

Abstract
HbSC disease is the second commonest form of sickle cell disease, with poorly understood pathophysiology and few treatments. We studied the role of K-Cl cotransport activity in determining clinical and laboratory features, and investigated its potential role as a biomarker. Samples were collected from 110 patients with HbSC disease and 41 with sickle cell anemia (HbSS). K-Cl cotransport activity was measured in the oxygenated (K-Cl cotransport100) and deoxygenated (K-Cl cotransport0) states, using radioactive tracer studies. K-Cl cotransport activity was high in HbSC and decreased significantly on deoxygenation. K-Cl cotransport activity correlated significantly and positively with the formation of sickle cells. On multiple regression analysis, K-Cl cotransport increased significantly and independently with increasing reticulocyte count and age. K-Cl cotransport activity was increased in patients who attended hospital with acute pain in 2011 compared to those who did not (K-Cl cotransport100: mean 3.87 versus 3.20, P=0.009, independent samples T-test; K-Cl cotransport0: mean 0.96 versus 0.68, P=0.037). On logistic regression only K-Cl cotransport was associated with hospital attendance. Increased K-Cl cotransport activity was associated with the presence of retinopathy, but this effect was confounded by age. This study links variability in a fundamental aspect of cellular pathology with a clinical outcome, suggesting that K-Cl cotransport is central to the pathology of HbSC disease. Increased K-Cl cotransport activity is associated with increasing age, which may be of pathophysiological significance. Effective inhibition of K-Cl cotransport activity is likely to be of therapeutic benefit.
Introduction
HbSC disease is the second commonest form of sickle cell disease in African populations, but both its pathophysiological and clinical aspects are poorly understood. It affects about 2% of the population in West Africa, with recorded incidences of up to 25% in some areas, such as Burkina Faso and Northern Ghana.1 It is less severe than sickle cell anemia (HbSS), with half the prevalence of acute pain,2 and life expectancy up to 20 years longer.3 However, serious complications occur: people with HbSC disease have a 100 times higher risk of stroke compared to the general population,4 and an increased risk of proliferative retinopathy, with up to 70% of adults affected.5 As with all types of sickle cell disease, there is marked, unexplained clinical variability: about 50% patients report no episodes of pain per year, whereas a minority suffer severe pain more than six times per year.2 Equally, steady-state hemoglobin levels vary widely, from 7 g/dL to 14 g/dL.6 Clinical management is inferred from studies of HbSS, with little evidence that this is appropriate, and there are no specific treatments for HbSC disease.
The pathophysiology of HbSC disease is very different from that of HbSS. For example, blood concentrations of hemoglobin and creatinine are significantly higher in HbSC than in HbSS, whereas concentrations of white cells, reticulocytes, lactate dehydrogenase and HbF are significantly lower.7 HbS polymerization, red cell dehydration and vaso-occlusion are central to the pathology of both HbSC and HbSS.8 However, the concentration of HbS in red cells from patients with HbSC is only 40–50%, significantly less than the 80–95% in HbSS. The rate of HbS polymerization is critically dependent on the intra-erythrocytic concentration of HbS, and so it is surprising that HbSC patients develop vaso-occlusion, given that sickle cell carriers (HbAS), with HbS levels of 30–40%, are largely asymptomatic.9
The explanation for the vaso-occlusive nature of HbSC erythrocytes is believed to center on the fact they leak cations and water, thereby increasing the concentration of HbS to critical levels. Oxygenated HbSC erythrocytes show an exaggerated potassium efflux in response to volume increase, and this is dependent on chloride ions, and stimulated by N-ethylmaleimide, which is characteristic of K-Cl cotransport (KCC).10 There is evidence that this dehydration is due to an interaction between the HbC and KCC molecules present in the red cell membrane, although the exact mechanism is unknown.9 HbC is known to bind to the cytoplasmic region of band 3,11 although it is unclear how this leads to erythrocyte dehydration.12
KCC activation has been established to be central to the pathophysiology of HbSC disease, with many experiments demonstrating its importance in vitro.13 In this study we investigated the hypothesis that variation in KCC activity is responsible for some of the phenotypic variation seen in HbSC disease, including differences in hospital attendance, retinopathy and the degree of anemia. We also assessed how KCC activity varies with age, sex and other parameters to try and understand how this variation occurs.
Methods
Patients
The study was approved by the National Research Ethics Committee London-East (reference 11/LO/0065). All participants gave written informed consent and the research was conducted in accordance with the Helsinki Declaration of 1975, as revised in 2008.
Patients were recruited from adults and children (>2 years old) attending clinics at King’s College Hospital in south London, UK. Approximately 300 patients with HbSC and 700 with HbSS are followed at our hospital. Patients with HbSC and HbSS were recruited sequentially as they attended clinic appointments, the latter for comparison to identify the specific significance of KCC in HbSC disease. All diagnoses of sickle cell disease were confirmed by family, hematologic and DNA studies, as appropriate. All patients were in the steady-state, with no acute symptoms requiring treatment for at least 7 days. Patients were excluded if they had received a blood transfusion in the preceding 4 months, had significant renal impairment, smoked or were on any medications known to alter red cell cation transport, including hydroxyurea. Following consent, clinical and laboratory data were recorded, including basic hematologic parameters, admissions to hospital and Emergency Room attendances with complications of sickle cell disease in 2011. Full blood counts were performed using a Siemens Advia system within 4 h of venesection. Retinopathy was screened for routinely every 12 – 24 months after the age of 14 by an experienced ophthalmologist, and was graded according to the Goldberg classification;14 younger children were only referred for formal ophthalmological review if they had relevant visual symptoms.
Measurement of K-Cl cotransport
Venous blood was taken into EDTA and analyzed within 24 h. Red cells were washed in MOPS-buffered saline, and suspended at a hematocrit of 30% in test saline. Oxygen tension was controlled using Eschweiler tonometers and a Wosthoff gas-mixing pump. Cell suspensions were equilibrated for 15 min at the required oxygen tension, before being diluted 10-fold into test tubes for flux measurement. Cl−-dependent K+ influx was measured using radioactive tracer assays for KCC activity, according to previously published methods.15 As the transport is known to be affected by oxygen tension in a way that differs between red cells from normal individuals and those from patients with sickle cell disease,15 KCC activity was measured at two different oxygen tensions (100 and 0 mmHg).
Measurement of red cell sickling
Red cells were washed and suspended in basic saline (NaCl 145 nM, MOPS 10 nM, glucose 5 nM, pH 7.4 adjusted with Tris base) at a hematocrit of 2%. The red cells were incubated in a tonometer in the absence of oxygen for 60 min and fixed by the addition of 0.3% glutaraldehyde. Within 10 min, 300–400 cells were counted under light microscopy using an improved Neubauer hemocytometer and the percentage of sickled cells (sickle0 %) was calculated.
Statistical analysis
An independent statistician gave advice. Data were normally distributed, as assessed by a Q-Q plot. Parametric tests were used as appropriate, including independent sample T-tests, correlation, and linear regression. Two-tailed P values were used, and values <0.05 were considered statistically significant. Analyses were performed using SPSS version 21 (IBM, Portsmouth, UK).
This study was funded by the MRC. The MRC had no direct input into the design, execution or interpretation of this study, nor the preparation of this manuscript.
Results
Basic parameters
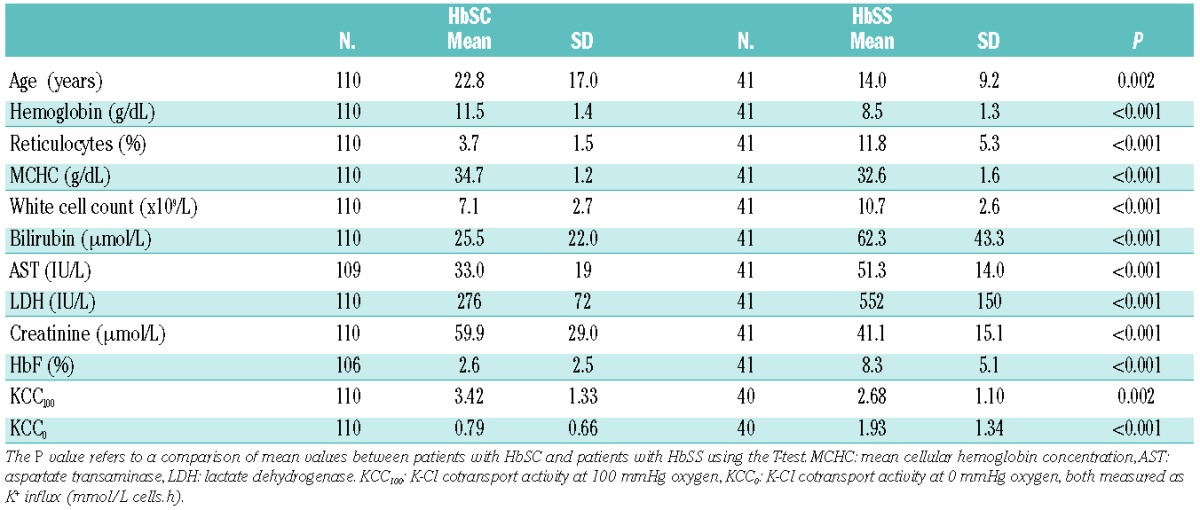
One hundred and eleven patients with HbSC disease consented to take part in the study. Their basic laboratory values are given in Table 1. Forty-eight (43.1%) were aged 16 years or younger. A comparison of distributions between male and female patients showed that male patients had significantly higher hemoglobin concentrations and white cell counts, with other parameters being equivalent across the sexes. Forty-one patients with HbSS were recruited, and are compared to the HbSC patients in Table 2. Twenty-nine (70.1%) HbSS patients were aged 16 years or younger. There were highly significant differences between the two types of sickle cell disease for all parameters, indicating that these are distinct disorders and cannot be analyzed together.
Table 1.
Basic laboratory features of patients with HbSC in the study.
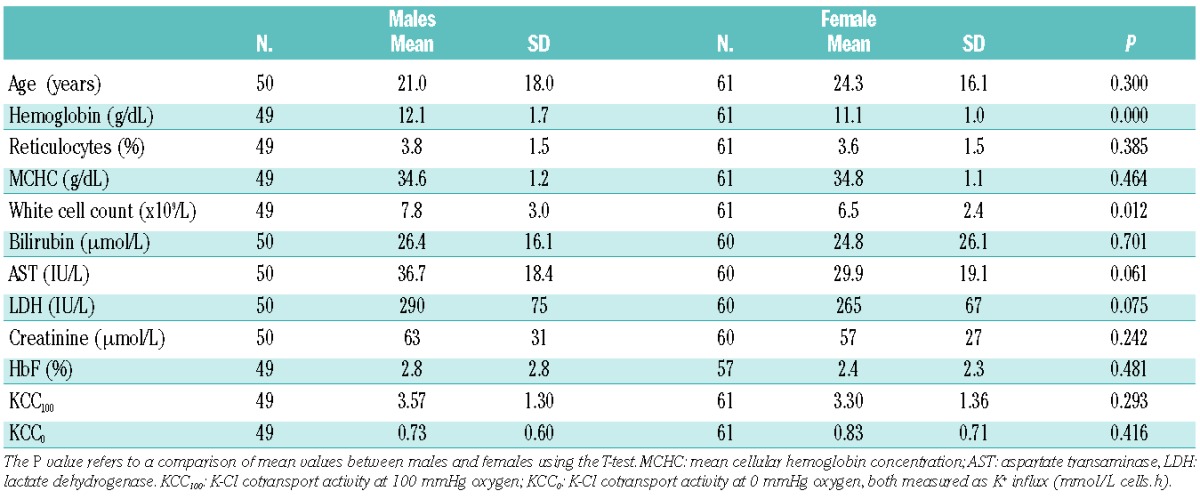
Table 2.
Comparison of laboratory parameters of patients with HbSS and HbSC.
Determinants of K-Cl cotransport activity in HbSC disease
There was no significant difference in erythrocyte KCC activity between male and female patients in either the oxygenated or deoxygenated state (Table 1); male and females were subsequently analyzed together. KCC activity was significantly greater in the oxygenated state (KCC100) than in the deoxygenated state (KCC0), with mean K+ influx of 3.42 mmol/L cells.h versus 0.79 mmol/L cells.h (paired-samples T-test, P<0.000).
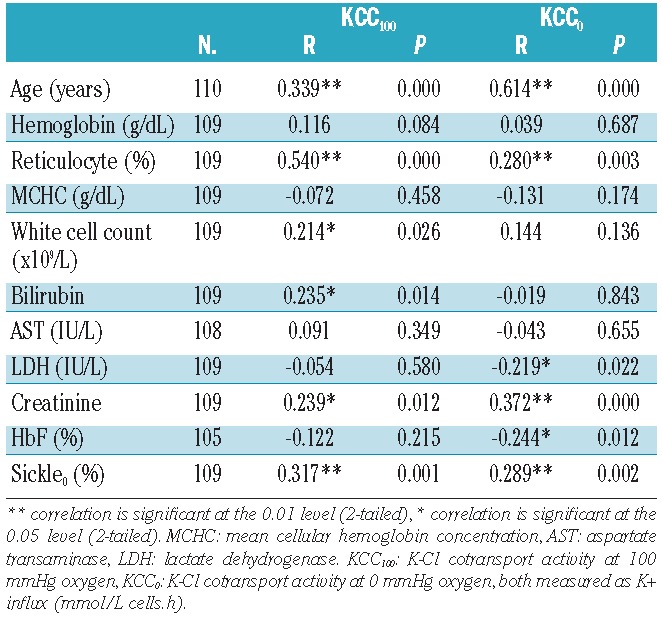
KCC activity was correlated against age, hemoglobin, reticulocyte percent, mean cellular hemoglobin concentration (MCHC), white blood cell (WBC) count, bilirubin, aspartate transaminase, lactate dehydrogenase (LDH), serum creatinine and hemoglobin F (HbF) levels (Table 3). KCC100 and KCC0 correlated with each other significantly (R=0.574, P=0.000). KCC100 correlated positively and significantly with age, reticulocyte percent, WBC count, bilirubin and creatinine; KCC0 correlated significantly and positively with age, reticulocyte percent, and creatinine, and negatively with LDH and HbF percent. Overall KCC activity increased with increasing age, reticulocytosis, and serum creatinine, and decreasing levels of LDH and HbF.
Table 3.
Pearson correlation of age, laboratory parameters and sickling against KCC100 and KCC0 in HbSC patients.
Linear regression was used to assess the most significant independent determinants of KCC100 and KCC0. For KCC100 the linear regression model included age, reticulocyte percent, WBC count, bilirubin and creatinine. This model explained 36% of the variability in KCC100 (R=0.60), and only age (Beta=0.249, P=0.024) and reticulocyte percent (Beta=0.314, P<0.001) maintained statistical significance.
Similarly, the significant independent determinants of KCC0 were assessed using linear regression against age, reticulocyte percent, creatinine, LDH and HbF percent. This model explained 42% of the variability in KCC0 (R=0.65). As for KCC100, only age (Beta=0.642, P<0.001) and reticulocyte percent (Beta=0.189, P=0.033) were independently significant.
Determinants of K-Cl cotransport activity in HbSS
The pattern of KCC activity differed significantly in the HbSS population studied, with a much smaller decrease in activity in the deoxygenated state compared to the oxygenated state (Table 2). KCC100 was significantly higher in HbSC patients than in HbSS ones, whereas KCC0 was significantly lower in HbSC patients. As for the HbSC population, KCC100 and KCC0 in patients with HbSS were correlated against age and relevant laboratory parameters. KCC100 correlated significantly only with age (R=0.705, P=0.002), whereas KCC0 correlated significantly with age (R=0.394, P=0.012) and HbF (R=−0.407, P=0.009).
Association between K-Cl cotransport activity and red cell sickling
The percentage of deoxygenated sickled cells correlated positively and significantly with KCC0 and KCC100 (Table 3). Variation in KCC0 accounted for 8.3% and variation in KCC100 explained 10% of the variation in sickle0%.
Association between K-Cl cotransport activity and hospital attendance in HbSC disease
The combined number of hospital admissions and Emergency Room attendances over 1 year was used as a surrogate marker of the frequency of acute pain. The number of these emergency attendances in 2011 varied from zero to five per patient, with a median of one and a mean of 0.95; 55 (49.5%) patients had no hospital attendances. Emergency hospital attendance was, therefore, analyzed as a binary variable with the two categories of ‘no attendances’ and ‘at least one attendance’. At least one attendance was associated with significantly increased activity of both KCC100 (mean 3.87 versus 3.20, P=0.009, independent samples T-test) and KCC0 (mean 0.96 versus 0.68, P=0.037) (Figure 1) compared to KCC activity in those patients with no admissions. There was no significant effect of age on the likelihood of attendance, which might have explained the link to KCC activity (mean age of patients with no attendances 16.4 years versus age of patients with at least one attendance 17.8 years; P=0.308).
Figure 1.
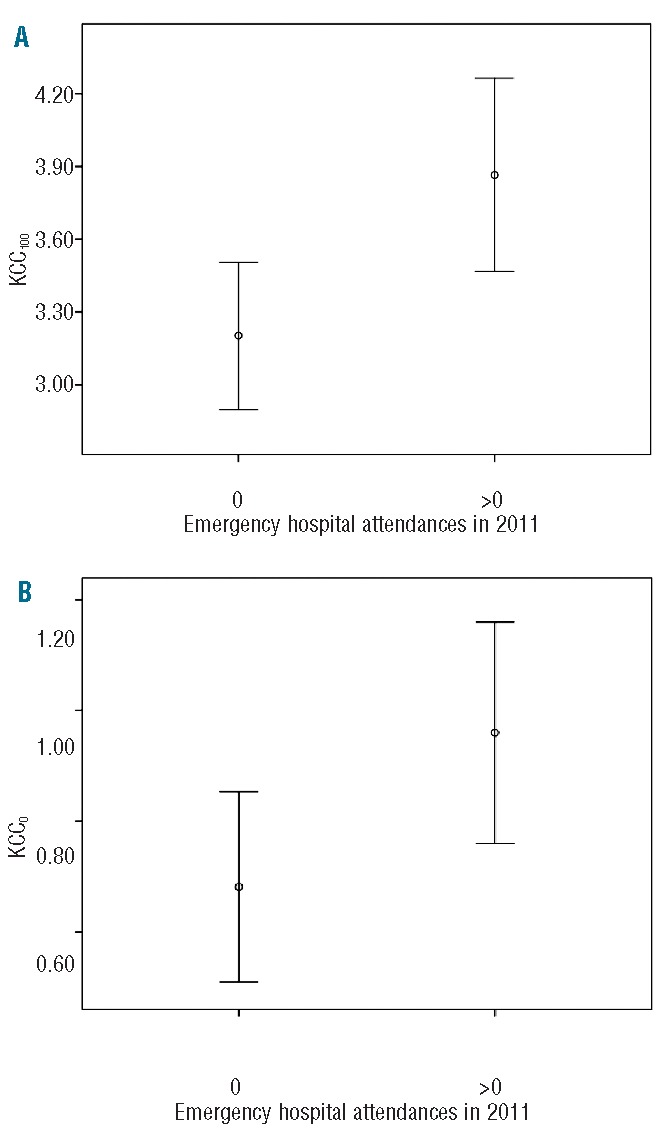
Error bar plots, showing mean and 95% confidence intervals, comparing KCC activity (mmol/L cells.h) in patients with no emergency hospital attendances and those who attended more than once at oxygen tensions of (A) 100 mmHg and (B) 0 mmHg.
Logistic regression analysis was used to ascertain the effects of KCC100, age, hemoglobin, reticulocyte percent, MCHC, WBC count, LDH, and HbF percent on the likelihood that a patient would attend hospital as an emergency in 2011. Only KCC100 was significantly associated with attendance (P=0.012), with increased activity associated with increased likelihood of attendance [Exp(B) =1.78, 95% CI 1.14 – 2.79].
Similarly, a logistic regression was used to analyze the effects of KCC0, age and laboratory parameters on the likelihood of emergency attendance at hospital. No factors were significant in this model, although KCC0 approached significance [P=0.052, Exp(B) 2.40, 95% CI 0.99 – 5.77].
Association between K-Cl cotransport activity and hospital attendance in HbSS
Unlike in HbSC, there was no association between emergency hospital attendance and KCC activity in HbSS patients. Twenty-one patients did not attend hospital urgently in 2011: these patients had a mean KCC100 activity of 2.50 and mean KCC0 activity of 1.71. These activities did not differ significantly from measurements in the 18 patients who attended the Emergency Room on one or more occasion, in whom the mean KCC100 was 2.90 (P=0.280) and mean KCC0 was 2.23 (P=0.240).
Association between K-Cl cotransport activity and retinopathy in HbSC disease
Eighty-six (77.5%) patients with HbSC disease were assessed for retinopathy. Sixty-five had no retinopathy, three had grade 1, two had grade 2, six had grade 3, nine had grade 4, and one had grade 5. Because of the small numbers, retinopathy was analyzed as a categorical variable comparing those without retinopathy (76%) against those with retinopathy (24%). The presence of retinopathy was associated with significantly increased activity of both KCC100 (mean 4.22 versus 3.15, P=0.002, independent samples T-test) and KCC0 (mean 1.18 versus 0.66, P=0.002). However, retinopathy was strongly associated with increasing age with the mean age of patients without retinopathy being 18.9 years compared to 35 years for the patients with this ocular complication (P<0.001); this is likely to be the explanation for the association between KCC and retinopathy.
Logistic regression analysis was used to ascertain the effects of KCC100, age, hemoglobin, reticulocyte percent, MCHC, WBC count, LDH, and HbF percent on the likelihood that a patient would have retinopathy. None of these factors was statistically significant in explaining the likelihood of retinopathy in this model. Similarly there were no significant effects on retinopathy noted when KCC0 was substituted into the regression.
Only a small number of patients with HbSS had significant retinopathy in this study, making statistical analysis inappropriate.
Discussion
KCC, first identified in erythrocytes,16 refers to the electroneutral-coupled transport of K+ and Cl− across membranes and is believed to be important in the regulation of cell volume.17 Cell swelling, mild acidity, magnesium depletion, protein kinase inhibitors and oxidative stress increase KCC activity; conversely, KCC activity is reduced by cell shrinkage, marked acidity, alkalinization and most bivalent cations. The gene responsible for this activity (SLC12A4) was cloned in 1996 and codes for the protein KCC1, which is similar to other cation-chloride cotransporters.18 Subsequently RNA transcripts from KCC3 (SLC12A6) and KCC4 (SLC12A7) were identified in erythrocytes, together with two splicing variants, although the exact role of these different entities is not clear.19
In normal erythrocytes, KCC activity is high in reticulocytes but largely absent in mature cells. In HbSS cells, activity is increased and this is thought to be one of the important mechanisms causing red cell dehydration and promoting HbS polymerization.20 However, KCC is particularly implicated in the unique pathophysiology of HbSC disease, with KCC100 activity being two to three times greater in HbSC cells compared to HbSS, although prior to this study, this interpretation was reliant on extrapolation of findings from only a small number of patients.9,10,21
In our study of 110 patients with HbSC disease, KCC activity varied widely between patients; KCC100 varied from 0.44 to 7.61 mmol/L cells.h, and KCC0 varied from 0 to 3.02 mmol/L cells.h. Genetic differences may account for some of this variability, but on linear regression, two factors were independently associated with activity of both KCC100 and KCC0: age and reticulocyte percent. The correlation of KCC with reticulocyte percent is expected based on the known increased KCC activity in younger red cells, and this explained 7.8% of the variability in KCC0 and 29% of the variability in KCC100.
However, the statistically significant increase of KCC activity with age is novel: age accounted for 38% of the variability of KCC0 and 11% of the variability of KCC100. Some of this increase with age may be related to falling HbF levels, or increasing creatinine, although neither of these was significant on multiple regression. This increase in KCC activity with age may contribute to the higher incidence of complications in older patients, including retinopathy22 and renal impairment,23 or may be a consequence of altered renal function.
It is also apparent from our study that oxygen affects KCC activity differently in red cells from patients with HbSS and HbSC disease. In oxygenated HbSC red cells, KCC activity is high, similar or greater than that in HbSS red cells; but at decreasing oxygen tensions, KCC activity is progressively inhibited, as in red cells from normal individuals. The reasons for these differences are not known, but the altered behavior of HbC compared to HbA or HbS is an obvious possibility. The concentration of soluble oxyHb and the presence of deoxyHb to interact with regulatory sites at the membrane have been proposed as key regulators of KCC activity.24 In this context, the presence of crystals of HbC25 at high oxygen tension and their dissolution at low oxygen tension may be important. The role of oxygen is clearly complicated in HbSC, and likely to differ depending on many dynamic factors, including pH, temperature and age of the red cell, but this study raises the possibility that oxygen supplementation may be harmful to patients with HbSC disease in some circumstances. Oxygen has no such potentially harmful effects in HbSS.
The most striking finding of our study is that patients with high KCC activity were more likely to present to hospital with acute vaso-occlusive problems. Red cell dehydration occurs at the beginning of the cascade of pathological events which ends in hospital attendance, and it is perhaps surprising that KCC shows such a clear relationship to acute complications. Most biomarkers identified in sickle cell disease measure epiphenomena occurring as a consequence of vaso-occlusion,26 and few reflect fundamental aspects of pathophysiology, such as red cell dehydration. The specific importance of KCC in HbSC disease is emphasized by the fact that the same relationship is not seen in HbSS patients. This effect is independent of age, and also cannot be assessed by measuring MCHC, which was not related to KCC activity in our study. Emergency hospital attendance is a difficult endpoint, with a lot of ‘noise’ unrelated to the pathophysiology of sickle cell disease. However, it is a highly relevant clinical endpoint and, although it may generate false negative results, it is much less likely to cause a false positive result. We, therefore, think the result in this study is likely to be real and important.
The presence of retinopathy was also strongly associated with increased KCC activity, although this association is confounded by the effects of age. Logistic regression suggests that age is responsible for both the increased prevalence of retinopathy and increased KCC activity, although further studies are needed to unpick this complicated association.
KCC measurements are potentially useful as prognostic biomarkers in HbSC disease, to identify those patients more likely to follow a severe course and benefit from more aggressive treatment. The assay used in this study requires specialist laboratory skills and equipment, although it may be possible to simplify the assay, as has been done for the similarly complicated assay which measures Psickle activity.27
This study also suggests that effective inhibition of KCC could reduce acute complications in HbSC disease. This approach was addressed in a randomized controlled trial involving hydroxyurea and magnesium pidolate, which was stopped early because of poor recruitment. Magnesium, used to inhibit KCC activity, showed no clinical benefit, although there was no measurable inhibition of KCC activity, reflecting that magnesium is a relatively weak inhibitor.28
Our study suggests that KCC is a clinically relevant measurement in HbSC; further investigations, including longitudinal studies, assessment of activity in different reticulocyte fractions and genetic studies to characterize inherited variation in activity, are required.
Acknowledgments
This study was funded by the Medical Research Council, UK Project Grant 92150.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Huntsman RG, Lehmann H. Treatment of sickle-cell disease. Br J Haematol. 1974;28(4): 437–444. [DOI] [PubMed] [Google Scholar]
- 2.Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325(1):11–16. [DOI] [PubMed] [Google Scholar]
- 3.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. [DOI] [PubMed] [Google Scholar]
- 4.Deane CR, Goss D, O’Driscoll S, et al. Transcranial Doppler scanning and the assessment of stroke risk in children with HbSC [corrected] disease. Arch Dis Child. 2008;93(2):138–141. [DOI] [PubMed] [Google Scholar]
- 5.Condon PI, Serjeant GR. Ocular findings in hemoglobin SC disease in Jamaica. Am J Ophthalmol. 1972;74(5):921–931. [DOI] [PubMed] [Google Scholar]
- 6.Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica. 2012;97(8):1136–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalibalta S, Ellory JC, Browning JA, Wilkins RJ, Rees DC, Gibson JS. Novel permeability characteristics of red blood cells from sickle cell patients heterozygous for HbS and HbC (HbSC genotype). Blood Cells Mol Dis. 2010;45(1):46–52. [DOI] [PubMed] [Google Scholar]
- 8.Brousse V, Makani J, Rees DC. Management of sickle cell disease in the community. BMJ. 2014;348:g1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev. 2003;17(3):167–178. [DOI] [PubMed] [Google Scholar]
- 10.Canessa M, Spalvins A, Nagel RL. Volume-dependent and NEM-stimulated K+,Cl− transport is elevated in oxygenated SS, SC and CC human red cells. FEBS Lett. 1986; 200(1):197–202. [DOI] [PubMed] [Google Scholar]
- 11.Reiss GH, Ranney HM, Shaklai N. Association of hemoglobin C with erythrocyte ghosts. J Clin Invest. 1982;70(5):946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brugnara C, Kopin AS, Bunn HF, Tosteson DC. Regulation of cation content and cell volume in hemoglobin erythrocytes from patients with homozygous hemoglobin C disease. J Clin Invest. 1985;75(5):1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lew VL, Bookchin RM. Ion transport pathology in the mechanism of sickle cell dehydration. Physiol Rev. 2005;85(1):179–200. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol. 1971;71(3):649–665. [DOI] [PubMed] [Google Scholar]
- 15.Gibson JS, Speake PF, Ellory JC. Differential oxygen sensitivity of the K+-Cl− cotrans-porter in normal and sickle human red blood cells. J Physiol. 1998;511(Pt 1):225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunham PB, Stewart GW, Ellory JC. Chloride-activated passive potassium transport in human erythrocytes. Proc Natl Acad Sci USA. 1980;77(3):1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adragna NC, Di Fulvio M, Lauf PK. Regulation of K-Cl cotransport: from function to genes. J Membr Biol 2004;201(3):109–137. [DOI] [PubMed] [Google Scholar]
- 18.Gillen CM, Brill S, Payne JA, Forbush B., 3rd Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem. 1996;271(27):16237–16244. [DOI] [PubMed] [Google Scholar]
- 19.Crable SC, Hammond SM, Papes R, et al. Multiple isoforms of the KC1 cotransporter are expressed in sickle and normal erythroid cells. Exp Hematol. 2005;33(6):624–631. [DOI] [PubMed] [Google Scholar]
- 20.Joiner CH, Rettig RK, Jiang M, Franco RS. KCl cotransport mediates abnormal sulfhydryl-dependent volume regulation in sickle reticulocytes. Blood. 2004;104(9): 2954–2960. [DOI] [PubMed] [Google Scholar]
- 21.Olivieri O, Vitoux D, Galacteros F, et al. Hemoglobin variants and activity of the (K+Cl−) cotransport system in human erythrocytes. Blood. 1992;79(3):793–797. [PubMed] [Google Scholar]
- 22.Clarkson JG. The ocular manifestations of sickle-cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481–504. [PMC free article] [PubMed] [Google Scholar]
- 23.Powars DR, Elliott-Mills DD, Chan L, et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med. 1991;115(8):614–620. [DOI] [PubMed] [Google Scholar]
- 24.Puchulu-Campanella E, Chu H, Anstee DJ, Galan JA, Tao WA, Low PS. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. J Biol Chem. 2013;288(2):848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch RE, Raventos-Suarez C, Olson JA, Nagel RL. Ligand state of intraerythrocytic circulating HbC crystals in homozygote CC patients. Blood. 1985;66(4):775–777. [PubMed] [Google Scholar]
- 26.Rees DC, Gibson JS. Biomarkers in sickle cell disease. Br J Haematol. 2012;156(4):433–445. [DOI] [PubMed] [Google Scholar]
- 27.Milligan C, Rees DC, Ellory JC, et al. A nonelectrolyte haemolysis assay for diagnosis and prognosis of sickle cell disease. J Physiol. 2013;591(Pt 6):1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol. 2011;152(6):771–776. [DOI] [PubMed] [Google Scholar]