 531 | 41 (124) | 29 | 94 | 274 (1101) | 99 (464) |
ACE Abundance-based coverage estimator;
Vaginal delivery;
Cesarean section delivery
Rarefaction analysis was then performed for each sample (Figure 1). This analysis clearly showed that in both mothers/neonates, placental samples showed the highest diversity, followed by fecal samples.
Figure 1).
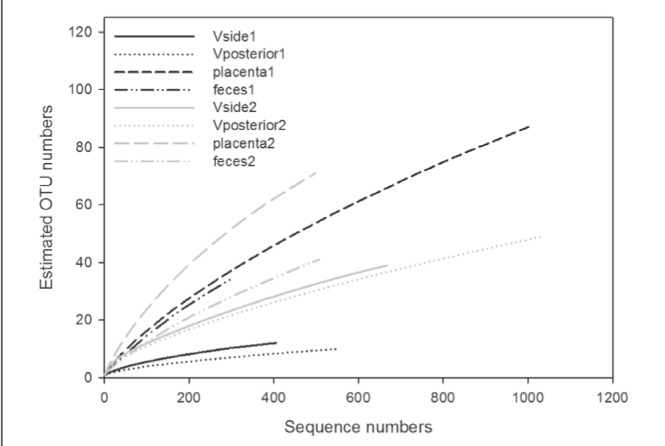
Rarefaction curves of the pyrosequencing sequences in samples from the mothers and neonates. The phylotypes were determined using a 97% similarity cutoff. OTU Operational taxonomic unit; V Vagina
Comparison of bacterial communities between neonatal first feces and mothers’ vaginas and placentae
To investigate the establishment of neonatal gut bacterial communities, the bacterial communities of each mother’s vagina and placenta were compared with the first feces of their neonates. The WPGMA tree showed that the bacteria in fecal and placental samples were distinct from those in the vaginal samples (Figure 2). Furthermore, fecal bacteria were more similar between the two infants than with each respective mother’s placenta, while bacteria in the vaginal samples showed individual characteristics.
Figure 2).
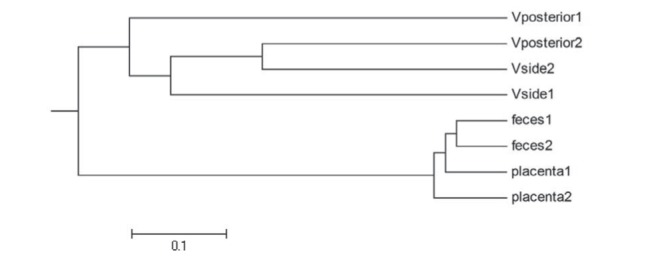
Weighted pair group method with arithmetic averages (WPGMA) cluster of the samples from the mothers and neonates. V Vagina
Figure 3 presents the PCA plots of the comparison of bacterial communities. These results clearly confirmed the WPGMA cluster. In the PCA analysis, the bacterial communities in fecal and placental samples were very similar.
Figure 3).
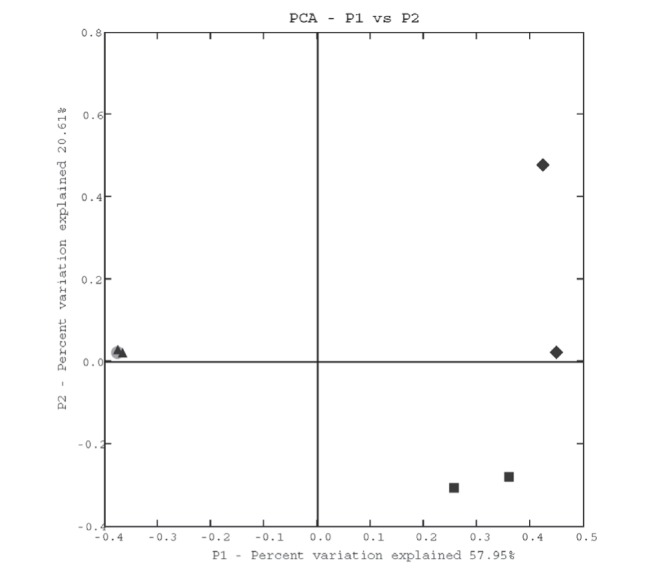
Principal coordinate analysis (PCA) plots based on UniFrac analysis of bacterial communities. The scatterplots were for the first two principal components. Diamond: vaginal samples from mother 1; square: vaginal samples from mother 2; triangle: fecal and placenta samples from mother/neonate 1; circle: fecal and placenta samples from mother/neonate 2
Taxonomic identification and phylogenetic analysis
Given the robust evidence that the bacterial community structures of the neonatal gut were similar to the mother’s placenta (Figures 1 and 2), the authors sought to identify the bacterial taxa that were contributing to this environment. According to the Silva taxonomic information, a total of eight phyla were detected in the present study, of which only four were detected in vaginal samples. More than 90% of the sequences could be assigned to the family level, and 37 families were observed (Figure 4). The major families in the vaginal samples were completely different from those in the feces and placental samples. In vaginal samples, the majority of 16S rRNA gene sequences belonged to the family Lactobacillaceae, followed by Micrococcineae. Approximately 1% of the sequences from the vagina of mother 2 were assigned to the family Bifidobacteriaceae, which was not detected in mother 1. As in the placental and fecal samples, the proportions of the families Lactobacillaceae and Micrococcineae were opposite to those in vaginal samples. In addition to the families mentioned above, the families Comamonadaceae, Pseudomonadaceae and Enterobacteriaceae were also found in placental and fecal samples.
Figure 4).
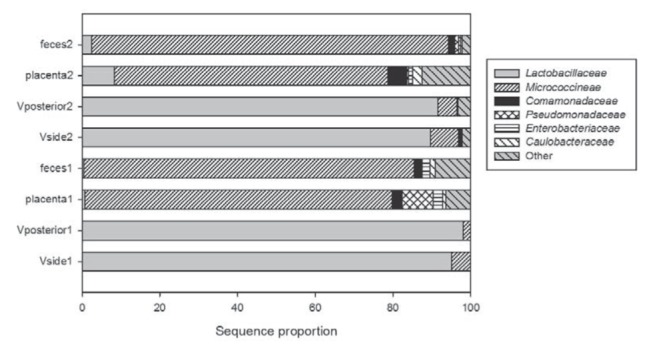
The sequence proportion of the major families (>1%) in samples from the mothers and neonates. V Vagina
A total of 343 OTUs from eight samples were identified based on a 3% cut-off. OTUs containing >1% of sequences in each sample were used to construct the phylogenetic tree (Figure 5). In vaginal samples, the most abundant OTUs in the samples from the two mothers were all similar to Lactobacillus crispatus (NR_041800), among which >90% of 16S rRNA gene sequences could be assigned to this group (OTUs Vside1_1, 96% and Vbehind1_1, 93%) in the vaginal samples from mother 1. In vaginal samples from mother 2, these proportions were 65% and 70%, respectively. In vaginal samples from mother 2, OTUs (Vside2_3, 8% and Vbehind2_2, 16%) were similar to Lactobacillus species detected in human vaginal samples. Twelve percent of sequences could be assigned to the OTU Vside2_2 with a 98% similarity to Lactobacillus hominis (FR681902). Placental and fecal samples showed similar bacterial community compositions. The OTUs of placenta1_1 (77%), placenta2_1 (68%), feces1_1 (84%) and feces2_1 (86%) showed more than 97% similarity to one another; these were assigned to Cellulosimicrobium species via phylogenetic analysis. Only one OTU, placenta2_3 (6%), was similar to the most abundant OTUs in the vaginal samples. The OTU feces2_3 (2%) was similar to Lactobacillus iners (NR_036982). All sequences were then classified using the online analysis tool EzTaxon (16). Approximately 3% of the 16S rRNA gene sequences were <97% similar to those of cultured bacteria.
Figure 5).

Phylogenetic tree of the 16S ribosomal RNA gene sequences (>1%) and their phylogenetic relatives. The tree was constructed using the neighbour-joining method. Numbers on nodes indicate bootstrap values (1000 resamplings). The scale bar indicates 0.05 nucleotide substitutions per nucleotide position. The operational taxonomic units obtained in the present study are shown in bold. V Vagina
DISCUSSION
The importance of human gastrointestinal microbiota and the establishment of gut microbiota in early infancy has received considerable attention in recent years (3,4,6,12,24,25). It is now known that fetal development does not occur in a sterile environment. Thus, the first bacterial colonization of the fetus may be from the placenta during gestation. A recent study characterized a low-abundance but metabolically rich microbiome composed of nonpathogenic commensal microbiota, which were most similar to the human oral microbiome (26). In this study, 16S rRNA gene-based OTU analyses revealed associations of the placental microbiome with a remote history of antenatal infection, such as urinary tract infection in the first trimester, as well as with preterm birth (26). Many studies have focused on the effects of the delivery modes on the gut microbiota of infants (11,12); the delivery mode was found to have no effect on the formation of infant gut microbiota in many studies (11,13). These studies provided rich information on the formation of the infant gut microbiota, and also confirmed that the developmental environment of the fetus was not sterile. In the present study, an analysis using bar-coded pyrosequencing was conducted to compare the bacterial communities among the first feces of neonates and the placentas and vaginas of the mothers who had given birth through either vaginal delivery or Cesarean section. Similar to previous studies, we found that the environment in which a human fetus develops is not sterile, and the bacterial specimens in the first feces of neonates were similar to the mothers’ placentae regardless of the method of delivery. The results provided useful information for further studies investigating the relationship between the neonatal gut microbiota and the mothers’ vaginal or placental communities.
Our results also revealed that placental bacterial communities had a higher diversity than vaginal bacterial communities (Figure 1), and the read numbers that were obtained may be sufficient to elucidate all of the bacterial communities in the placentae in the present study, according to the rarefaction curves and coverage. The bacterial communities in the first feces of the neonates also showed higher diversity than those of the vaginas.
The bacterial community composition indicated that the bacteria in the feces may have come from the mothers’ placentae. However, the bacterial composition in fecal and placental samples were unlike either of the vaginas (27–29) or infant fecal samples from previous studies (4,12,13,30). The neonatal first feces and mothers’ placentae had their own unique bacterial community composition. In vaginal bacterial communities, lactobacilli are recognized as bioindicators in healthy women, and bacterial vaginosis is characterized by a loss of most Lactobacillus species (31,32). In the fecal samples in the present study, only 2% of sequences of neonate 2 were classified as L iners strain DSM 13335 (NR_036982) (33), which was first isolated from human urine. In addition, 6% of placental sequences from neonate 2 could be assigned to L crispatus strain ATCC33820 (NR_041800). Both of these strains were recognized as normal flora in a healthy vagina (27,34). In previous studies investigating the infant fecal bacterial community, the phyla Firmicutes and Bacteroides were recognized as dominant in the human gut (4,24). However, the majority of fecal and placental bacteria were identified as Cellulosimicrobium cellulans or Cellulosimicrobium funkei belonging to the phylum Actinobacteria, which was considered to have cellulose decomposition capabilities (35) as well as alkaline protease production (36). Human placentae provide nutrition and food to fetuses in utero, and bacterial communities may be one sources of these nutrients. Similar to other vaginal studies (27–29,31,32,34,37), the vaginal bacterial community composition observed in the present study was dominated by Lactobacillus species.
Because the results of the present study have further confirmed that the fetal developmental environment is not sterile, we compared the bacterial communities among the first feces of neonates and the mothers’ vaginas and placentae to understand the origin of the microbial communities in the first feces of neonates. There were some limitations to the present study. The lower number of samples compared with other studies investigating human vaginal or fecal bacterial communities limited the results of the present study. In future studies, a sufficient number of samples should be analyzed to compare the influencing factors of placental and neonatal fecal bacteria composition and structures. In addition, many studies have shown that even full-length 16S rRNA gene sequences were not suitable to identify sequences at the species level, especially in the family Lactobacillaceae (5,38). In the present study, similar results were observed; 16S rRNA gene sequences could be only identified accurately at the genus level. Thus, in future studies, a combination of other molecular markers is necessary to clarify the exact bacterial composition and community succession. However, our results have demonstrated that the family Lactobacillaceae is not the dominant bacteria in the first feces of neonates; thus, the role of the family Micrococcineae in the neonatal gut should receive more attention in future studies. It would be interesting to confirm whether Micrococcineae lead to preterm labour and neonatal sepsis, or are helpful for the reconstruction of neonatal intestinal flora.
REFERENCES
- 1.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A. 2002;99:15451–5. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–5. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li M, Wang B, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A. 2008;105:2117–22. doi: 10.1073/pnas.0712038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trosvik P, Stenseth NC, Rudi K. Convergent temporal dynamics of the human infant gut microbiota. ISME J. 2010;4:151–8. doi: 10.1038/ismej.2009.96. [DOI] [PubMed] [Google Scholar]
- 5.Penders J, Thijs C, Vink C, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118:511–21. doi: 10.1542/peds.2005-2824. [DOI] [PubMed] [Google Scholar]
- 6.Rastall RA. Bacteria in the gut: Friends and foes and how to alter the balance. J Nutr. 2004;134:2022S–2026S. doi: 10.1093/jn/134.8.2022S. [DOI] [PubMed] [Google Scholar]
- 7.Wassenaar TM, Panigrahi P. Is a foetus developing in a sterile environment? Lett Appl Microbiol. 2014;59:572–9. doi: 10.1111/lam.12334. [DOI] [PubMed] [Google Scholar]
- 8.Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342:1500–7. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- 9.Heron M. Deaths: Leading causes for 2010. Natl Vital Stat Rep. 2013;62:1–96. [PubMed] [Google Scholar]
- 10.Stout MJ, Conlon B, Landeau M, et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol. 2013;208:226, e1–7. doi: 10.1016/j.ajog.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–5. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gronlund MM, Lehtonen OP, Eerola E, Kero P. Fecal microflora in healthy infants born by different methods of delivery: Permanent changes in intestinal flora after cesarean delivery. J Pediatr Gastroenterol Nutr. 1999;28:19–25. doi: 10.1097/00005176-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Favier CF, de Vos WM, Akkermans AD. Development of bacterial and bifidobacterial communities in feces of newborn babies. Anaerobe. 2003;9:219–29. doi: 10.1016/j.anaerobe.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt TM, DeLong EF, Pace NR. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 1991;173:4371–8. doi: 10.1128/jb.173.14.4371-4378.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao D-P, Zhou Q, Chen C-Y, Quan Z-X. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 2012;12:66. doi: 10.1186/1471-2180-12-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chun J, Lee J-H, Jung Y, et al. EzTaxon: A web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol. 2007;57:2259–61. doi: 10.1099/ijs.0.64915-0. [DOI] [PubMed] [Google Scholar]
- 17.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 18.Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 19.Chao A, Lee SM. Estimating the number of classes via sample coverage. J Am Statist Ass. 1992;87:210–7. [Google Scholar]
- 20.Chao A, Hwang W-H, Chen Y-C, Kuo C-Y. Estimating the number of shared species in two communities. Stat Sin. 2000;10:227–46. [Google Scholar]
- 21.Lee SM, Chao A. Estimating population size via sample coverage for closed capture-recapture models. Biometrics. 1994;50:88–97. [PubMed] [Google Scholar]
- 22.Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40:237–64. [Google Scholar]
- 23.Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–7. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gosalbes MJ, Durbán A, Pignatelli M, et al. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS ONE. 2011;6:e17447. doi: 10.1371/journal.pone.0017447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6:237ra65. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsson T, Forsum U. Lactobacillus iners: A marker of changes in the vaginal flora? J Clin Microbiol. 2007;45:3145–5. doi: 10.1128/JCM.00558-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oakley BB, Fiedler TL, Marrazzo JM, Fredricks DN. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl Environ Microbiol. 2008;74:4898–909. doi: 10.1128/AEM.02884-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aagaard K, Riehle K, Ma J, et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE. 2012;7:e36466. doi: 10.1371/journal.pone.0036466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albesharat R, Ehrmann MA, Korakli M, Yazaji S, Vogel RF. Phenotypic and genotypic analyses of lactic acid bacteria in local fermented food, breast milk and faeces of mothers and their babies. Syst Appl Microbiol. 2011;34:148–55. doi: 10.1016/j.syapm.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimura K, Morotomi N, Fukuda K, et al. Intravaginal microbial flora by the 16S rRNA gene sequencing. Am J Obstet Gynecol. 2011;205:235, e1–9. doi: 10.1016/j.ajog.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 32.Srinivasan S, Hoffman NG, Morgan MT, et al. Bacterial communities in women with bacterial vaginosis: High resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS One. 2012;7:e37818. doi: 10.1371/journal.pone.0037818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falsen E, Pascual C, Sjoden B, Ohlen M, Collins MD. Phenotypic and phylogenetic characterization of a novel Lactobacillus species from human sources: Description of Lactobacillus iners sp. nov. Int J Food Microbiol. 1999;49:217–21. doi: 10.1099/00207713-49-1-217. [DOI] [PubMed] [Google Scholar]
- 34.Vasquez A, Jakobsson T, Ahrne S, Forsum U, Molin G. Vaginal Lactobacillus flora of healthy Swedish women. J Clin Microbiol. 2002;40:2746–9. doi: 10.1128/JCM.40.8.2746-2749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang S, Sheng P, Zhang H. Isolation and identification of cellulolytic bacteria from the gut of Holotrichia parallela larvae (Coleoptera: Scarabaeidae) Int J Mol Sci. 2012;13:2563–77. doi: 10.3390/ijms13032563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferracini-Santos L, Sato HH. Production of alkaline protease from Cellulosimicrobium cellulans. Braz J Microbiol. 2009;40:54–60. doi: 10.1590/S1517-83822009000100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marrazzo JM. Interpreting the epidemiology and natural history of bacterial vaginosis: Are we still confused? Anaerobe. 2011;17:186–90. doi: 10.1016/j.anaerobe.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X-R, Ma E-B, Yan L-Z, et al. Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. Int J Food Microbiol. 2011;146:31–7. doi: 10.1016/j.ijfoodmicro.2011.01.030. [DOI] [PubMed] [Google Scholar]