

Abstract
The molecule of the title compound, C5H8O2, a low-melting α,β-unsaturated carboxylic acid, is essentially planar [maximum displacement = 0.0239 (13) Å]. In the crystal, molecules are linked into centrosymmetric dimers via pairs of O—H⋯O hydrogen bonds.
Keywords: crystal structure, hydrogen bond, dimer, unsaturated carboxylic acid
Related literature
For the synthesis of unsaturated carboxylic acids including the title compound, see: Shabtai et al. (1981 ▸); Gastaminza et al. (1984 ▸); Outurquin & Paulmier (1989 ▸). For crystal structure determinations of acrylic acid, see: Higgs & Sass (1963 ▸); Chatani et al. (1963 ▸); Boese et al. (1999 ▸); Oswald & Urquhart (2011 ▸). For the structure of crotonic acid, see: Shimizu et al. (1974 ▸). For the structure of related hexenoic acid cocrystals, see: Aakeröy et al. (2003 ▸); Stanton & Bak (2008 ▸).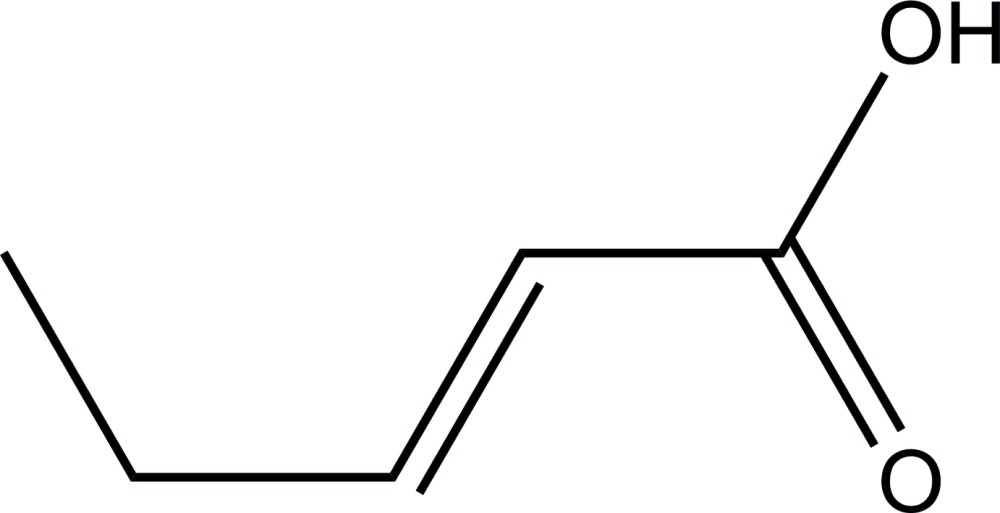
Experimental
Crystal data
C5H8O2
M r = 100.11
Triclinic,
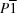
a = 6.7336 (13) Å
b = 6.7821 (13) Å
c = 7.2349 (14) Å
α = 67.743 (2)°
β = 75.518 (2)°
γ = 64.401 (2)°
V = 274.29 (9) Å3
Z = 2
Mo Kα radiation
μ = 0.09 mm−1
T = 150 K
0.51 × 0.35 × 0.27 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2014 ▸) T min = 0.81, T max = 0.97
7544 measured reflections
1323 independent reflections
1122 reflections with I > 2σ(I)
R int = 0.026
Refinement
R[F 2 > 2σ(F 2)] = 0.037
wR(F 2) = 0.113
S = 1.10
1323 reflections
69 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.36 e Å−3
Δρmin = −0.19 e Å−3
Data collection: APEX2 (Bruker, 2014 ▸); cell refinement: SAINT (Bruker, 2013 ▸); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▸); program(s) used to refine structure: SHELXL2014 (Sheldrick, 2015 ▸); molecular graphics: SHELXL2014; software used to prepare material for publication: SHELXL2014.
Supplementary Material
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S2056989015007203/rz5155sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015007203/rz5155Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015007203/rz5155Isup3.cml
. DOI: 10.1107/S2056989015007203/rz5155fig1.tif
The molecular structure of the title compound with displacement ellipsoids drawn at 50% probability level.
ORTEP . DOI: 10.1107/S2056989015007203/rz5155fig2.tif
ORTEP representation of a dimer formed by intermolecular O—H⋯O hydrogen bonds.
CCDC reference: 1058870
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| O1H1O2i | 0.95(2) | 1.69(2) | 2.6322(13) | 173.3(19) |
Symmetry code: (i) 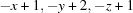 .
.
Acknowledgments
The authors thank P. Thiele (University of Rostock) for DSC measurements and Professor Dr J. G. de Vries (LIKAT) for helpful support.
supplementary crystallographic information
S1. Synthesis and crystallization
Malonic acid (24.8 g, 237.8 mmol, 1eq) was dissolved in dry pyridine (37.6 g, 475.7 mmol, 2 eq) at room temperature in a three-necked flask equipped with a magnetic stir bar and a reflux condenser under a mild flow of argon. Propanal (13.8 g, 240.2 mmol, 1 eq) was then added in one portion and the resulting clear solution further stirred for 72 h at room temperature under argon. Afterwards, the resulting light yellow to orange solution was brought to an acidic pH value by adding phosphoric acid at 0°C (42.5 wt.-%, 582.7 mmol, 2.45 eq). The resulting two layers were extracted three times with 150 mL portions of ethyl acetate and reduced to a volume of ca. 150 mL. To remove impurities from aldol condensation the raw acid was converted into the corresponding sodium salt by addition of an aqueous solution of sodium carbonate (18.9 g, 178.4 mmol, 0.75 eq in 200 mL). After stirring for 30 minutes the water phase was separated und extracted three times with 150 mL portions of ethyl acetate. The water phase was then acidified with concentrated hydrochloric acid (35.2 g, 356.7 mmol, 1.5 eq), the organic phase was separated and the water phase was again extracted three times with 150 mL portions of ethyl acetate. The combined organic phases were dried over Na2SO4 and evaporated to dryness under diminished pressure. The resulting raw product was further purified by distillation in vacuo yielding the product in purity >99% (GC). M. p. 10°C. 1H NMR (400 MHz, CDCl3): δ = 12.35 (br s, 1H, OH); 7.14 (dt, 3J = 15.6 Hz, 3J = 6.3 Hz, 1H, -CH-); 5.82 (dt, 3J = 15.6 Hz, 4J = 1.7 Hz, 1H, -CH-); 2.30-2.21 (m, 2H, -CH2-); 1.08 (t, 3J = 7.4 Hz, 3H, -CH3-). 13C NMR (100 MHz, CDCl3): δ = 172.69 (CO); 153.77 (CH); 119.76 (CH); 25.54 (CH2); 21.10 (CH3). MS (EI, 70 eV): m/z = 100 (M+, 50), 83 (13), 82 (23), 81 (10), 58 (11), 57 (17), 56 (23), 55 (100), 54 (43), 53 (35), 52 (12), 51 (25), 50 (28), 45 (77), 41 (36), 40 (13), 39 (99), 38 (25), 37 (11), 29 (61). HRMS (ESI-TOF/MS): calculated for C5H8O2 (M+) 99.04515, found 99.04529. Elemental analysis for C5H8O2 % (calc.): C 59.99 (59.98); H 8.05 (8.05). Suitable single crystals were grown by slow evaporation of an ethanolic solution at -30 °C over one week.
S2. Refinement
The carboxylic H atom could be found in a difference Fourier map and was refined freely. All other H atoms were placed in idealized positions with d(C—H) = 0.95 Å (CH), 0.99 Å (CH2), 0.98 Å (CH3) and refined using a riding model with Uiso(H) fixed at 1.2 Ueq(C) for CH and CH2 and 1.5 Ueq(C) for CH3. A rotating model was used for the methyl group.
Figures
Fig. 1.
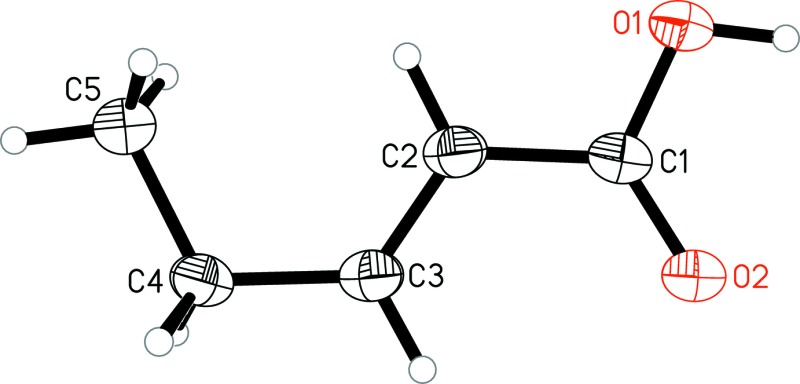
The molecular structure of the title compound with displacement ellipsoids drawn at 50% probability level.
Fig. 2.
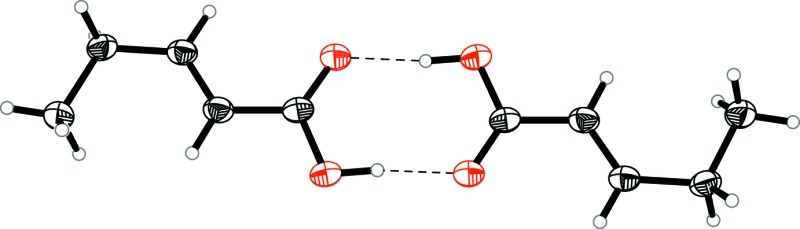
ORTEP representation of a dimer formed by intermolecular O—H···O hydrogen bonds.
Crystal data
| C5H8O2 | Z = 2 |
| Mr = 100.11 | F(000) = 108 |
| Triclinic, P1 | Dx = 1.212 Mg m−3 |
| a = 6.7336 (13) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 6.7821 (13) Å | Cell parameters from 4399 reflections |
| c = 7.2349 (14) Å | θ = 3.1–28.7° |
| α = 67.743 (2)° | µ = 0.09 mm−1 |
| β = 75.518 (2)° | T = 150 K |
| γ = 64.401 (2)° | Prism, colourless |
| V = 274.29 (9) Å3 | 0.51 × 0.35 × 0.27 mm |
Data collection
| Bruker APEXII CCD diffractometer | 1323 independent reflections |
| Radiation source: fine-focus sealed tube | 1122 reflections with I > 2σ(I) |
| Detector resolution: 8.3333 pixels mm-1 | Rint = 0.026 |
| φ and ω scans | θmax = 28.0°, θmin = 3.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2014) | h = −8→8 |
| Tmin = 0.81, Tmax = 0.97 | k = −8→8 |
| 7544 measured reflections | l = −9→9 |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.037 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.113 | w = 1/[σ2(Fo2) + (0.0562P)2 + 0.0602P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.10 | (Δ/σ)max < 0.001 |
| 1323 reflections | Δρmax = 0.36 e Å−3 |
| 69 parameters | Δρmin = −0.19 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.36613 (18) | 0.77456 (19) | 0.59878 (16) | 0.0284 (3) | |
| C2 | 0.25933 (18) | 0.6065 (2) | 0.66526 (16) | 0.0295 (3) | |
| H2 | 0.1507 | 0.6289 | 0.5893 | 0.035* | |
| C3 | 0.31028 (18) | 0.4243 (2) | 0.82842 (16) | 0.0290 (3) | |
| H3 | 0.4190 | 0.4066 | 0.9018 | 0.035* | |
| C4 | 0.2111 (2) | 0.2446 (2) | 0.90640 (17) | 0.0323 (3) | |
| H4A | 0.1414 | 0.2401 | 1.0450 | 0.039* | |
| H4B | 0.3320 | 0.0930 | 0.9135 | 0.039* | |
| C5 | 0.0389 (2) | 0.2791 (2) | 0.78294 (19) | 0.0372 (3) | |
| H5A | −0.0864 | 0.4244 | 0.7812 | 0.056* | |
| H5B | −0.0133 | 0.1517 | 0.8430 | 0.056* | |
| H5C | 0.1055 | 0.2834 | 0.6452 | 0.056* | |
| O1 | 0.30172 (15) | 0.93766 (15) | 0.42861 (12) | 0.0357 (3) | |
| O2 | 0.50223 (14) | 0.76619 (15) | 0.69113 (12) | 0.0361 (3) | |
| H1 | 0.376 (3) | 1.039 (4) | 0.395 (3) | 0.071 (6)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0277 (5) | 0.0275 (6) | 0.0251 (5) | −0.0060 (4) | −0.0070 (4) | −0.0056 (4) |
| C2 | 0.0270 (5) | 0.0326 (6) | 0.0272 (5) | −0.0093 (5) | −0.0077 (4) | −0.0066 (4) |
| C3 | 0.0265 (5) | 0.0325 (6) | 0.0257 (5) | −0.0086 (4) | −0.0067 (4) | −0.0071 (4) |
| C4 | 0.0321 (6) | 0.0332 (6) | 0.0267 (5) | −0.0121 (5) | −0.0087 (4) | −0.0010 (4) |
| C5 | 0.0378 (6) | 0.0382 (7) | 0.0355 (6) | −0.0177 (5) | −0.0135 (5) | −0.0013 (5) |
| O1 | 0.0406 (5) | 0.0339 (5) | 0.0295 (4) | −0.0147 (4) | −0.0153 (3) | 0.0015 (3) |
| O2 | 0.0402 (5) | 0.0345 (5) | 0.0332 (5) | −0.0155 (4) | −0.0166 (4) | −0.0005 (3) |
Geometric parameters (Å, º)
| C1—O2 | 1.2337 (14) | C4—C5 | 1.5239 (16) |
| C1—O1 | 1.3223 (13) | C4—H4A | 0.9900 |
| C1—C2 | 1.4723 (16) | C4—H4B | 0.9900 |
| C2—C3 | 1.3301 (16) | C5—H5A | 0.9800 |
| C2—H2 | 0.9500 | C5—H5B | 0.9800 |
| C3—C4 | 1.4981 (16) | C5—H5C | 0.9800 |
| C3—H3 | 0.9500 | O1—H1 | 0.95 (2) |
| O2—C1—O1 | 122.75 (11) | C5—C4—H4A | 108.4 |
| O2—C1—C2 | 123.99 (10) | C3—C4—H4B | 108.4 |
| O1—C1—C2 | 113.26 (10) | C5—C4—H4B | 108.4 |
| C3—C2—C1 | 122.03 (10) | H4A—C4—H4B | 107.5 |
| C3—C2—H2 | 119.0 | C4—C5—H5A | 109.5 |
| C1—C2—H2 | 119.0 | C4—C5—H5B | 109.5 |
| C2—C3—C4 | 125.63 (10) | H5A—C5—H5B | 109.5 |
| C2—C3—H3 | 117.2 | C4—C5—H5C | 109.5 |
| C4—C3—H3 | 117.2 | H5A—C5—H5C | 109.5 |
| C3—C4—C5 | 115.33 (10) | H5B—C5—H5C | 109.5 |
| C3—C4—H4A | 108.4 | C1—O1—H1 | 108.7 (12) |
| O2—C1—C2—C3 | −3.05 (19) | C1—C2—C3—C4 | −179.72 (10) |
| O1—C1—C2—C3 | 176.98 (11) | C2—C3—C4—C5 | 0.45 (18) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2i | 0.95 (2) | 1.69 (2) | 2.6322 (13) | 173.3 (19) |
Symmetry code: (i) −x+1, −y+2, −z+1.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: RZ5155).
References
- Aakeröy, C. B., Beatty, A. M., Helfrich, B. A. & Nieuwenhuyzen, M. (2003). Cryst. Growth Des. 3, 159–165.
- Boese, R., Bläser, D., Steller, I., Latz, R. & Bäumen, A. (1999). Acta Cryst. C55, IUC9900006.
- Bruker (2013). SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2014). APEX2 and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Chatani, Y., Sakata, Y. & Nitta, I. (1963). J. Polym. Sci. B Polym. Lett. 1, 419–421.
- Gastaminza, A. E., Ferracutti, N. N. & Rodriguez, N. M. (1984). J. Org. Chem. 49, 3859–3860.
- Higgs, M. A. & Sass, R. L. (1963). Acta Cryst. 16, 657–661.
- Oswald, I. D. H. & Urquhart, A. J. (2011). CrystEngComm, 13, 4503–4507.
- Outurquin, F. & Paulmier, C. (1989). Synthesis, pp. 690–691.
- Shabtai, J., Ney-Igner, E. & Pines, H. (1981). J. Org. Chem. 46, 3795–3802.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Shimizu, S., Kekka, S., Kashino, S. & Haisa, M. (1974). Bull. Chem. Soc. Jpn, 47, 1627–1631.
- Stanton, M. K. & Bak, A. (2008). Cryst. Growth Des. 8, 3856–3862.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S2056989015007203/rz5155sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015007203/rz5155Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015007203/rz5155Isup3.cml
. DOI: 10.1107/S2056989015007203/rz5155fig1.tif
The molecular structure of the title compound with displacement ellipsoids drawn at 50% probability level.
ORTEP . DOI: 10.1107/S2056989015007203/rz5155fig2.tif
ORTEP representation of a dimer formed by intermolecular O—H⋯O hydrogen bonds.
CCDC reference: 1058870
Additional supporting information: crystallographic information; 3D view; checkCIF report