

Abstract
The antimalarial agents artemisinin and arterolane act via initial reduction of a peroxide bond in a process likely mediated by ferrous iron sources in the parasite. Here, we report the synthesis and antiplasmodial activity of arterolane-like 1,2,4-trioxolanes specifically designed to release a tethered drug species within the malaria parasite. Compared to our earlier drug delivery scaffolds, these new arterolane-inspired systems are of significantly reduced molecular weight and possess superior metabolic stability. We also demonstrate the use of the aminonucleoside antibiotic puromycin as a chemo/biomarker to validate successful drug release in live P. falciparum parasites.
Keywords: antimalarials, drug delivery, targeted prodrugs, trioxolanes, puromycin
Peroxidic antimalarials such as the artemisinins, trioxolanes (e.g., OZ439[1] and arterolane[2,3], 1), and tetraoxanes[4], exert their therapeutic effects via initial reduction of a hindered peroxide bond. While the point is still debated, it seems probable that this reduction is mediated by ferrous iron heme liberated during intra-parasitic proteolysis of haemoglobin.[5–11] It appears then that these agents lack a ‘target’ in the traditional sense and instead exploit an aberrant chemical environment produced through host-parasite interaction. Our group and others have shown that this reducing environment in the parasite can be exploited for parasite-selective drug delivery from suitably engineered endoperoxides[12], trioxolanes[13–15], or tetraoxanes.[16][17] Unlike ‘hybrid antimalarials’[18][19] these drug delivery systems release their payload in an untethered form, free from any linker. Accordingly, this approach can be used to deliver existing antimalarial drugs with improved selectivity and reduced off-target toxicity.
In our earlier studies, we employed trioxolane conjugate 2 to establish the concept of trioxolane-mediated, ferrous iron-dependent drug delivery.[13,15] The potent antimalarial ML4118S[20], an inhibitor of the parasite cysteine protease dipeptidyl peptidase-1 (DPAP1), was employed as the drug species to be delivered from 2. Using activity-based probes, we established that conjugate 2 efficiently releases ML4118S in live P. falciparum parasites with a half-life estimated at 1.5 hrs.[13] In the P. berghei in vivo model, we observed more sustained DPAP1 inhibition in mice receiving 2 than in mice administered ML4118S directly, and also saw greatly reduced off-target effects in 2-treated mice.[15] The use of non-peroxidic control compounds in this study established that the beneficial effects realized with 2 were indeed a result of trioxolane-mediated, parasite-selective drug delivery.
Although first-generation molecules like 2 proved invaluable to establish the concept, their high molecular weight and large number of rotatable bonds predicts less then optimal drug-like properties. Herein, we describe a new drug delivery scaffold 3 with greatly reduced molecular weight and generally superior drug-like properties. We also describe the use of the aminonucleoside antibiotic puromycin as a chemical-biological probe to validate drug delivery from 3 in live P. falciparum parasites.
As in our earlier systems, drug delivery from trioxolane 3 is achieved by the coupling of two reaction processes. First, reduction of the peroxide bond in 3 and trioxolane fragmentation leads to ketone 4 in which drug is tethered at the β position. Release of free drug (5) from 4 then occurs by spontaneous retro-Michael reaction and decarboxylation (Scheme 1). Significantly, the carbamate linkage in 3 is stable, but becomes labile upon unmasking of the ketone function in intermediate 4. Compared to 2, second-generation conjugates 3 are ~150 Da. lower in molecular weight and structurally more closely related to successful drug candidates such as arterolane and OZ439. We therefore anticipated that conjugates 3 should possess drug-like properties superior to 2.
Scheme 1.
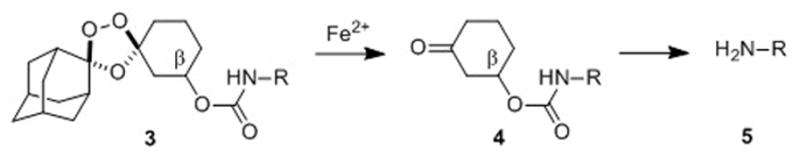
Fe(II)-mediated reduction of trioxolane 3 leads to retro-Michael intermediate 4 which subsequently releases free drug 5 after β-elimination and decarboxylation.
While 1,2,4-trioxolanes have been widely studied as antimalarial agents, analogs substituted at the 3-position of the cyclohexane ring (as in 3) have been scarecely explored.[21][22] We therefore sought to develop a general synthetic approach to prepare such analogs. Gratifyingly, we found that the partially protected diketone 6 participated in Griesbaum co-ozonolysis with O-methyl 2-adamantanone oxime to afford the desired trioxolane 7 in nearly quantitative yields (Scheme 2). Notably, the use of ≥ 2 equivalents of oxime in this reaction was essential to achieve high chemical yields. Deprotection of ketal 7 with ferric chloride hexahydrate in acetone-dichloromethane proceeded smoothly to afford ketone 8 in excellent yield. Using a modification of the conditions reported previously[23] for reductions of 4″-keto trioxolanes, we successfully reduced 3″-keto trioxolane 8 to the desired alcohol intermediate 9 in 67% yield. This reduction proceeded with little diastereofacial selectivity, affording 9 as a roughly equal mixture of cis and trans diastereoisomers (both racemic). The synthesis of 9 reported here (3 steps, ~63% overall yield) compares favorably with our previous synthesis[13] of the analogous first-generation alcohol 10 (3 steps, 12% overall yield) used to prepare 2. Although we have recently developed a diastereoselective synthesis of trans-9 (to be described elsewhere), all compounds reported herein were prepared from the diastereomeric mixture of cis and trans-9.
Scheme 2.
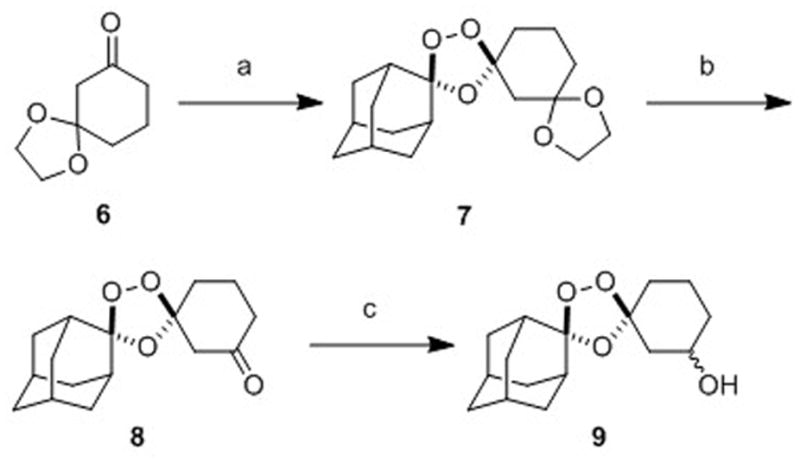
Reagents and conditions: (a) 2.4 equiv O-methyl 2-adamantaone oxime, CCl4, O3, 2.5 h, 0°C, 97%; (b) 3 equiv FeCl3•6H2O, acetone/CH2Cl2, 0°C-rt, 1.5 h, 96%; (c) NaBH4, EtOH/THF, -78 to 0°C, 5 h, 67%.
To compare the first- and second-generation trioxolane scaffolds, we prepared congeneric conjugates of alcohols 9 and 10 with ethylamine and 2,5-dichloroaniline. The ethyl carbamates were prepared to evaluate intrinsic antimalarial potency and in vitro ADME properties while conjugates of 2,5-dichloroaniline were employed to study in vitro drug release rates (the aniline moiety affording a chromophore for spectroscopic detection). For the first generation analogs, reaction of alcohol 10 with ethyl or 2,5-dichlorophenyl isocyanate afforded the desired carbamates 11 and 12 (Scheme 3). Second-generation comparators 13 and 14 were prepared from alcohol 9 under similar conditions and in comparable chemical yields (Scheme 4).
Scheme 3.
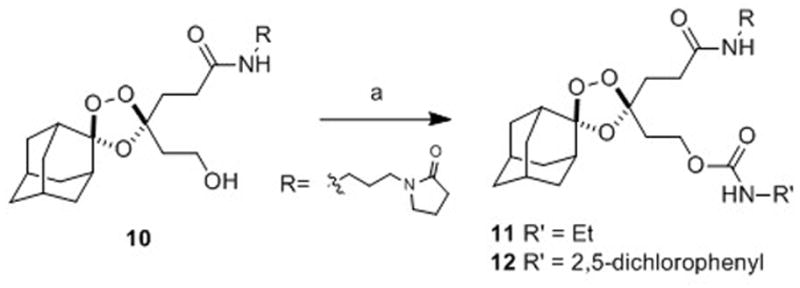
Reagents and Conditions: (a) R’NCO, pyridine, toluene, 50 °C, 42 h, 71% (for 11), 63% (R = for 12).
Scheme 4.
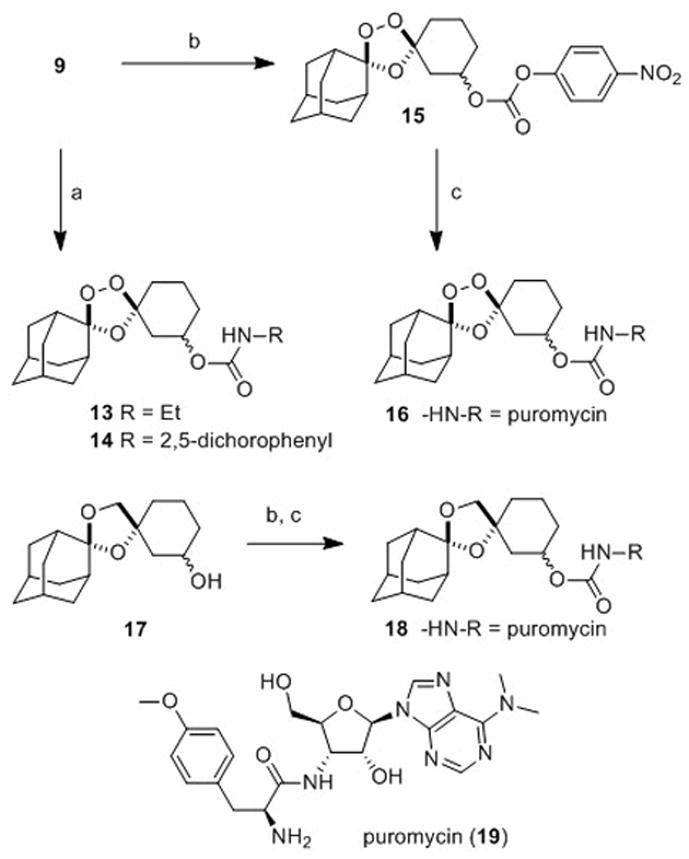
Reagents and Conditions: (a) RNCO, pyridine, toluene, 50 °C, 18–72 h, 74% (for 13), 55–79% (for 14); (b) p-NO2C6H4OC(O)Cl, i-Pr2NEt, DMAP, CH2Cl2, 25 min, 0 °C-rt, 94% (for 15); (c) 19, i-Pr2NEt, DMAP, DMF, rt, 44 h, 61% (for 16); 52% over two steps (for 18).
We compared the in vitro antiplasmodial activities and in vitro ADME properties of first-generation conjugates 11 and 12 with their second-generation congeners 13 and 14 (Table 1). All four compounds exhibited low nanomolar effects on the growth of cultured W2 P. falciparum parasites as determined using a flow cytometry-based growth inhibition assay.[24] Second-generation analogs 13 and 14 thus behave as typical trioxolane antimalarials and are presumably reduced in the parasite via the canonical iron(II)-mediated process. First- and second-generation ethyl carbamates 11 and 13 were evaluated for stability in the presence of cultured liver microsomes from human, rat, and mouse. In all cases, analog 13 exhibited significantly improved metabolic stability as compared to first-generation conjugate 11. The aqueous solubility of 11 and 13 was superior to 1, while stability of the compounds in human and mouse plasma was comparable to 1 (Supplementary Table S1). With favorable ADME properties and significantly (~150 Da.) lower molecular weight, second-generation trioxolane conjugates derived from 9 should have superior prospects for oral bioavailability and in general improved drug-like properties.
Table 1.
Antiplasmodial activity, MW, microsome stability, and drug release kinetics of trioxolane conjugates and controls.
| Cmpd | EC50 (nM) W2[a] | 95% Cl[b] | Rat CL[c] | In vitro Drug Release T1/2 | |
|---|---|---|---|---|---|
| ACN/H2O[d] | DMEM/FBS[e] | ||||
| ART | 7.1 | 6.1–8.4 | --- | --- | --- |
| 1 | 4.8 | 2.7–8.4 | 59 | --- | --- |
| 11 | 16 | 11–24 | 694 | --- | --- |
| 12 | 0.39 | 0.32–0.46 | --- | 5.3 h | --- |
| 13 | 12 | 11–14 | 155 | --- | --- |
| 14 | 13 | 11–15 | --- | 170 h | 9 min |
| 16 | 10 | 9.1–12 | --- | --- | --- |
| 18 | >1,000 | n.a. | --- | --- | --- |
| 19 | 42 | 32–56 | --- | --- | --- |
Potency against W2 P. falciparum parasites in cell culture, average of three determinations;
95% confidence interval for the EC50 values;
rat liver microsome stability, expressed as clearance (CL) in units of μL/min/mg;
100 equiv. FeBr2, 0.3 mM in H2O-CH3CN (1:1);
100 equiv FeBr2, 0.3 mM in high glucose DMEM, 10% FBS.
Next, we employed the aryl carbamates 12 and 14 to study trioxolane fragmentation and drug release in vitro. Using LC/MS instrumentation with evaporative light-scattering and mass detection, we were able to follow the parent compounds 12 and 14 as well as their corresponding retro-Michael intermediates and the liberated 2,5-dichloroaniline (a surrogate for released drug). Intrinsic reactivity with ferrous iron was evaluated using the in vitro conditions first recommended by Charman.[25] Hence, 12 and 14 were subjected to reaction with a 100-fold molar excess of FeBr2 in 1:1 water-acetonitrile at 37 °C. Both 12 and 14 were reduced rapidly under these conditions, with second-generation analogue 14 exhibiting a longer half-life of ~16 min, as compared to t1/2 ~2 min for 12 (Table 1).
While ferrous iron-promoted reduction of 12 and 14 was rapid in acetonitrile-water, we found that subsequent β-elimination and release of 2,5-dichloroaniline was very slow (hours to days) in this organic-aqueous solvent mixture (Table 1). These sluggish rates of β-elimination were not altogether reconcilable with our earlier findings that drug release from 2 occurs on therapeutically relevant time scales (t1/2 ~1.5 h), both in cultured parasites and in animal models.[13,15] We therefore sought to identify more biologically relevant reaction media for in vitro studies of drug release from these systems. Interestingly, we found that reaction with FeBr2 in a common cell-culture media comprising high-glucose Dulbecco’s modified eagle medium (DMEM) and 10% fetal bovine serum (FBS) resulted in drug release rates more in accord with our previous in vivo and cell culture results. Thus, in DMEM/FBS release of 2,5-dichloroaniline from 14 was complete within 60–90 minutes, with a half-life of ~9 minutes (Table 1, Figure 2). Trioxolane activation occurs too rapidly in this media to determine a half-life for the iron-promoted reduction step. While release rates may be under-estimated in water-acetonitrile and over-estimated in DMEM/FBS, we found under both conditions that retro-Michael reaction is the rate-determining step in trioxolane-mediated drug delivery.
Figure 2.
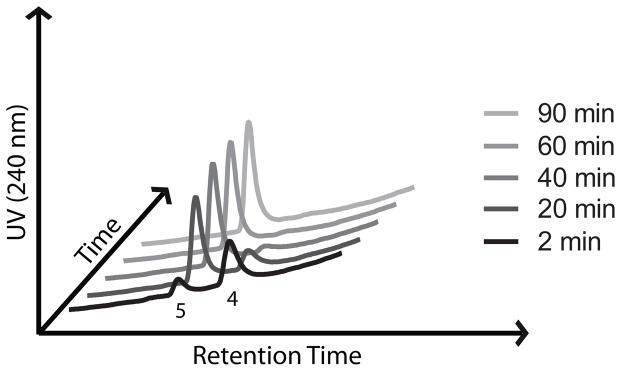
Chromatograms showing generation of the retro-Michael intermediate 4 and subsequent release of 2,5-dichloroanilne (5) following reaction of 14 with FeBr2 in DMEM/FBS media.
In our previous studies with 2, we employed activity-based probes to demonstrate the efficient release of tethered ML4118S in parasites. Here we report another approach to study drug release in parasites by using trioxolane conjugates of the aminonucleoside antibiotic puromycin (19, Scheme 4). Puromycin acts by mimicking the 3′ end of tyrosine tRNA and becomes incorporated into growing polypeptides at the ribosome, eventually causing premature chain termination and the release of puromycin-containing peptides. Notably, these puromycin-polypeptides can be detected with α-puromycin antibodies. Previously, this activity of puromycin was used to validate protease-activated prodrugs in mammalian cancer cell lines and xenograft models.[26] Puromycin is known to be toxic to P. falciparum in vitro and the gene for puromycin N-acetyltransferase has been used as a selectable marker for manipulating the parasite genetically.[27] Thus, we became intrigued by the notion of using trioxolane-puromycin conjugates to study drug release in live P. falciparum parasites.
The requisite trioxolane-puromycin conjugate 16 was prepared in two steps and 59% overall yield from alcohol 9 (Scheme 4). The non-peroxidic puromycin conjugate 18 was prepared from alcohol 17 by an analogous procedure and serves as an important control (Scheme 4 and Supporting Information). The α-amine of puromycin is carbamoylated in 16 and 18 and so the intact conjugates should be incapable of incorporation into parasite polypeptides. Exposure to ferrous iron in the parasite however is expected to release free puromycin from 16 (but not from 18). Thus, the detection of puromycin incorporation in parasites treated with 16 (but not in those treated with 18) would provide strong evidence for the trioxolane-mediated release of puromycin from 16. As detailed below, this is precisely what we observed with conjugates 16 and 18.
First, we evaluated the antiplasmodial activity of puromycin itself (19) as well as its two conjugates 16 and 18. As expected, puromycin and its trioxolane conjugate 16 exhibited low nM activity against W2 parasites, whereas non-peroxidic conjugate 18 was ~100-fold less potent (Table 1). This result confirms that the toxicity of puromycin is largely ablated when conjugated at the α-amine, as expected. Next, we employed puromycin (19), trioxolane-puromycin conjugate 16, and non-peroxidic control 18 to study peroxide-dependent drug release in live P. falciparum parasites. Synchronized trophozoite stage parasites, the erythrocytic stage when protein synthesis is most robust, were incubated with equimolar concentrations (400 nM) of compounds 19, 16, or 18 for up to 12 hours. Parasites were periodically released from erythrocytes by saponin lysis and the isolated parasites were analysed for total puromycin incorporation in parasite proteins using a “dot blot” analysis with α-puromycin antibody (Supplementary Figure S1). Protein samples run on an SDS-PAGE gel and analyzed by Western blotting with α-puromycin antibody confirmed that puromycin is incorporated broadly into the parasite proteome (Supplementary Figure S2).
The results of the puromycin studies were unambiguous and fully consistent with peroxide-dependent release of puromycin from 16. Thus, parasites treated with either puromycin (19) or its trioxolane-conjugate 16 showed a time-dependent increase in puromycin incorporation in parasite protein (Figure 3 and Supplementary Figure S1). In contrast, parasites treated with dioxolane-puromycin conjugate 18 showed negligible puromycin incorporation over the course of the experiment. This finding both confirms the peroxide-dependence of puromycin release from 16 and also demonstrates that puromycin action and toxicity is largely ablated while in conjugated forms (as revealed also by the ~100-fold higher EC50 value for 18 vs 19). The ability to block the activity/toxicity of a drug species prior to release in the parasite is a key advantage of trioxolane-mediated drug delivery and distinguishes this approach from hybrid antimalarials, which must necessarily remain active/toxic in their conjugated forms.
Figure 3.
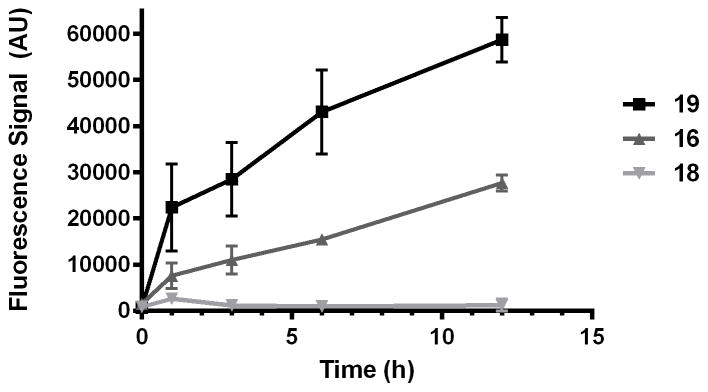
Quantification of puromycin incorporation in proteins of P. falciparum parasites treated with 19, 16, or 18. Negligible puromycin incorporation is observed for dioxolane conjugate 18, indicating that puromycin release from 16 is peroxide-dependent.
Also of note from these studies was the fact that puromycin incorporation appeared to be greater in parasites treated directly with puromycin (19) than in those treated with 16. This difference appears to be a consequence of more rapid puromycin incorporation during the first hour in 19-treated parasites (Figure 3). After this initial burst, rates of incorporation are similar (nearly equivalent slopes) between parasites treated with 19 or 16. The difference in the early stages of treatment may reflect a lag in puromycin release from 16, which is not unexpected given the results of our in vitro drug release studies, where complete release of free drug required 60–90 minutes (Figure 2). Alternatively, the difference may simply reflect the toxic effects of a trioxolane-based insult that is conferred to parasites by 16 but not 19. Thus, the application of trioxolane 16 in excess of its EC50 value might reasonably be expected to affect rates of parasite growth and protein synthesis, leading to slower puromycin incorporation initially. Whatever the case, the dot blot studies with 19, 16, and 18 combined with the in vitro drug release studies described herein serve to validate 3 as a competent scaffold for drug delivery to P. falciparum parasites.
In conclusion, we have described an improved chemical scaffold for trioxolane-mediated drug delivery. The new molecules 3 are of significantly lower molecular weight, and exhibit superior drug-like properties when compared to our earlier systems. An efficient, concise and scale-able synthesis of the key synthetic intermediate 9 is described and should facilitate additional studies of these molecules in various applications. Finally, we have demonstrated the utility of the aminonucleoside puromycin as a chemical-biological marker to study drug release in P. falciparum. We anticipate that the use of puromycin as a drug surrogate will enable future studies of trioxolane-mediated drug delivery in malaria and other disease models.
Supplementary Material
Figure 1.
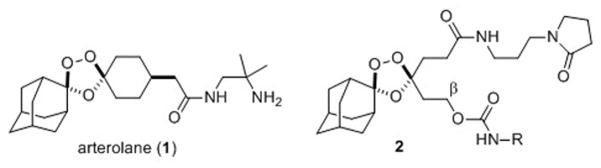
Structures of the antimalarial agent arterolane (1) and trioxolane conjugate 2, which confers parasite-selective delivery of a tethered DPAP1 inhibitor (ML4118S, or NH2-R in 2).
Acknowledgments
ARR acknowledges the support of the US National Institutes of Health (NIH) (R21AI0944233 and R01AI105106) and the Bill and Melinda Gates Foundation (USA). We thank Prof. Matthew Bogyo for helpful discussions and suggestions.
Footnotes
Supporting information for this article is available on the WWW
References
- 1.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FCK, Chollet J, Craft JC, Creek DJ, et al. Proc Natl Acad Sci U S A. 2011;108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FCK, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, et al. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 3.Dong Y, Wittlin S, Sriraghavan K, Chollet J, Charman SA, Charman WN, Scheurer C, Urwyler H, Santo Tomas J, Snyder C, et al. J Med Chem. 2010;53:481–91. doi: 10.1021/jm901473s. [DOI] [PubMed] [Google Scholar]
- 4.Marti F, Chadwick J, Amewu RK, Burrell-Saward H, Srivastava A, Ward Sa, Sharma R, Berry N, O’Neill PM. Medchemcomm. 2011;2:661. [Google Scholar]
- 5.O’Neill PM, Posner GH. J Med Chem. 2004;47:2945–2964. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Dong Y, Wang X, Sriraghavan K, Wood JK, Vennerstrom JL. J Org Chem. 2005;70:5103–5110. doi: 10.1021/jo050385+. [DOI] [PubMed] [Google Scholar]
- 7.Haynes RK, Chan WC, Lung CM, Uhlemann AC, Eckstein U, Taramelli D, Parapini S, Monti D, Krishna S. ChemMedChem. 2007;2:1480–1497. doi: 10.1002/cmdc.200700108. [DOI] [PubMed] [Google Scholar]
- 8.Creek DJ, Charman WN, Chiu FCK, Prankerd RJ, Dong Y, Vennerstrom JL, Charman Sa. Antimicrob Agents Chemother. 2008;52:1291–1296. doi: 10.1128/AAC.01033-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Neill PM, Barton VE, Ward SA. Molecules. 2010;15:1705–1721. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartwig CL, Lauterwasser EMW, Mahajan SS, Hoke JM, Cooper RA, Renslo AR. J Med Chem. 2011;54:8207–8213. doi: 10.1021/jm2012003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haynes RK, Cheu KW, Chan HW, Wong HN, Li KY, Tang MMK, Chen MJ, Guo ZF, Guo ZH, Sinniah K, et al. ChemMedChem. 2012;7:2204–2226. doi: 10.1002/cmdc.201200383. [DOI] [PubMed] [Google Scholar]
- 12.O’Neill PM, Stocks PA, Pugh MD, Araujo NC, Korshin EE, Bickley JF, Ward SA, Bray PG, Pasini E, Davies J, et al. Angew Chem Int Ed Engl. 2004;43:4193–4197. doi: 10.1002/anie.200453859. [DOI] [PubMed] [Google Scholar]
- 13.Mahajan SS, Deu E, Lauterwasser EMW, Leyva MJ, Ellman Ja, Bogyo M, Renslo AR. ChemMedChem. 2011;6:415–419. doi: 10.1002/cmdc.201100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahajan SS, Gut J, Rosenthal PJ, Renslo AR. Future Med Chem. 2012;4:2241–2249. doi: 10.4155/fmc.12.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deu E, Chen I, Lauterwasser EMW, Valderramos J, Li H, Edgington L, Renslo AR, Bogyo M. Proc Natl Acad Sci U S A. 2013;110:18244–18249. doi: 10.1073/pnas.1312782110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliveira R, Newton AS, Guedes RC, Miranda D, Amewu RK, Srivastava A, Gut J, Rosenthal PJ, O’Neill PM, Ward Sa, et al. ChemMedChem. 2013;8:1528–1536. doi: 10.1002/cmdc.201300202. [DOI] [PubMed] [Google Scholar]
- 17.Oliveira R, Guedes RC, Meireles P, Albuquerque IS, Gonçalves LM, Pires E, Bronze MR, Gut J, Rosenthal PJ, Prudêncio M, et al. J Med Chem. 2014;57:4916–4923. doi: 10.1021/jm5004528. [DOI] [PubMed] [Google Scholar]
- 18.Meunier B. Acc Chem Res. 2008;41:69–77. doi: 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- 19.Feng TS, Guantai EM, Nell M, van Rensburg CEJ, Ncokazi K, Egan TJ, Hoppe HC, Chibale K. Biochem Pharmacol. 2011;82:236–47. doi: 10.1016/j.bcp.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 20.Deu E, Leyva MJ, Albrow VE, Rice MJ, Ellman JA, Bogyo M. Chem Biol. 2010;17:808–819. doi: 10.1016/j.chembiol.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong Y, Chollet J, Matile H, Charman Sa, Chiu FCK, Charman WN, Scorneaux B, Urwyler H, Santo Tomas J, Scheurer C, et al. J Med Chem. 2005;48:4953–4961. doi: 10.1021/jm049040u. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Q, Vargas M, Dong Y, Zhou L, Wang X, Sriraghavan K, Keiser J, Vennerstrom JL. J Med Chem. 2010;53:4223–4233. doi: 10.1021/jm100226t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang Y, Dong Y, Karle JM, DiTusa Ca, Vennerstrom JL. J Org Chem. 2004;69:6470–3. doi: 10.1021/jo040171c. [DOI] [PubMed] [Google Scholar]
- 24.Sijwali PS, Kato K, Seydel KB, Gut J, Lehman J, Klemba M, Goldberg DE, Miller LH, Rosenthal PJ. Proc Natl Acad Sci U S A. 2004;101:8721–6. doi: 10.1073/pnas.0402738101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Creek D, Charman W, Chiu FCK, Prankerd RJ, Mccullough KJ, Dong Y, Vennerstrom JL, Charman SA. J Pharm Sci. 2007;96:2945–2956. doi: 10.1002/jps.20958. [DOI] [PubMed] [Google Scholar]
- 26.Ueki N, Lee S, Sampson NS, Hayman MJ. Nat Commun. 2013;4:2735. doi: 10.1038/ncomms3735. [DOI] [PubMed] [Google Scholar]
- 27.de Koning-Ward TF, Waters AP, Crabb BS. Mol Biochem Parasitol. 2001;117:155–160. doi: 10.1016/s0166-6851(01)00344-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.