

While attempting to synthesize a cyclopentadienyl iridium complex by the reaction between IrCl3·xH2O in methanol, several well-shaped crystals formed from the reaction mixture. Surprisingly, the crystals were of di-μ-chlorido-bis[dichloridobis(methanol-κO)iridium(III)] dihydrate, [Ir2Cl6(CH3OH)4]·2H2O. This is a surprising result in that, while many reactions of iridium chloride hydrate are carried out in alcoholic solvents, especially methanol and ethanol, this is the first structure of a chlorido-iridium compound with only methanol ligands.
Keywords: crystal structure, iridium, chlorido bridge, methanol ligand, hydrogen bonding
Abstract
The reaction between IrCl3·xH2O in methanol led to the formation of small amounts of the title compound, [Ir2Cl6(CH3OH)4]·2H2O, which consists of two IrCl4O2 octahedra sharing an edge via chloride bridges. The molecule lies across an inversion center. Each octahedron can be envisioned as being comprised of four chloride ligands in the equatorial plane with methanol ligands in the axial positions. A lattice water molecule is strongly hydrogen-bonded to the coordinating methanol ligands and weak interactions with coordinating chloride ligands lead to the formation of a three-dimensional network. This is a surprising structure given that, while many reactions of iridium chloride hydrate are carried out in alcoholic solvents, especially methanol and ethanol, this is the first structure of a chloridoiridium compound with only methanol ligands.
Chemical context
The use of alcoholic solvents with IrCl3·xH2O for the formation of cyclopentadienyl or olefin iridium complexes is exceedingly common (Herde et al., 2007 ▸; Liu et al., 2008 ▸, 2011 ▸; Morris et al., 2014 ▸). Lately, we have been investigating the syntheses of half-sandwich iridium complexes with varying tetramethylalkylcyclopentadienyl ligands (Morris et al., 2014 ▸). In all cases, the reaction takes place between IrCl3·xH2O and the tetramethylalkylcyclopentadiene in methanol, either under thermal or microwave conditions. In most cases, the yields of of Cp*R iridium chlorido-bridged dimers are good to excellent. Several reactions to synthesize Cp*R iridium complexes with R = long-chain alkyls such as n-hexyl, n-heptyl and n-octyl produced good yields of the desired [Cp*RIrCl2]2 compounds but, in one instance, only produced a few crystals which turned out to be those of the title compound. Given the number of reactions that are carried out with IrCl3·xH2O in methanol, that this is the first time this compound has been seen by us or by any others active in the field is surprising.
Structural commentary
The title structure (Fig. 1 ▸) consists of two iridium-centered octahedra sharing one edge via chloride bridges. For each octahedron, there are two terminal chloride ligands in the same plane as the bridging chloride ligands. The axial positions that complete the octahedra are occupied by O-bonded methanol ligands. One of the methanol ligands on each iridium atom is hydrogen-bonded to a lattice water. The two iridium-centered octahedra are related by an inversion center. The Ir—Cl bridges are symmetrical with identical Ir—Cl bond lengths of 2.385 (1) Å with two of the Ir—Cl bonds equivalent by symmetry and the unique bonds coincidentally equivalent [2.3847 (10) and 2.3846 (11) Å]. The only structure similar to the title compound currently in the Cambridge Structural Database (CSD version 5.35 with updates, Groom & Allen, 2014 ▸) is CCDC: CLESIR, bis(μ-chlorido)tetrachloridotetrakis(diethylsulfide)diiridium(III) (Williams et al., 1980 ▸). The structural similarities between CLESIR and the title compound are that both contain octahedrally coordinated iridium atoms with the octahedra sharing one edge via Ir–Cl–Ir bridges. There are also two terminal chloride ligands on each iridium for both compounds. In the case of CLESIR, however, the remaining ligands on the iridium are diethylsulfido ligands. An additional difference is that, for the title compound, all chloride ligands are in the equatorial plane with methanol ligands occupying axial positions. For CLESIR, the diethylsulfido ligands on one iridium atom occupy axial positions but occupy equatorial positions on the second iridium.
Figure 1.

The full molecular unit of the title compound with hydrogen-bonded lattice water molecules [symmetry code (i) −x + 1, −y + 1, −z + 2]. Displacement ellipsoids are drawn at the 50% probability level.
Supramolecular features
Each lattice water molecule forms four hydrogen bonds linking four different iridium-centered dimers. Table 1 ▸ lists the various parameters describing the hydrogen bonding. As a donor, the water participates in two O—H⋯Cl bonds to chloride ligands on different molecules while, as acceptor, the water participates in two O—H⋯O bonds to methanol oxygen atoms on two additional molecules. A search of the CSD for O—H⋯Cl bonds between lattice water and chloride attached to any transition metal followed by analysis in Mercury (Macrae et al., 2008 ▸) show that the O⋯Cl distances have a mean of 3.151 Å with a mean deviation of 0.055 Å. The two O⋯Cl distances of 3.208 (4) and 3.285 (3) Å for this structure places the distances at the high end of the range. However, when acting as acceptors, the lattice water displays O(methanol)—O(water) distances of 2.752 (5) and 2.647 (5) Å. A search of the CSD with analysis by Mercury (Macrae et al., 2008 ▸) uncovers a mean O(donor)⋯O(acceptor) distance of 2.742 Å with a mean deviation of 0.085 Å, putting the donor–acceptor distances at the mean and slightly below the mean of these types of hydrogen bonds. Fig. 2 ▸ shows the hydrogen-bonding network that is created throughout the lattice of the title compound with the methanol methyl groups removed for clarity.
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| O1H1O3i | 0.86(1) | 1.96(3) | 2.752(5) | 151(4) |
| O2H2O3 | 0.87(1) | 1.78(1) | 2.647(5) | 179(2) |
| O3H3ACl2ii | 0.85 | 2.39 | 3.208(4) | 160 |
| O3H3BCl1iii | 0.85 | 2.45 | 3.285(3) | 166 |
Symmetry codes: (i)  ; (ii)
; (ii)  ; (iii)
; (iii) 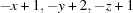 .
.
Figure 2.

Packing diagram of the title compound showing the hydrogen-bonding (dashed lines) network. Displacement ellipsoids are drawn at the 50% probability level.
Database survey
A survey of the CSD found only 11 structures of iridium with methanol ligands (methoxide ligands were excluded from the search but they added only an additional eight structures to the result). Analysis with Mercury (Macrae et al., 2008 ▸) found that Ir—O bonds in this small subset ranged from 2.185 to 2.317 Å with a mean of 2.251 Å and a standard deviation of 0.042 Å. The Ir—O bond lengths of the title compound of 2.066 (3) and 2.057 (3) Å are significantly smaller than the low end of this range. The small number of samples and the variety of structures available for comparison do not permit any clear conclusions as to the significance of these distances. All of the structures are of iridium(III) but the title compound is the only one with chloride as the sole other ligand set on each metal. While this structure determination was carried out at 100 K compared with room temperature for most of the other compounds with methanol ligands, such a significant bond shortening would not be expected based solely on temperature (Macchi & Sironi, 2004 ▸).
Synthesis and crystallization
IrCl3·xH2O and 1-heptyl, 2,3,4,5-tetramethylcyclopentadiene were mixed in a round-bottom flask with 15 mL of MeOH and the reaction mixture was refluxed for two days. This procedure has been successfully used to synthesize a number of pentaalkyliridium chloride compounds in the past. After cooling to room temperature, the round-bottom flask was placed into a freezer overnight. There was no evidence of any product crystallization. The reaction mixture was then evaporated to dryness, yielding a tarry mixture. The tarry mixture was dissolved in diethyl ether and allowed to evaporate slowly. After the ether had evaporated, the mixture was again very tarry in appearance, but this time with a few crystals obvious in the flask. The structure of the title compound was determined from one of those crystals. It is unclear why this reaction did not proceed normally.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸. H atoms bonded to C atoms were included in calculated positions with C—H = 0.96 Å and U iso(H) = 1.5U eq(C). H atoms bonded to water O atoms were included in calculated positions with O—H = 0.85 Å and U iso(H) = 1.5U eq(C). The H atoms bonded to methanol O atoms were refined independently with isotropic displacement parameters.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | [Ir2Cl6(CH4O)4]2H2O |
| M r | 380.68 |
| Crystal system, space group | Triclinic, P

|
| Temperature (K) | 100 |
| a, b, c () | 7.1445(4), 7.4876(5), 8.6362(7) |
| , , () | 73.597(6), 75.596(5), 89.404(5) |
| V (3) | 428.37(5) |
| Z | 2 |
| Radiation type | Mo K |
| (mm1) | 16.46 |
| Crystal size (mm) | 0.14 0.11 0.09 |
| Data collection | |
| Diffractometer | Agilent Xcalibur Eos Gemini ultra |
| Absorption correction | Gaussian (CrysAlis PRO; Agilent, 2014 ▸) |
| T min, T max | 0.184, 0.342 |
| No. of measured, independent and observed [I > 2(I)] reflections | 7987, 2817, 2575 |
| R int | 0.040 |
| (sin /)max (1) | 0.750 |
| Refinement | |
| R[F 2 > 2(F 2)], wR(F 2), S | 0.028, 0.055, 1.03 |
| No. of reflections | 2817 |
| No. of parameters | 93 |
| No. of restraints | 6 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| max, min (e 3) | 1.67, 1.94 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989015006672/lh5757sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015006672/lh5757Isup2.hkl
CCDC reference: 1057748
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The open-access fee was provided by the Virginia Tech Open Access Subvention Fund.
supplementary crystallographic information
Crystal data
| [Ir2Cl6(CH4O)4]·2H2O | Z = 2 |
| Mr = 380.68 | F(000) = 348 |
| Triclinic, P1 | Dx = 2.951 Mg m−3 |
| a = 7.1445 (4) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.4876 (5) Å | Cell parameters from 3445 reflections |
| c = 8.6362 (7) Å | θ = 4.0–32.1° |
| α = 73.597 (6)° | µ = 16.46 mm−1 |
| β = 75.596 (5)° | T = 100 K |
| γ = 89.404 (5)° | Prism, clear light orange |
| V = 428.37 (5) Å3 | 0.14 × 0.11 × 0.09 mm |
Data collection
| Agilent Xcalibur Eos Gemini ultra diffractometer | 2817 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source, Agilent | 2575 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.040 |
| Detector resolution: 16.0122 pixels mm-1 | θmax = 32.2°, θmin = 4.0° |
| ω scans | h = −10→10 |
| Absorption correction: gaussian (CrysAlis PRO; Agilent, 2014) | k = −11→11 |
| Tmin = 0.184, Tmax = 0.342 | l = −12→12 |
| 7987 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.028 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.055 | w = 1/[σ2(Fo2) + (0.0194P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.03 | (Δ/σ)max = 0.001 |
| 2817 reflections | Δρmax = 1.67 e Å−3 |
| 93 parameters | Δρmin = −1.94 e Å−3 |
| 6 restraints |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Ir1 | 0.64307 (2) | 0.65568 (2) | 0.81418 (2) | 0.01042 (5) | |
| Cl1 | 0.82335 (16) | 0.93834 (15) | 0.74853 (14) | 0.0167 (2) | |
| Cl2 | 0.72146 (16) | 0.65918 (17) | 0.53518 (13) | 0.0177 (2) | |
| Cl3 | 0.45411 (15) | 0.36814 (14) | 0.89543 (12) | 0.01298 (19) | |
| O1 | 0.9022 (5) | 0.5412 (5) | 0.8390 (4) | 0.0169 (7) | |
| H1 | 1.0118 (19) | 0.603 (3) | 0.785 (5) | 0.025* | |
| O2 | 0.3880 (4) | 0.7770 (5) | 0.7908 (4) | 0.0151 (6) | |
| H2 | 0.328 (5) | 0.759 (4) | 0.720 (4) | 0.023* | |
| C1 | 0.9356 (7) | 0.3485 (7) | 0.8412 (6) | 0.0208 (10) | |
| H1A | 0.9287 | 0.3317 | 0.7366 | 0.031* | |
| H1B | 0.8388 | 0.2669 | 0.9302 | 0.031* | |
| H1C | 1.0615 | 0.3197 | 0.8584 | 0.031* | |
| C2 | 0.3605 (8) | 0.9702 (7) | 0.7877 (7) | 0.0274 (12) | |
| H2A | 0.4497 | 1.0503 | 0.6912 | 0.041* | |
| H2B | 0.3835 | 0.9897 | 0.8868 | 0.041* | |
| H2C | 0.2303 | 0.9984 | 0.7831 | 0.041* | |
| O3 | 0.2082 (5) | 0.7157 (5) | 0.5754 (4) | 0.0182 (7) | |
| H3A | 0.2516 | 0.6327 | 0.5284 | 0.027* | |
| H3B | 0.2060 | 0.8178 | 0.5013 | 0.027* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Ir1 | 0.01073 (8) | 0.01068 (8) | 0.00999 (8) | −0.00075 (6) | −0.00180 (6) | −0.00389 (6) |
| Cl1 | 0.0172 (5) | 0.0126 (5) | 0.0189 (5) | −0.0039 (4) | −0.0030 (4) | −0.0034 (4) |
| Cl2 | 0.0209 (5) | 0.0215 (6) | 0.0104 (5) | 0.0007 (4) | −0.0018 (4) | −0.0058 (4) |
| Cl3 | 0.0167 (5) | 0.0117 (5) | 0.0113 (4) | −0.0021 (4) | −0.0022 (4) | −0.0056 (4) |
| O1 | 0.0149 (15) | 0.0173 (17) | 0.0199 (16) | 0.0019 (13) | −0.0048 (13) | −0.0074 (13) |
| O2 | 0.0152 (15) | 0.0185 (17) | 0.0161 (15) | 0.0035 (13) | −0.0073 (12) | −0.0092 (13) |
| C1 | 0.018 (2) | 0.020 (2) | 0.025 (2) | 0.0057 (19) | −0.0036 (19) | −0.009 (2) |
| C2 | 0.028 (3) | 0.017 (2) | 0.036 (3) | 0.003 (2) | −0.009 (2) | −0.006 (2) |
| O3 | 0.0196 (17) | 0.0180 (17) | 0.0184 (16) | 0.0041 (14) | −0.0076 (13) | −0.0051 (14) |
Geometric parameters (Å, º)
| Ir1—Cl1 | 2.3400 (11) | O2—C2 | 1.452 (6) |
| Ir1—Cl2 | 2.3267 (11) | C1—H1A | 0.9600 |
| Ir1—Cl3i | 2.3847 (10) | C1—H1B | 0.9600 |
| Ir1—Cl3 | 2.3846 (11) | C1—H1C | 0.9600 |
| Ir1—O1 | 2.066 (3) | C2—H2A | 0.9600 |
| Ir1—O2 | 2.057 (3) | C2—H2B | 0.9600 |
| Cl3—Ir1i | 2.3847 (10) | C2—H2C | 0.9600 |
| O1—H1 | 0.864 (10) | O3—H3A | 0.8500 |
| O1—C1 | 1.456 (6) | O3—H3B | 0.8498 |
| O2—H2 | 0.867 (9) | ||
| Cl1—Ir1—Cl3i | 92.90 (4) | C1—O1—H1 | 105.1 (16) |
| Cl1—Ir1—Cl3 | 177.08 (4) | Ir1—O2—H2 | 121.2 (16) |
| Cl2—Ir1—Cl1 | 91.47 (4) | C2—O2—Ir1 | 121.9 (3) |
| Cl2—Ir1—Cl3i | 175.59 (4) | C2—O2—H2 | 104.7 (15) |
| Cl2—Ir1—Cl3 | 91.44 (4) | O1—C1—H1A | 109.5 |
| Cl3—Ir1—Cl3i | 84.19 (4) | O1—C1—H1B | 109.5 |
| O1—Ir1—Cl1 | 83.45 (10) | O1—C1—H1C | 109.5 |
| O1—Ir1—Cl2 | 90.01 (9) | H1A—C1—H1B | 109.5 |
| O1—Ir1—Cl3i | 91.08 (9) | H1A—C1—H1C | 109.5 |
| O1—Ir1—Cl3 | 96.79 (10) | H1B—C1—H1C | 109.5 |
| O2—Ir1—Cl1 | 94.91 (10) | O2—C2—H2A | 109.5 |
| O2—Ir1—Cl2 | 90.73 (9) | O2—C2—H2B | 109.5 |
| O2—Ir1—Cl3 | 84.81 (10) | O2—C2—H2C | 109.5 |
| O2—Ir1—Cl3i | 88.30 (9) | H2A—C2—H2B | 109.5 |
| O2—Ir1—O1 | 178.22 (13) | H2A—C2—H2C | 109.5 |
| Ir1—Cl3—Ir1i | 95.81 (4) | H2B—C2—H2C | 109.5 |
| Ir1—O1—H1 | 121.2 (16) | H3A—O3—H3B | 109.5 |
| C1—O1—Ir1 | 121.8 (3) |
Symmetry code: (i) −x+1, −y+1, −z+2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O3ii | 0.86 (1) | 1.96 (3) | 2.752 (5) | 151 (4) |
| O2—H2···O3 | 0.87 (1) | 1.78 (1) | 2.647 (5) | 179 (2) |
| O3—H3A···Cl2iii | 0.85 | 2.39 | 3.208 (4) | 160 |
| O3—H3B···Cl1iv | 0.85 | 2.45 | 3.285 (3) | 166 |
Symmetry codes: (ii) x+1, y, z; (iii) −x+1, −y+1, −z+1; (iv) −x+1, −y+2, −z+1.
References
- Agilent (2014). CrysAlis PRO. Agilent Technologies Ltd, Yarnton, England.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Groom, C. R. & Allen, F. H. (2014). Angew. Chem. Int. Ed. 53, 662–671. [DOI] [PubMed]
- Herde, J. L., Lambert, J. C., Senoff, C. V. & Cushing, M. A. (2007). Inorganic Syntheses, pp. 18–20. New York: John Wiley & Sons Inc.
- Liu, Z., Habtemariam, A., Pizarro, A. M., Fletcher, S. A., Kisova, A., Vrana, O., Salassa, L., Bruijnincx, P. C. A., Clarkson, G. J., Brabec, V. & Sadler, P. J. (2011). J. Med. Chem. 54, 3011–3026. [DOI] [PubMed]
- Liu, J., Wu, X., Iggo, J. A. & Xiao, J. (2008). Coord. Chem. Rev. 252, 782–809.
- Macchi, P. & Sironi, A. (2004). Acta Cryst. A60, 502–509. [DOI] [PubMed]
- Macrae, C. F., Bruno, I. J., Chisholm, J. A., Edgington, P. R., McCabe, P., Pidcock, E., Rodriguez-Monge, L., Taylor, R., van de Streek, J. & Wood, P. A. (2008). J. Appl. Cryst. 41, 466–470.
- Morris, D. M., McGeagh, M., De Peña, D. & Merola, J. S. (2014). Polyhedron, 84, 120–135.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Williams, A. F., Flack, H. D. & Vincent, M. G. (1980). Acta Cryst. B36, 1204–1206.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989015006672/lh5757sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015006672/lh5757Isup2.hkl
CCDC reference: 1057748
Additional supporting information: crystallographic information; 3D view; checkCIF report