

Abstract
GluN2B subunit containing NMDARs (GluN2B-NMDARs) mediate pathophysiological effects of acutely applied amyloid beta (Aβ), including impaired long-term potentiation (LTP). However, in transgenic Alzheimer’s disease (AD) mouse models which feature gradual Aβ accumulation, the function of GluN2B-NMDARs and their contribution to synaptic plasticity are unknown. Therefore, we examined the role of GluN2B-NMDARs in synaptic function and plasticity in the hippocampus of PS2APP transgenic mice. Although LTP induced by theta burst stimulation (TBS) was normal in PS2APP mice, it was significantly reduced by the selective GluN2B-NMDAR antagonist Ro25-6981 (Ro25) in PS2APP mice, but not wild type (wt) mice. While NMDARs activated by single synaptic stimuli were not blocked by Ro25, NMDARs recruited during burst stimulation showed larger blockade by Ro25 in PS2APP mice. Thus, the unusual dependence of LTP on GluN2B-NMDARs in PS2APP mice suggests that non-synaptic GluN2B-NMDARs are activated by glutamate that spills out of synaptic cleft during the burst stimulation used to induce LTP. While long-term depression (LTD) was normal in PS2APP mice, and Ro25 had no impact on LTD in wt mice, Ro25 impaired LTD in PS2APP mice, again demonstrating aberrant GluN2B-NMDAR function during plasticity. Together these results demonstrate altered GluN2B-NMDAR function in a model of early AD pathology that has implications for the therapeutic targeting of NMDARs in AD.
Keywords: NMDA receptor, GluN2B, NR2B, PS2APP, Alzheimer’s disease, Synaptic plasticity, LTP, LTD, Perisynaptic, Ro25-6981
Introduction
Amyloid beta (Aβ), the major constituent of the hallmark plaques found in AD patients’ brains, plays a causative role in AD: Mutations in the amyloid precursor protein (APP) or the secretases that cleave APP that result in elevated Aβ levels cause dominantly inherited AD (St George-Hyslop, 2000), while APP mutations that reduce Aβ production confer a decreased risk for sporadic AD (Jonsson et al., 2012). At the same time, the failure of clinical trials aimed at reducing Aβ burden in the later stages of AD has motivated current therapeutic efforts to intervene early in disease progression, before irreversible neuronal loss has occurred (Aisen et al., 2013; Callaway, 2012). Multiple lines of evidence suggest that the deleterious effects of Aβ prior to neuronal loss can be mediated by NMDARs, especially GluN2B-NMDARs. First, impairment of LTP by acute application of exogenous Aβ is mitigated or prevented by antagonists that are selective for GluN2B-NMDARs (Li et al., 2011; Olsen and Sheng, 2012; Rammes et al., 2011; Ronicke et al., 2011). Second, GluN2B-NMDAR antagonists prevent the synapse loss induced by incubation with exogenous Aβ (Ronicke et al., 2011). Third, GluN2B-NMDAR antagonists can also block other effects of exogenous Aβ application to neurons including disruption of intracellular calcium homeostasis (Ferreira et al., 2012) and endoplasmic reticulum oxidative stress (Costa et al., 2012). Furthermore, a significant amount of GluN2B-NMDARs are located in extrasynaptic regions of adult excitatory neurons (Hanson et al., 2013; Papouin et al., 2012). Consistent with the idea that blocking extrasynaptic NMDARs could be beneficial for AD, memantine, an NMDAR antagonist approved for the treatment of AD, preferentially inhibits extrasynaptic NMDARs (Xia et al., 2010), suggesting that the benefit of memantine in AD could involve blockade of extrasynaptic GluN2B-NMDARs.
To our knowledge, all previous studies of the effects of GluN2B antagonists on synaptic plasticity in the context of AD have exclusively relied on acute application of high concentrations of Aβ. In contrast, the gradual accumulation of Aβ that occurs in AD could have very distinct effects from those observed with acute application. To determine the impact of gradual Aβ accumulation on GluN2B-NMDAR function in synaptic transmission and plasticity, we used the PS2APP mouse model of AD that expresses both APP (Swedish mutation) and presenilin 2 (N141I mutation) transgenes, and accumulates Aβ starting at an early age (Ozmen et al., 2009; Richards et al., 2003). GluN2B NMDARs were blocked using Ro25, an antagonist that can be used to block di-heteromeric GluN1/GluN2B/GluN2B NMDARs with little effect on di-heteromeric GluN1/GluN2A/GluN2A or tri-heteromeric GluN1/GluN2A/GluN2B NMDARs (Hansen et al., 2014; Hatton and Paoletti, 2005).
We found that while the magnitude of synaptic plasticity was normal in PS2APP mice, Ro25 resulted in partial impairment of LTP and LTD in PS2APP mice, despite not affecting wt synaptic plasticity. This unusual reliance of LTP and LTD on GluN2B-NMDARs appears to be mediated by a pool of non-synaptic NMDARs that are activated selectively during burst stimulation. We also observed evidence of other abnormalities in GluN2B-NMDAR function which could include presynaptic alterations. The unexpected contribution of GluN2B-NMDARs to synaptic function at an early stage of pathology in PS2APP mice provides new insights regarding potential pharmacological manipulation of NMDARs in the treatment of AD.
Methods
Ethics statement
All animal experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Genentech Institutional Animal Care and Use Committee.
Animals
Twelve to 16 week old male mice homozygous for the PS2APP transgenes and their wt littermate controls were used (Ozmen et al., 2009; Richards et al., 2003). All experiments were approved by the Genentech IUCAC and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Drugs
Brain slice experiments used: 3 μM Ro25-6981, 50 μM D-(−)-2-amino-5-phosphonopentanoic acid (AP5), 20 μMMK-801(Dizocilpine), 10 μM 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) disodium salt, and 100 μM Picrotoxin (PTX) from Tocris.
Brain slices
400 μm horizontal slices containing hippocampus were prepared with a vibrating sectioning system(Leica, Germany) and were recorded in oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM) 127 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.25 Na2HPO4, 25 NaHCO3 and 25 glucose. Slices were prepared in ice-cold oxygenated ACSF with the MgSO4 concentration elevated to 7 mM, NaCl replaced with 110 mM choline and with 11.6 mM Na-ascorbate and 3.1 mM Na-pyruvate.
Field recordings
Excitatory postsynaptic potentials (EPSPs) were measured from the stratum radiatum of CA1 in response to stimulation of Schaffer collateral inputs. NMDAR EPSPs were measured with the Mg2+ concentration in ACSF reduced to 0.5 mM and in the presence of PTX and NBQX. LTP was induced using 1, 2 or 3 bouts of theta burst stimulation (TBS) separated by 20 s, each bout consisting of 5 pulses at 100 Hz repeated 10 times at 200 ms intervals. The subthreshold LTD induction protocol used 15 min of 1 Hz stimulation with 200 ms interval paired stimuli in the presence of normal ACSF. Effective LTD was induced in adult slices using 10 min of 1 Hz stimulation with the Ca2+ concentration in ACSF increased to 4 mM. Ro25 was applied to brain slices for >30 min prior to inducing LTP or LTD. All experiments with Ro25 vs. vehicle treatment of wt and PS2APP mice were analyzed using two-factor ANOVAs followed by assessment with Holm–Sidak tests. Comparisons of phenotypes in PS2APP mice vs. wt mice in experiments with no drug treatment were performed using a Student’s t-test.
Western blot analysis
Western blot analysis of NMDAR subunits was performed as described previously (Hanson et al., 2013). Antibodies were used at the following dilutions: anti-NMDAR1 (1:1000; Abcam); anti-NMDAR2A (1:1000; Novus Biologicals); anti-NMDAR2B (1:1000; NeuroMab); and anti-β-actin (1:2000; Cell Signaling Technology). The expression of the target protein and the corresponding internal control (β-actin) were measured by quantifying the total intensity of enhanced chemiluminescence signals captured by a CCD camera within its linear range using Quantity One software (Bio-Rad).
Electron microscopy immunostaining
Deeply anesthetized mice were transcardially perfusion-fixed with cold oxygenated ACSF followed by a fixative containing 0.1% glutaraldehyde and 4% paraformaldehyde in phosphate buffer (PB; 0.1M, pH 7.4). The brain was removed and post-fixed in 4% paraformaldehyde overnight before washing with phosphate buffer (PB) and sectioned into 60 μm coronal sections using a vibrating microtome. Sections at the level of CA1 in the hippocampus were selected, incubated in a solution of 1% sodium borohydride in PBS for 20 min then washed repeatedly in PBS. The sections were then placed in a cryoprotectant solution (PB, 0.05 M, pH 7.4, containing 25% sucrose and 10% glycerol), frozen at −80 °C, thawed and returned to a graded series of cryoprotectant (100%, 70%, 50%, 30%, 0%) diluted in PBS. The sections were then incubated in PBS containing 5% nonfat dry milk and then washed in TRIS-buffered saline with gelatin (TBS-gelatin; 0.02 M Tris, 0.15 M NaCl, and 1 μl/ml fish gelatin, pH 7.6) to block non-specific labeling. Well-characterized, highly specific, primary monoclonal mouse anti-NMDAR1 antibodies (Neuro-Mab/UC Davis, Cat# 75-272) (Cheng et al., 2013; DeParis et al., 2012; Murata and Constantine-Paton, 2013) were diluted in TBS-gelatin containing 1% nonfat dry milk at a 1:100 dilution. Sections were incubated at room temperature overnight, rinsed 3 × 10 min in TBS-gelatin and incubated in the secondary antibody solution consisting of goat anti-mouse coupled to 1.4 nm gold (Nanoprobes, cat# 2002) at a 1:100 dilution in TBS-gelatin containing 1% nonfat dry milk for 2 h at room temperature. The sections were then washed in TBS-gelatin 2 × 10 min, and acetate buffer (2% aqueous solution, pH 7.0) 1 × 10 min, followed by silver enhancement of gold particles with the HQ silver kit (Nanoprobes Inc., Cat# 2012). Sections were then processed for electron microscopy and ultrathin sections were cut and collected on single slot copper grids as previously described (Galvan et al., 2006; Gonzales et al., 2013; Hanson and Smith, 1999). As a control for the specificity of the immunolabeling, omission of the primary antibody from the incubation solution virtually abolished the immunostaining.
Analysis of EM material
Electron microscopic analysis was performed using a JEOL model 1011 transmission electron microscope. Blocks of tissue from the CA1 region of the hippocampus were examined and data were collected exclusively from sections on the surface of the blocks to ensure optimal antibody penetration into the tissue. Series of 50 random electron micrographs of immunostained tissue were taken from each of 3 wt and 3 PS2APP mice at 25,000×. In each of these micrographs, immunogold-labeled elements were categorized as pre- or post-synaptic structures based on their ultrastructural features (Peters and Palay, 1996). Only gold particles located in structures that could be clearly identified were quantified, resulting in an average of 787 ± 85 gold particles quantified per animal. The average percentages of pre-versus post-synaptic labeled elements from each group were then calculated and statistically compared using a Student’s t-test.
Results
Reliance of LTP on GluN2B-NMDARs in PS2APP mice
To assess potential alterations to synaptic transmission and plasticity that might occur in early stages of AD, we used PS2APP mice at 3–4 months of age when transgenic Aβ 1–40 and 1–42 are already detectable, but significant plaque load has not yet accumulated (Ozmen et al., 2009; Richards et al., 2003). We first examined the input–output (I–O) relationship in area CA1 of the hippocampus by stimulating Schaffer collateral inputs to pyramidal neurons with increasing stimulus intensities and measuring field EPSPs. These recordings showed no change in the I–O relationship indicating unaltered AMPAR-mediated basal synaptic transmission in PS2APP mice (Fig. 1A). When LTP was induced by 1–3 bouts of TBS under physiological recording conditions, there was no difference between wt and PS2APP mice (Fig. 1B). Since many previous studies have shown that GluN2B-NMDAR antagonists rescue impairment of LTP caused by acute application of Aβ (Li et al., 2011; Olsen and Sheng, 2012; Rammes et al., 2011; Ronicke et al., 2011), we determined whether the LTP in PS2APP mice could be altered by the selective GluN2B antagonist Ro25. To our surprise, while Ro25 did not affect wt LTP, a significant reduction in LTP was seen in slices from PS2APP mice in the presence of Ro25 (Fig. 1C, D, F). On the other hand, post-tetanic potentiation (PTP) was enhanced by Ro25 in slices from wt, but not PS2APP mice (Fig. 1C–E). The selective impairment of LTP in PS2APP mice is unlikely due to effects of Ro25 treatment on basal synaptic transmission because Ro25 did not result in any changes to the I–O relationship between PS2APP and wt mice (Fig. 1G). These results indicate that GluN2B-NMDARs provide an essential contribution to LTP induction in PS2APP mice that is normally absent in wt mice.
Fig. 1.

Ro25 treatment selectively impairs LTP in PS2APP mice. (A) Basal synaptic transmission as assessed by the relationship of stimulus intensity to EPSP magnitude was normal in PS2APP mice (n = 5 wt, 7 PS2APP). (B) LTP was induced in CA1 using 1, 2, or 3 bouts of TBS (arrows). Both wt and PS2APP mice exhibited robust LTP when recorded in the presence of vehicle (DMSO; n = 5 wt, 7 PS2APP). Sample EPSPs during the baseline period and at the end of the experiment for wt and PS2APP mice are shown inset. (C) Robust LTP with enhanced post-tetanic potentiation (PTP) was induced in the presence of Ro25 in wt mice (n = 5). Sample EPSPs during the baseline period and at the end of the experiment are shown inset. (D) LTP induced in the presence of Ro25 was reduced in PS2APP mice (n = 8)while there was no significant effect of Ro25 on PTP. Sample EPSPs during the baseline period and at the end of the experiment are shown inset. (E) PTP was quantified as the average EPSP slope during the first minute after the first bout of TBS. Assessment by ANOVA revealed an interaction between treatment and genotype (p < 0.05), and post hoc tests showed significant effects of treatment within wt slices (p < 0.05) and genotype within Ro25 treated slices (p < 0.05). (F) The magnitude of LTP was quantified as the average normalized EPSP slope during the final 5 min of the experiment. Assessment by ANOVA revealed a significant effect of genotype (p < 0.05), and post hoc tests showed significant effects of treatment within PS2APP slices (p < 0.05) and genotype within Ro25 treated slices (p < 0.05). (G) Basal synaptic transmission was normal in PS2APP mice in the presence of Ro25 (n = 5 wt, 8 PS2APP). All data are shown as mean ± SEM and all scale bars are 1 mV and 5 ms.
Enhanced contribution of GluN2B-NMDARs during LTP induction in PS2APP mice
The unexpected reliance of LTP on GluN2B-NMDARs in PS2APP mice could be caused by the enhanced activation of these receptors during LTP induction. To determine whether this is the case, we examined the responses during the TBS that was used to induce LTP, which consisted of 10 bursts of 5 stimuli at 100 Hz given at 200 ms intervals. Because these responses were measured under physiological recording conditions (including 1.3 mM extracellular Mg2+), they represent mixed AMPAR/NMDAR EPSPs, with progressively larger contribution from NMDARs later in the response due to alleviation of NMDAR Mg2+ block by depolarization, and due to the longer decay time of NMDAR EPSPs. The EPSP initial slopes during the TBS were not different between wt and PS2APP mice and were not affected by Ro25 (data not shown), which is consistent with the unaltered AMPAR-mediated synaptic transmission, since the early synaptic responses are mainly mediated by AMPARs. However, the area of the tail portion of the averaged burst responses was significantly reduced by Ro25 in slices from PS2APP mice, but not in wt slices (Fig. 2). These results suggest an enhanced contribution of GluN2B-NMDARs during TBS in PS2APP mice which could explain the impaired LTP induction in the presence of Ro25.
Fig. 2.

Ro25 treatment reduces EPSP tail area during LTP induction in PS2APP mice. (A) Sample average TBS-induced composite EPSPs (AMPAR+NMDAR) during LTP induction are shown for Ro25 treated slices from wt and PS2APP mice. The red line indicates the initial slope and the gray area indicates tail area that was used for quantification. Scale bars are 1 mV and 20 ms. (B) The area of the average EPSP tail during TBS for vehicle and Ro25 treated wt slices is shown (n = 5 veh, 5 Ro25). Data are normalized to the initial slope of each recording. (C) The area of the average EPSP tail during TBS for vehicle and Ro25 treated PS2APP slices is shown (n = 7 veh, 8 Ro25), wt vehicle data from panel A are re-plotted in gray for comparison. (D) Quantification of the average tail area normalized to the initial slope is shown for the first 3 bursts (prior to significant rundown of the burst responses on the later bursts). Assessment by ANOVA revealed an effect of treatment (p < 0.05) and post hoc tests showed a significant effect of treatment within PS2APP mice (p < 0.05). All data are shown as mean ± SEM.
Synaptically-activated NMDARs do not contain significant GluN2B-NMDARs in wt or PS2APP mice
We and others have previously shown that GluN2B-NMDARs make a significant contribution to isolated synaptic NMDAR responses in pyramidal neurons in 2 week-old but not 3 month-old wt mice (Hanson et al., 2013; Harris and Pettit, 2007). Thus one possibility is that the enhanced contribution of GluN2B-NMDARs in 3 month old PS2APP mice could be due to a failure to undergo the normal developmental switch from synaptic GluN2B- to GluN2A-containing NMDARs. This would predict enhanced sensitivity of synaptic NMDAR responses to GluN2B-NMDAR antagonists. As commonly described in the literature, synaptic NMDARs are operationally defined here as being activated by single synaptic stimulations. We first examined the relative magnitude of synaptic NMDAR responses by measuring the NMDA/AMPA ratio (the initial slope of NMDAR responses over the initial slope of AMPAR responses) and found no alteration in the PS2APP mice (Fig. 3A, B). Taken together with the normal magnitude of AMPAR responses (Fig. 1A), this indicates that the total magnitude of synaptic NMDAR function is not grossly altered in PS2APP mice. We then tested the Ro25-sensitivity of synaptic NMDARs. These experiments showed that as in wt mice, synaptic NMDARs were not blocked by Ro25 in PS2APP mice (Fig. 3C, D). Thus, excessive synaptic GluN2B NMDAR contribution is not present in the PS2APP mice, arguing against a failure to undergo the developmental switch in synaptic NMDAR subunit composition in PS2APP mice. As an additional test of the relative abundance of NMDAR subunits, we performed Western blot analysis of GluN1, GluN2A, and GluN2B proteins in forebrain homogenates from PS2APP and WT mice. These experiments showed no significant differences between PS2APP and WT mice in levels of any of these NMDAR subunits (Supp. Fig. 1). This provides confirmation that 3 month PS2APP mice have undergone normal developmental changes in GluN2 subunits.
Fig. 3.

NMDARs activated by single synaptic stimuli are not blocked by Ro25 in PS2APP mice. (A) EPSPs were recorded first under conditions designed to measure AMPAR EPSPs (physiological ACSF with 1.3 mM Mg2+; scale bars are 1 mV and 10 ms) followed by isolated NMDAR conditions (0.5 mM Mg2+; with NBQX scale bars are 1 mV and 50 ms). The sample traces are from a wt brain slice. (B) The NMDAR to AMPAR ratio was quantified using the initial slopes of the AMPAR and NMDAR EPSPs, and was normalized to the average value in wt slices. No significant differences were seen between wt and PS2APP mice (n = 11 wt, 10 PS2APP). (C) In a separate set of experiments, NMDAR EPSPs were measured during a stable baseline period and following application of Ro25. Sample average NMDAR EPSPs before (black) and after Ro25 application (blue) are shown for wt and PS2APP mice. Scale bars are 0.5 mV and 50 ms. (D) While Ro25 did not significantly alter NMDAR EPSPs in wt or PS2APP mice, there was a trend towards an increase in NMDAR EPSP initial slope in PS2APP mice compared to wt mice following Ro25 application (p = 0.06, n = 5 wt, 5 PS2APP). All data are shown as mean ± SEM.
Enhanced activation of GluN2B-NMDARs recruited during burst stimulation in PS2APP mice
During TBS, the rapid increase in synaptic glutamate concentration could overwhelm the capacity of glutamate transporters and result in spill-over of glutamate and activation of glutamate receptors outside the synapse. To isolate the NMDAR pool that is selectively activated by burst stimulation, we first blocked synaptic NMDARs activated by single synaptic stimulations with the use-dependent irreversible NMDAR antagonist MK-801 (Fig. 4A). After washing out MK-801, we measured NMDAR responses elicited by burst stimulation (Fig. 4B). This protocol revealed significant NMDAR responses selectively recruited by burst stimulation. We operationally define this pool of NMDARs as non-synaptic. There was no change in the size of this NMDAR pool in PS2APP mice compared to wt mice as assessed by normalizing the burst-elicited NMDAR EPSP area to the single pulse-elicited area (Fig. 4C). We were not able to directly test the Ro25 sensitivity of this non-synaptic NMDAR pool due to a substantial run-down of the isolated NMDAR responses during repetitive burst stimuli under these recording conditions (low Mg2+ ACSF with AMPARs blocked). However, because run-down does not happen to mixed AMPAR/NMDAR responses to burst stimulation recorded in regular ACSF, we assessed the Ro25-sensitivity of burst-recruited NMDARs recorded under this condition. The NMDAR component during burst stimulation could be estimated by subtracting the AP5-insensitive component measured at the end of each experiment from the mixed AMPAR/NMDAR EPSPs (Fig. 4D). These experiments revealed only a very small reduction of the burst-elicited NMDAR EPSP by Ro25 in wt slices, but a significantly larger reduction in slices from PS2APP mice (Fig. 4E). This demonstrates a larger contribution of GluN2B-NMDARs to the non-synaptic NMDAR pool in PS2APP mice. Taken together with the normal synaptic NMDAR function, this suggests an increased contribution of non-synaptic GluN2B-NMDARs during burst stimulation could underlie the reliance of LTP on GluN2B-NMDARs in PS2APP mice.
Fig. 4.

NMDARs activated during bursts of stimuli are excessively sensitive to Ro25 in PS2APP mice. (A) After obtaining stable isolated NMDAR EPSP responses to single stimuli (black trace), MK-801 was applied to the slice and single pulse stimulation was continued to block the synaptic NMDAR response. Following 300 stimuli in the presence of MK-801 there was only a small residual component of the response remaining (blue). The single pulse evoked NMDAR EPSP area was quantified by subtracting this residual component from the baseline response (red). Scale bars are 1 mV and 50 ms. (B) After blocking the single pulse evoked NMDAR response, MK-801 was washed from the slice for 35 min. At this time point a single stimulus still failed to evoke an EPSP indicating no recovery of synaptic NMDARs (not shown). However, when synapses were activated with bursts of 5 stimuli at 100 Hz a substantial NMDAR EPSP was evoked (black). This burst-evoked response could be blocked by AP5 application (blue). The burst evoked NMDAR EPSP area was quantified by subtracting the residual component in the presence of AP5 from the initial burst response (red). Scale bars are 1 mV and 50 ms. (C) The ratio of single pulse-evoked to burst-evoked NMDAR EPSP area was not significantly different between wt and PS2APP mice (n = 12 wt, 9 PS2APP). (D) To avoid rundown during burst responses, composite EPSPs (AMPAR + NMDAR) were recorded under physiological conditions in response to burst stimulation. Top: After a stable baseline was acquired (black), Ro25 was applied to the slice for 40 min and the remaining response was measured (red). Subsequently AP5 was added to block the NMDAR component of the composite EPSP, leaving only the AMPAR component (blue). Scale bars for composite traces are 1 mV and 100 ms. Bottom: The NMDAR component during the baseline period (black) and after Ro25 application (red) was determined by subtracting the EPSP recorded in the presence of AP5. Sample subtracted traces from wt and PS2APP mice are shown with stimulus artifacts removed. Scale bars for subtracted traces are 0.5 mV and 100 ms. (E) While Ro25 caused only a small reduction in the burst-evoked NMDAR EPSP in wt mice, there was a significantly greater reduction in PS2APP mice (p < 0.05, n = 10 wt, 12 PS2APP). All data are shown as mean ± SEM.
Aberrant dependence of LTD on GluN2B NMDARs in PS2APP mice
Previous work has shown that acute application of Aβ enhances LTD (Li et al., 2009). Thus, while LTD is normally difficult to induce in brain slices from mature mice using standard protocols, it’s possible that normally subthreshold protocols could elicit LTD in 3 month PS2APP mice due to the elevated Aβ levels. However, contrary to this hypothesis, we did not see induction of LTD in 3 month old wt or PS2APP mice using a subthreshold induction protocol (1 Hz stimulation in normal ACSF; Supp. Fig. 2). While Ro25 treatment didn’t change the lack of response to the subthreshold induction protocol in either genotype, Ro25 treatment did impair the frequency facilitation (FF) of EPSPs seen during the onset of 1 Hz stimulation in wt mice. Interestingly, FF was impaired in PS2APP mice compared to wt mice, which occluded any potential effect of Ro25 (Supp. Fig. 2). This is consistent with a normal contribution of GluN2B-NMDARs to FF in wt mice that is disrupted in PS2APP mice, potentially suggesting altered presynaptic NMDAR function (see Discussion).
While there was no effect of genotype or Ro25 on the response to the subthreshold LTD induction protocol, we wanted to test if like LTP, LTD involved an unusual dependence on GluN2B NMDARs in PS2APP mice. To this end, we successfully induced LTD using 1 Hz stimulation in the presence of elevated external Ca2+ (4 mM), a manipulation previously found to allow induction of LTD in adult rodents (Norris et al., 1996). Using this protocol, robust LTD was induced with similar magnitude in both wt and PS2APP mice, indicating that, as with LTP, there is no deficit in this form of plasticity in PS2APP mice (Fig. 5A). While the dependence of LTD on GluN2B NMDARs has been controversial and can depend on the details of experimental conditions (Bartlett et al., 2011; Liu et al., 2004; Morishita et al., 2007), under our experimental conditions, we observed no impact of Ro25 on LTD in wt mice (Fig. 5B). However, Ro25 resulted in significant impairment of LTD in PS2APP mice (Fig. 5C, D). This unusual dependence of LTD on GluN2B NMDARs in PS2APP mice mirrors the results with LTP and reinforces the conclusion of aberrant contribution of GluN2B receptors to synaptic plasticity in PS2APP mice at this early stage of pathology.
Fig. 5.

Ro25 treatment selectively impairs LTD in PS2APP mice. (A) LTD was induced in both wt and PS2APP mice using 600 stimuli delivered at 1 Hz for 10 min in the presence 4 mM external Ca2+ (n = 10 wt, 12 PS2APP). (B) Ro25 treatment had no impact on LTD magnitude in wt mice (n = 10 DMSO, 14 Ro25). (C) In PS2APP mice Ro25 treatment resulted in partial impairment of LTD (n = 12 DMSO, 15 Ro25). (D) LTD was quantified as the average EPSP slope during the last 10 min of the experiment normalized to the baseline slope. Assessment by ANOVA revealed a significant effect of genotype (p < 0.01) that was difficult to interpret due to a potential interaction between treatment and genotype (p = 0.06). Post-hoc tests showed significant effects of treatment within PS2APP slices (p < 0.05), and genotype within Ro25 treated slices (p < 0.001). All data are shown as mean ± SEM.
Discussion
Our study using PS2APP mice at an age with increased soluble Aβ, but prior to significant plaque accumulation, revealed altered and aberrant contributions of GluN2B-NMDARs to synaptic function and plasticity in the hippocampus. Strikingly, we observed a reliance of LTP on GluN2B-NMDARs in the PS2APP mice that was absent in wt mice. Excessive GluN2B-NMDAR function in PS2APP mice was recruited by bursts of stimulation but not single stimulations, implicating non-synaptic NMDARs that aren’t reached by the glutamate released during a single stimulation. While the magnitude of LTD and ability to induce LTD was normal in PS2APP mice, as with LTP, there was an unusual dependence of LTD on of GluN2B-NMDARs. Overall our results indicate alterations to GluN2B-NMDAR function in PS2APP mice that include excessive functional contributions by a non-synaptic pool of NMDARs.
The role of GluN2B-NMDARs in long-term plasticity
Our findings on the contribution of GluN2B-NMDARs to long-term plasticity are in stark contrast with the previous literature showing that acute application of Aβ impairs LTP and enhances LTD, and that the effects of Aβ on synaptic plasticity are mediated by GluN2B-NMDARs (Li et al., 2009, 2011; Olsen and Sheng, 2012; Rammes et al., 2011; Ronicke et al., 2011). Rather, our results suggest that at an early stage of pathology in these mice with gradually accumulating Aβ production, the system is able to maintain normal plasticity, but does so by engaging GluN2B-NMDARs that are not normally involved in LTP or LTD. The normal expression of LTP under standard recording conditions is consistent with a previous study of these PS2APP mice (Richards et al., 2003), and is a common observation in transgenic mouse models of AD (Marchetti and Marie, 2011). The discrepancy between impaired LTP with acute Aβ application and normal LTP in transgenic models likely reflects differential impacts of a sudden elevation of Aβ concentration to an excessive level in an otherwise normal system vs. the chronic and gradual accumulation of Aβ. We propose that the latter situation involves adaptive changes that preserve synaptic plasticity, but result in reliance on abnormal mechanisms.
Precedent for dependence of synaptic plasticity on unusual mechanisms comes from other studies of AD models using pharmacological perturbations: In the 3xTg-AD model, blockade of ryanodine receptors unmasks excessive LTD and reduces LTP (Chakroborty et al., 2009, 2012), and in mutant PS1 KI mice, the normal enhancement of LTP by cholinergic activation instead results in an impairment of LTP (Wang et al., 2009). In PS2APP mice, the net impact of Aβ-induced changes and any homeostasis is maintenance of the normal ability to induce LTP and LTD. However, the abnormal GluN2B receptor function that is able to support synaptic plasticity could have negative consequences in other respects, as discussed below regarding the potential involvement of GluN2B-NDMARs in cell damage. In this respect, the system may be thought of as under an allostatic load where maintenance of normal physiological function could come at the expense of long-term health.
Enhanced function of GluN2B-NMDARs in a non-synaptic pool of NMDARs in PS2APP mice
Our measurements of NMDAR function in postsynaptic neurons implicate enhanced function of non-synaptic GluN2B-NMDARs. Specifically, based on the way the NMDARs are activated, we identified two distinct pools, 1) a pool that is activated by single synaptic stimuli—operationally defined as synaptic, and, 2) a pool that is activated selectively by burst stimuli—operationally defined as non-synaptic. Because the NMDARs in the non-synaptic pool cannot be activated by single stimuli, they likely require glutamate spilling beyond the synaptic cleft during bursts of activity. Our current data agree with previous studies showing that the synaptic pool does not contain significant GluN2B-NMDARs in wt adult excitatory neurons (Hanson et al., 2013; Harris and Pettit, 2007; Papouin et al., 2012), and we also did not see a GluN2B-NMDAR contribution to the synaptic pool in PS2APP mice. On the other hand, while there is only a very small contribution of GluN2B-NMDARs to the non-synaptic pool in wt mice, GluN2B-NMDARs make a significantly larger contribution to the non-synaptic pool in PS2APP mice.
Regarding the anatomical correlates of these receptor pools, we propose a model in which, in addition to synaptic NMDARs within the post-synaptic density, there are perisynaptic NMDARs which are in the regions just outside the synapse and can be activated only during burst stimulation in both genotypes. This non-synaptic pool is likely perisynaptic rather than extrasynaptic, as previous studies using exogenous glutamate or NMDA application in wt mice have shown a significant presence of GluN2B-NMDARs in the extrasynaptic pool (Harris and Pettit, 2007; Papouin et al., 2012). Normally, this perisynaptic receptor pool does not contain many GluN2B-NMDARs but in PS2APP mice their presence/activation is enhanced (Fig. 6). An excessive contribution of non-synaptic GluN2B NMDARs has also been shown in mouse models of Huntington’s disease (Milnerwood et al., 2010; Okamoto et al., 2009), indicating increased non-synaptic GluN2B function could be a shared feature of distinct neurodegenerative diseases.
Fig. 6.
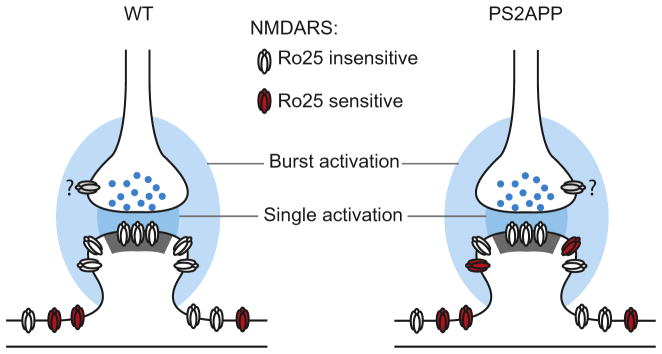
Model of aberrant GluN2B function in PS2APP mice. Glutamate from single synaptic activations (dark blue) only reaches synaptic NMDARs while bursts of synaptic activation lead to glutamate reaching perisynaptic pools of NMDARs (light blue). In wt mice, these perisynaptic NMDARs are generally insensitive to Ro25 (white), while in PS2APP mice, these peri-synaptic NMDARs are more sensitive to Ro25 (red). Extrasynaptic NMDARs located farther from the synapse are distinct from the other pools. Question marks indicate the unknown Ro25 sensitivity of presynaptic NMDARs (gray). While an altered physical presence of GluN2B-NMDARs is depicted, the observed functional phenotypes could also result from altered activation caused by impaired glutamate transport and greater spread of glutamate from the synapses during burst activation.
Additional potential sites of altered GluN2B NMDAR function
Some phenotypes in the PS2APP mice are suggestive of disrupted GluN2B-NMDARs at sites in addition to postsynaptic locations on pyramidal neurons. For example the lack of enhancement of PTP by Ro25 in PS2APP mice as seen in wt mice, could be due to altered function in presynaptic excitatory inputs, or in the interneurons that gate plasticity induction (Hanson et al., 2013). In addition, GluN2B-NMDARs are required for FF in wt mice and these GluN2B-NMDARs could be located at presynaptic terminals. Since FF is impaired in PS2APP mice and Ro25 has no effect on FF, this could reflect altered presynaptic GluN2B-NMDARs in PS2APP mice.
To assess potential gross alterations in presynaptic NMDAR localization, we used electron microscopy to determine the relative prevalence of presynaptic vs. postsynaptic expression. Because pilot studies with available NMDAR subunit-specific antibodies established conditions for specific labeling of GluN1 in our hands, we focused on assessing localization of NMDARs using immunogold labeling of these receptors. These experiments confirmed previous reports (Aoki et al., 1994; Berg et al., 2013; Paquet and Smith, 2000; Wang and Pickel, 2000) that NMDARs are presynaptically and postsynaptically expressed in adult wt mice, and significant presynaptic and postsynaptic GluN1 labeling was also found in PS2APP mice (Supp. Fig. 3). However, although there was a trend towards greater presynaptic localization in PS2APP mice, there was no significant difference in the relative localization of GluN1 labeling in presynaptic vs. postsynaptic compartments between wt and PS2APP mice (Supp. Fig. 3). Nonetheless, because our EM studies were limited to GluN1 localization, a subunit present in all NMDARs, it remains possible that the fraction of presynaptic NMDARs containing GluN2B is altered in PS2APP mice and this could contribute to the observed phenotypes. Ultimately, future studies will be needed to explore the nature of potential presynaptic and/or interneuron NMDAR phenotypes in these mice.
Implications for therapeutic targeting of GluN2B-NMDARs in AD
Preclinical research in a variety of neurodegenerative disease models raise hopes for neuroprotection with GluN2B antagonists (Beinat et al., 2010; Gogas, 2006; Mony et al., 2009). In particular, GluN2B antagonists protect against excitotoxic neuronal death (Liu et al., 2007) and against Aβ-induced synapse loss (Ronicke et al., 2011). In adult rodents, GluN2B-NMDARs are preferentially located extrasynaptically on excitatory neurons and extrasynaptic receptors are especially implicated in neuronal cell death (Hardingham and Bading, 2010; Hardingham et al., 2002). Therefore it seems likely that extrasynaptic GluN2B-NMDARs could mediate cell damage during chronic activation that may occur under pathological conditions such as excessive glutamate release or reduced glutamate clearance in conditions including AD. As discussed above our results suggest up-regulated perisynaptic GluN2B NMDARs (non-synaptic but close enough to synapses to be activated during burst stimulation) in PS2APP mice. This raises the possibility that this pool of GluN2B-NMDARs could have deleterious effects on cell health even during normal physiological activation such as the theta burst activity that can induce LTP.
If neurodegeneration in AD in fact involves excessive activation of non-synaptic GluN2B-NMDARs, then GluN2B-NMDAR inhibitors could have a neuroprotective benefit. On the other hand, the aberrant GluN2B-NMDAR function in PS2APP mice appears to be playing an essential role in synaptic plasticity induction. If similar GluN2B-NMDAR gain-of-function were to also occur as a compensatory adaptation in AD, then potential benefits of blocking GluN2B-NMDARs to prevent excitotoxicity might be opposed by negative effects on physiological function. In this context, we recently found that chronic in vivo GluN2B antagonism failed to protect PS2APP mice against amyloid plaque-associated spine loss, and failed to prevent deficits in cognitive function (Hanson et al., 2014). Given the present results, we can’t exclude the possibility that chronic GluN2B antagonism exerted some beneficial effects by blocking extrasynaptic NMDARs, but negative consequences of blocking perisynaptic GluN2B receptors, which appear to be playing essential physiological roles in the PS2APP mice, may have masked any impact of these benefits on the phenotypes that were examined. Thus to the extent that this preclinical model might reflect the impacts of ongoing Aβ impacts in AD patient brains, our results raise the possibility that aberrant GluN2B-NMDAR function could complicate efforts to achieve therapeutic benefits via neuroprotection with GluN2B-NMDAR antagonists.
Supplementary Material
Acknowledgments
The authors would like to thank William J. Meilandt for providing PS2APP and wt brain homogenates for protein analysis and would like to thank Morgan Sheng and members of the Genentech Neurophysiology group for comments and discussion on the content of the manuscript. YS and J-FP received financial support from the NIH base grant to the Yerkes Primate Center (RR00165). The authors declare no competing financial interests.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2014.11.017.
References
- Aisen PS, Vellas B, Hampel H. Moving towards early clinical trials for amyloid-targeted therapy in Alzheimer’s disease. Nat Rev Drug Discov. 2013;12:324. doi: 10.1038/nrd3842-c1. [DOI] [PubMed] [Google Scholar]
- Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM. Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci. 1994;14:5202–5222. doi: 10.1523/JNEUROSCI.14-09-05202.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett TE, Lu J, Wang YT. Slice orientation and muscarinic acetylcholine receptor activation determine the involvement of N-methyl D-aspartate receptor subunit GluN2B in hippocampal area CA1 long-term depression. Mol Brain. 2011;4:41. doi: 10.1186/1756-6606-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinat C, Banister S, Moussa I, Reynolds AJ, McErlean CS, Kassiou M. Insights into structure-activity relationships and CNS therapeutic applications of NR2B selective antagonists. Curr Med Chem. 2010;17:4166–4190. doi: 10.2174/092986710793348572. [DOI] [PubMed] [Google Scholar]
- Berg LK, Larsson M, Morland C, Gundersen V. Pre- and postsynaptic localization of NMDA receptor subunits at hippocampal mossy fibre synapses. Neuroscience. 2013;230:139–150. doi: 10.1016/j.neuroscience.2012.10.061. [DOI] [PubMed] [Google Scholar]
- Callaway E. Alzheimer’s drugs take a new tack. Nature. 2012;489:13–14. doi: 10.1038/489013a. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgo J, Stutzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J Neurosci. 2012;32:8341–8353. doi: 10.1523/JNEUROSCI.0936-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Liu W, Duffney LJ, Yan Z. SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors. J Physiol. 2013;591:3935–3947. doi: 10.1113/jphysiol.2013.255075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RO, Lacor PN, Ferreira IL, Resende R, Auberson YP, Klein WL, Oliveira CR, Rego AC, Pereira CM. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell. 2012;11:823–833. doi: 10.1111/j.1474-9726.2012.00848.x. [DOI] [PubMed] [Google Scholar]
- DeParis S, Caprara C, Grimm C. Intrinsically photosensitive retinal ganglion cells are resistant to N-methyl-D-aspartic acid excitotoxicity. Mol Vis. 2012;18:2814–2827. [PMC free article] [PubMed] [Google Scholar]
- Ferreira IL, Bajouco LM, Mota SI, Auberson YP, Oliveira CR, Rego AC. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium. 2012;51:95–106. doi: 10.1016/j.ceca.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Galvan A, Kuwajima M, Smith Y. Glutamate and GABA receptors and transporters in the basal ganglia: what does their subsynaptic localization reveal about their function? Neuroscience. 2006;143:351–375. doi: 10.1016/j.neuroscience.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006;6:68–74. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Gonzales KK, Pare JF, Wichmann T, Smith Y. GABAergic inputs from direct and indirect striatal projection neurons onto cholinergic interneurons in the primate putamen. J Comp Neurol. 2013;521:2502–2522. doi: 10.1002/cne.23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Smith Y. Group I metabotropic glutamate receptors at GABAergic synapses in monkeys. J Neurosci. 1999;19:6488–6496. doi: 10.1523/JNEUROSCI.19-15-06488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Weber M, Meilandt WJ, Wu T, Luu T, Deng L, Shamloo M, Sheng M, Scearce-Levie K, Zhou Q. GluN2B antagonism affects interneurons and leads to immediate and persistent changes in synaptic plasticity, oscillations, and behavior. Neuropsychopharmacology. 2013;38:1221–1233. doi: 10.1038/npp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Meilandt WJ, Gogineni A, Reynen P, Herrington J, Weimer RM, Scearce-Levie K, Zhou Q. Chronic GluN2B antagonism disrupts behavior in wild-type mice without protecting against synapse loss or memory impairment in Alzheimer’s disease mouse models. J Neurosci. 2014;34:8277–8288. doi: 10.1523/JNEUROSCI.5106-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–519. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jonsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C, Marie H. Hippocampal synaptic plasticity in Alzheimer’s disease: what have we learned so far from transgenic models? Rev Neurosci. 2011;22:373–402. doi: 10.1515/RNS.2011.035. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Mony L, Kew JN, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol. 2009;157:1301–1317. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Murata Y, Constantine-Paton M. Postsynaptic density scaffold SAP102 regulates cortical synapse development through EphB and PAK signaling pathway. J Neurosci. 2013;33:5040–5052. doi: 10.1523/JNEUROSCI.2896-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Korol DL, Foster TC. Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J Neurosci. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen KM, Sheng M. NMDA receptors and BAX are essential for Abeta impairment of LTP. Sci Rep. 2012;2:225. doi: 10.1038/srep00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozmen L, Albientz A, Czech C, Jacobsen H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener Dis. 2009;6:29–36. doi: 10.1159/000170884. [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Paquet M, Smith Y. Presynaptic NMDA receptor subunit immunoreactivity in GABAergic terminals in rat brain. J Comp Neurol. 2000;423:330–347. doi: 10.1002/1096-9861(20000724)423:2<330::aid-cne10>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL. The morphology of synapses. J Neurocytol. 1996;25:687–700. doi: 10.1007/BF02284835. [DOI] [PubMed] [Google Scholar]
- Rammes G, Hasenjager A, Sroka-Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology. 2011;60:982–990. doi: 10.1016/j.neuropharm.2011.01.051. [DOI] [PubMed] [Google Scholar]
- Richards JG, Higgins GA, Ouagazzal AM, Ozmen L, Kew JN, Bohrmann B, Malherbe P, Brockhaus M, Loetscher H, Czech C, Huber G, Bluethmann H, Jacobsen H, Kemp JA. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci. 2003;23:8989–9003. doi: 10.1523/JNEUROSCI.23-26-08989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronicke R, Mikhaylova M, Ronicke S, Meinhardt J, Schroder UH, Fandrich M, Reiser G, Kreutz MR, Reymann KG. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging. 2011;32:2219–2228. doi: 10.1016/j.neurobiolaging.2010.01.011. [DOI] [PubMed] [Google Scholar]
- St George-Hyslop PH. Molecular genetics of Alzheimer’s disease. Biol Psychiatry. 2000;47:183–199. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- Wang H, Pickel VM. Presence of NMDA-type glutamate receptors in cingulate corticostriatal terminals and their postsynaptic targets. Synapse. 2000;35:300–310. doi: 10.1002/(SICI)1098-2396(20000315)35:4<300::AID-SYN8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Wang Y, Greig NH, Yu QS, Mattson MP. Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging. 2009;30:1061–1068. doi: 10.1016/j.neurobiolaging.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.