

Abstract
Inherited bone marrow failure syndromes are a group of rare, heterogeneous genetic disorders with a risk of clonal and malignant myeloid transformation including clonal marrow cytogenetic abnormalities, myelodysplastic syndrome and acute myeloid leukemia. The clinical characteristics, risk classification, prognostic factors and outcome of clonal and malignant myeloid transformation associated with inherited bone marrow failure syndromes are largely unknown. The aims of this study were to determine the impact of category, cytopathology and cytogenetics, the three components of the “Category Cytology Cytogenetics” classification of pediatric myelodysplastic syndrome, on the outcome of clonal and malignant myeloid transformation associated with inherited bone marrow failure. We used data from the Canadian Inherited Marrow Failure Registry. Among 327 patients with inherited bone marrow failure syndrome enrolled in the registry, the estimated risk of clonal and malignant myeloid transformation by the age of 18 years was 37%. The risk of clonal and malignant myeloid transformation varied according to the type of inherited bone marrow failure syndrome but was highest in Fanconi anemia. The development of clonal and malignant myeloid transformation significantly affected overall survival. Mortality varied based on cytopathological group. The largest group of patients had refractory cytopenia. Clonal marrow cytogenetic abnormalities were identified in 87% of patients with clonal and malignant myeloid transformation, and different cytogenetic groups had different impacts on disease progression. We conclude that category, cytopathology and cytogenetics in cases of clonal and malignant myeloid transformation associated with inherited bone marrow failure syndromes have an important impact on outcome and that the classification of such cases should incorporate these factors.
Introduction
Inherited bone marrow failure syndromes (IBMFS) are a group of rare genetic disorders with single or multi-lineage cytopenia resulting from impaired hematopoiesis and a variable degree of predisposition to cancer. These disorders carry a risk of clonal and malignant myeloid transformation (CMMT), which includes isolated clonal marrow cytogenetic abnormalities (CMCA), myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).1–8 There are multiple reports of cases of IBMFS with early CMMT changes that eventually progressed to either severe bone marrow failure, advanced MDS and/or AML.9,10
The precise definition of adult and pediatric de novo CMMT is still evolving, but it is widely accepted that it should be based on peripheral blood cell numbers and types, and on bone marrow blasts, cellularity, cytogenetics and presence of dysplasia. Unfortunately, there is no widely accepted definition of IBMFS-associated CMMT.11 For example, Hasle et al. defined pediatric MDS based on peripheral blood counts, marrow morphologic dysplasia, CMCA and blasts.12 These are measurable criteria and potentially excellent tools for defining MDS. However, the ability of these criteria to include all cases of IBMFS-associated MDS without falsely including cases without true transformation has never been tested.
Apart from challenges in defining IBMFS-associated CMMT, the classification and grading of this disorder presents another challenge, because some of the clinical and biological features that are used to characterize de novo CMMT (e.g. prominent dysplasia and increased marrow cellularity) are less common in IBMFS-associated CMMT. In 2002 we developed the “Category Cytology Cytogenetics” (CCC) classification for pediatric MDS, which aimed to address aspects not only of de novo MDS, but also of therapy-related and IBMFS-associated MDS.11 In the present study we examined the three components of the CCC classification in a large cohort of patients with CMMT from one comprehensive population-based IBMFS registry and hypothesized that they have prognostic utility.
Methods
The Canadian Inherited Marrow Failure Registry
The Canadian Inherited Marrow Failure Registry and its inclusion and exclusion criteria are described elsewhere,13,14 and are summarized in the Online Supplementary Methods.
Definition and diagnostic criteria
CMMT was defined as having bone marrow failure and at least one of the following: (i) an isolated CMCA, (ii) MDS or (iii) AML.
CMCA was defined as the presence of at least two hematopoietic cells with the same cytogenetic abnormality detected by metaphase cytogenetics or the presence of positive cells, as determined by fluorescence in situ hybridization, at a percentage higher than the reference value.
The diagnostic criteria for pediatric MDS proposed by Hasle et al. were used. Thus, at least two of the following criteria had to be met: (i) sustained unexplained cytopenia, (ii) at least bilineage prominent morphological myelodysplasia (>10% of the cells in each lineage), (iii) acquired clonal cytogenetic abnormality in hematopoietic cells, and (iv) 5%–29% blast cells.12 MDS cases also could not have CMCA that are pathognomonic for AML, nor could they have very rapid leukemic blast cell growth as indicated by repeat bone marrow testing 2–3 weeks after diagnosis. Information on all cases was reviewed centrally, and the diagnosis of MDS could be modified based on these criteria. Clinical cases of MDS that did not meet all diagnostic criteria were carefully reviewed to determine whether the existing diagnostic criteria needed to be modified.
AML was defined as ≥30% myeloid leukemic blasts in the bone marrow or pathognomonic AML-type CMCA and very rapid leukemic blast cell growth, as indicated in repeat bone marrow testing 2–3 weeks after diagnosis.
Classification of cases with clonal and malignant myeloid transformation
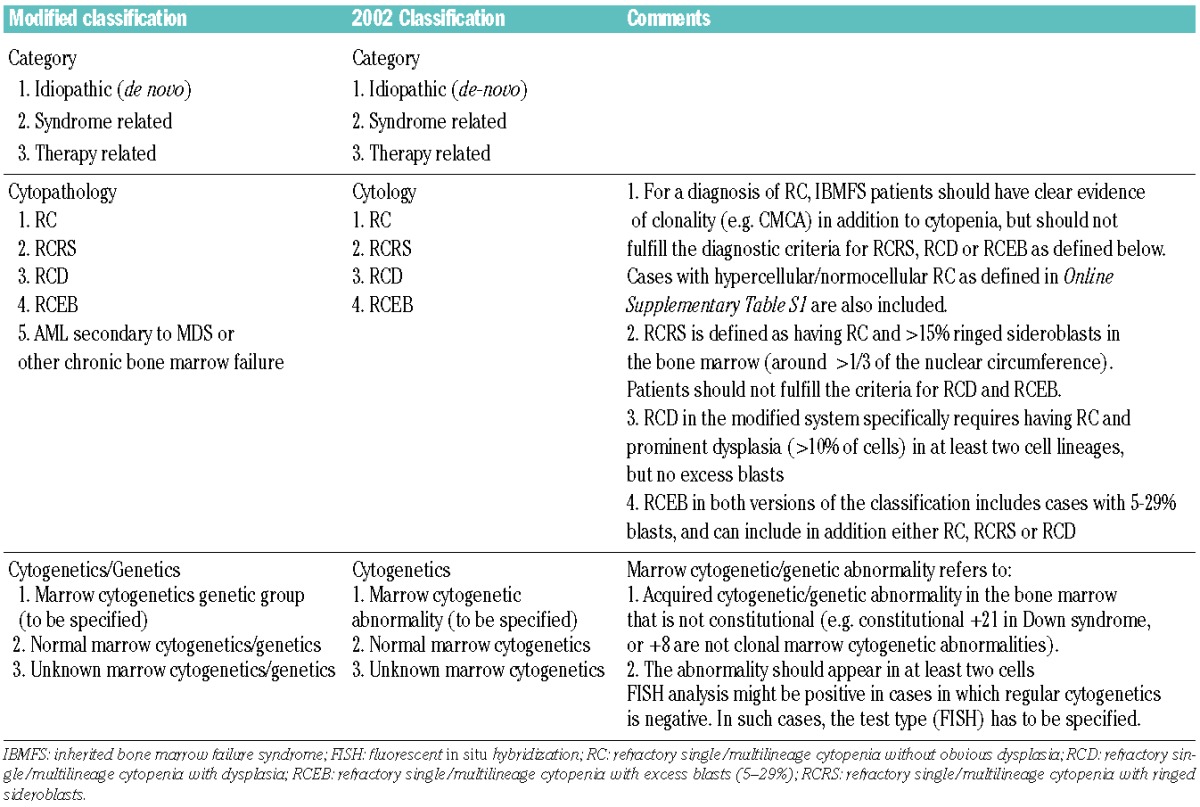
Grading or classification of CMMT was based on the CCC classification for pediatric MDS 2002.11 We made minor modifications to the system that we published in 2002 (Table 1). First, we changed the term ‘cytology’ to ‘cytopathology’ to reflect changes found on bone marrow biopsy testing. Second, due to the natural progression of MDS to AML, we added the category of AML secondary to MDS or chronic bone marrow failure syndrome. Third, some degree of dysplasia is a feature of the IBMFS bone marrow morphology and does not necessarily indicate malignancy,1,15,16 while single lineage dysplasia is an inherent feature of certain inherited disorders. We, therefore, included only patients who had prominent dysplasia (>10% of cells) in each lineage and in at least two lineages to qualify for a diagnosis of refractory cytopenia with dysplasia (RCD). Fourth, emerging genomics data can be incorporated in the future to characterize and understand IBMFS-associated CMMT; we, therefore, changed the heading ‘Cytogenetics’ to “Cytogenetics/Genetics”.
Table 1.
The modified “category, cytopatology, cytogenetics” classification of childhood MDS.
Definition of disease progression
For patients who underwent HSCT, the duration of disease progression was calculated as the time from CMMT diagnosis to the day before the transplant preparatory therapy was started. There were no deaths that were unrelated to CMMT or transplant. HSCT was applied at the discretion of the treating physician. The indications for transplant included at least one of the following: severe cytopenia, progressive cytopenia approaching severely low counts and excess blasts.
Disease progression was defined by the development of one or more of the following three criteria:
(i) one of the following cytopathological changes: (a) refractory cytopenia (RC) or refractory cytopenia with ringed sideroblasts (RCRS) to refractory cytopenia with dysplasia (RCD) or refractory cytopenia with excess blasts (RCEB) or AML; (b) RCD to RCEB or AML; (c) RCEB to AML;
(ii) one of the following cytogenetic changes: (a) new cytogenetic abnormality in patients with normal cytogenetics at baseline; (b) new cytogenetic abnormality in those with a single cytogenetic abnormality [such as del(20q) and trisomy 8, or i(7q) in Shwachman-Diamond syndrome, SDS];
(iii) one of the following changes in the severity of bone marrow failure: (a) single-lineage severe cytopenia to bilineage or trilineage severe cytopenia; (b) bilineage severe cytopenia to trilineage severe cytopenia. The various lineages were defined as severely reduced if the platelet count was less than 20×109/L or transfusion was required, if the hemoglobin concentration was less than 70 g/L or transfusion was required and if the absolute neutrophil count was less than 0.5×109/L or granulocyte-colony stimulating factor therapy was required.
Data analysis
Survival and risk were estimated by Kaplan-Meier analysis and the Wilcoxon test was used to determine significant differences. P values <0.05 were considered statistically significant. Some analyses (e.g. risk of CMMT) were stopped at the age of 18 years, due to the possibility of referral bias of patients with CMMT who were older than 18 years and were not treated at pediatric centers. We did not include solid tumors in this study because of the small number of patients with this complication. We chose IBMFS categories that had more than ten patients for our analyses. We also only included categories that had more than three patients with CMMT to assess the impact of category on progression and overall survival of patients with CMMT. We, therefore, included the categories of Fanconi anemia (FA), SDS and unclassified-IBMFS. We analyzed only cytopathological groups and cytogenetic groups with more than three patients to determine how cytopathology and cytogenetics affect progression and overall survival. The cytogenetic groups included were: (i) complex cytogenetics (≥3 cytogenetic abnormalities), (ii) del(7), del(7q) and deletion or translocation at areas 7q32–34, (iii) i(7q) and (iv) normal cytogenetics (constitutional abnormalities without CMCA were included). Statistical analyses were performed using XLSTAT and Microsoft Excel software.
Results
Patients with inherited bone marrow failure syndromes have a high risk of developing clonal and malignant myeloid transformation
As of August 31st, 2011, 327 patients were enrolled in the registry. Seven patients were excluded because of missing information so a total of 320 patients were analyzed. The distribution of specific categories of IBMFS is presented in Table 2. Forty-four patients met the criteria of MDS or had frank AML. An additional patient with the MDS/AML-predisposition syndrome (constitutional trisomy 8) had cytopenia and a hypercellular marrow without dysplasia, CMCA or excess blasts, and did not fit criteria for any other blood dyscrasia but MDS, and thus was also included. This gave a crude CMMT prevalence of 14.1% among IBMFS patients. Kaplan-Meier analysis showed a risk of CMMT of 37% by the age of 18 years (Figure 1A). The peak incidence of CMMT was between the ages of 5–10 years (Online Supplementary Figure S1).
Table 2.
Categories of the IBMFS-associated cases of CMMT.
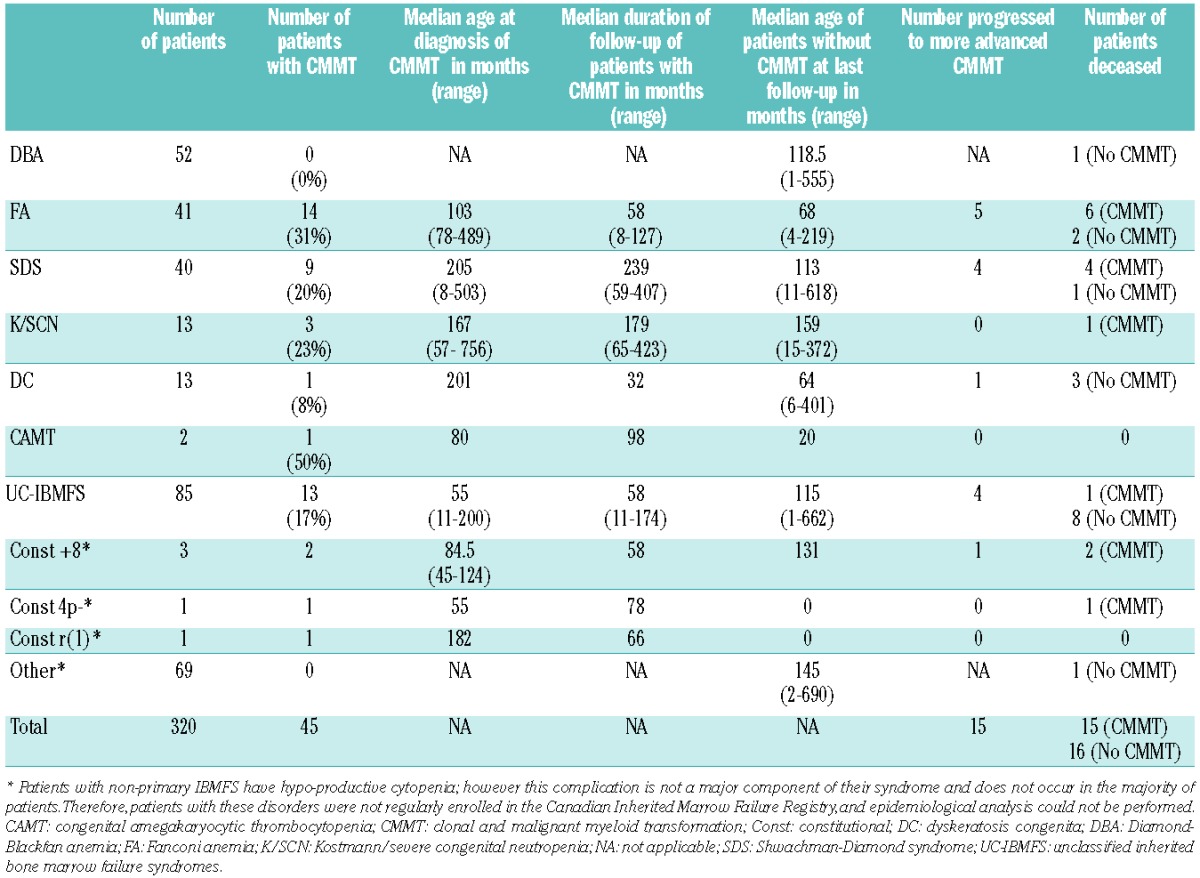
Figure 1.

Characteristics and actuarial risk of clonal or malignant myeloid transformation (CMMT) among patients with IBMFS. (A) Actuarial risk of CMMT was estimated by Kaplan-Meier analysis. Censoring was performed at last follow-up or at the time of hematopoietic stem cell transplantation for patients who had undergone such a procedure. The figure represents risk with 95% confidence intervals. (B) Risk of progression to more advanced transformational stage among patients with CMMT. The figure represents risk with 95% confidence intervals. (C) Overall survival analysis of patients with and without CMMT up to the age of 18 years. (D-I) Actuarial risk of CMMT among the common categories of inherited bone marrow failure syndromes: Fanconi anemia (FA), Shwachman-Diamond syndrome (SDS), dyskeratosis congenital (DC), Kostmann/severe congenital neutropenia (K/SCN), unclassified-IBMFS (UC-IBMFS), and comparison between FA, SDS and DBA. The figures represent risk with 95% confidence intervals.
Forty-seven percent of patients were diagnosed with CMMT at the time of diagnosis of their IBMFS. The median age at diagnosis of IBMFS in those with CMMT was 61.5 months (range, 0–376 months). The median age at diagnosis of CMMT was 104 months (range, 8–756 months), compared to a median age of diagnosis of about 70 years in the general population of western countries. The incidence of MDS in the United States in people aged 0–18 years is reported to be 1 per 1,000,000 per year and the risk of developing AML before the age of 18 years is 8 per 1,000,000 per year.17–20 Given that the risk of developing CMMT in our study was 37% by the age of 18 years, the risk of CMMT (MDS and AML combined) during childhood among subjects with IBMFS is estimated to be 2284-fold higher than the risk in the general population.
Twenty-five of the patients with CMMT (55.6%) were males and 20 (44.4%) were females. Most patients (28/45, 62.2%) had physical malformations along with their blood dyscrasia. Thirteen patients (28.9%) had physical anomalies involving three or more non-hematologic systems (data not shown).
The 45 patients with CMMT were followed for a total of 394 person-years with a median duration of 66 months per patient (range, 1–423 months). Fifteen out of 45 patients (33.3%) progressed. Of the 30 patients who did not progress, 14 required HSCT due to significant cytopenia or advanced MDS at presentation with CMMT; the remaining 16 patients were followed for a total of 153 person-years with a median duration of 75 months per patient (range, 1–309 months). The predicted risk of progression within 10 years of diagnosis with CMMT was 60% (Figure 1B).
Impact of clonal and malignant myeloid transformation on survival
Overall mortality among the 45 patients with CMMT was 33.3% (15/45), while overall mortality among the non-CMMT group was 5.8% (16/275). Kaplan-Meier analysis predicted that by the age of 18 years, the survival rate in the CMMT group would be 56% compared to 92% in those without CMMT (P=0.02) by 18 years of age (Figure 1C). The median survival after diagnosis of CMMT was 32 months (range, 1–174 months). Causes of death in the CMMT group included: post-transplant complications in eight patients (4 therapy-related, 2 nonengraftment and 2 secondary graft failure), failure to achieve remission (1 patient), and CMMT-related complications such as infections and bleeding complications (3 patients). The median age of death among patients with CMMT was 12.8 years (range, 5.2–43.1 years) compared to 7.1 years (range, 0.2–34 years) among those without CMMT.
The impact of category of inherited bone marrow failure syndrome on clonal and malignant myeloid transformation risk and outcome
Among the whole group of patients in the registry, CMMT was most common in patients with FA (14 patients), followed by SDS (9 patients) and unclassified-IBMFS (13 patients) (Table 2). Other categories included Kostmann/severe congenital neutropenia (K/SCN), dyskeratosis congenita, and congenital amegakaryocytic thrombocytopenia. There were two patients with constitutional mosaic trisomy 8, one with constitutional 4p-syndrome1 and one with constitutional supernumerary ring chromosome 1 among the patients with non-primary IBMFS who had CMMT.
FA patients had the highest actuarial risk of 75% of developing CMMT by 18 years of age. This was followed by patients with dyskeratosis congenita, unclassified- IBMFS, K/SCN and SDS with risks of about 25%, 24%, 24% and 20%, respectively, by 18 years of age (Figure 1D–H). None of the patients with Diamond-Blackfan anemia (DBA) developed CMMT. When we compared the three categories with the largest numbers of patients, DBA, FA and SDS, the difference in risk of developing CMMT by 18 years of age was statistically significant (P=0.005)(Figure 1I). This difference was still significant (P=0.02) when we compared DBA, FA, SDS, unclassified-IBMFS, dyskeratosis congenita and K/SCN (data not shown). The earliest age of onset of CMMT was 78 months in FA, 8 months in SDS, 201 months in dyskeratosis congenita, 57 months in K/SCN and 11 months in unclassified-IBMFS.
The risk of progression within 10 years of diagnosis with CMMT in patients with FA, SDS, and unclassified-IBMFS was not significantly different between categories, and was estimated to be 41%, 72% and 43%, respectively (Figure 2A–E). The overall survival at 10 years from diagnosis with CMMT in FA, SDS and unclassified-IBMFS was also not significantly different between categories, and was estimated at 57%, 25%, and 91%, respectively (Figure 2F–J).
Figure 2.
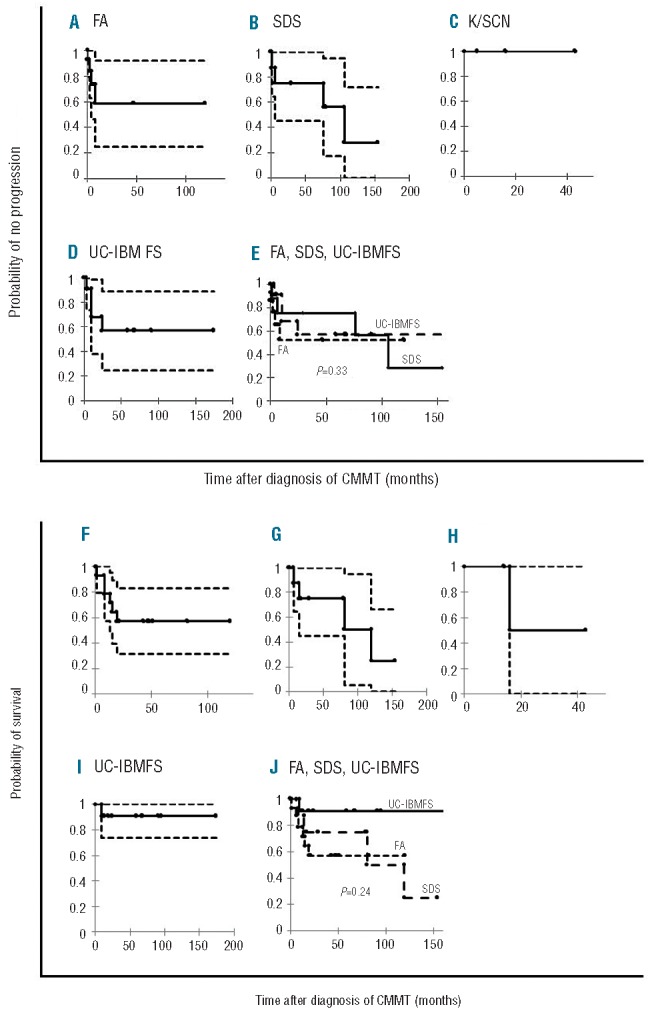
Actuarial risk of progression and survival of clonal or malignant myeloid transformation (CMMT) among the various categories of inherited bone marrow failure syndromes. (A) Fanconi anemia (FA), (B) Shwachman-Diamond syndrome (SDS), (C) Kostmann/severe congenital neutropenia (K/SCN), (D) Unclassified-IBMFS (UC-IBMFS). (E) Comparative analysis between FA, SDS and UC-IBMFS. The figures represent risk with 95% confidence intervals. (F) Fanconi anemia (FA), (G) Shwachman-Diamond syndrome (SDS), (C) Kostmann/severe congenital neutropenia (K/SCN), (H) Unclassified-IBMFS (UC-IBMFS), (I) Comparative analysis between FA, SDS, UC-IBMFS all groups with more than three patients with CMMT, (J) Comparative analysis of three categories (without K/SCN). The figures represent risk with 95% confidence intervals.
We did not find a correlation between IBMFS category and bone marrow failure severity, specific cytopathology, or cytogenetics at presentation with CMMT (data not shown).
Cytopathology of clonal and malignant myeloid transformation associated with inherited bone marrow failure syndromes and impact on outcome
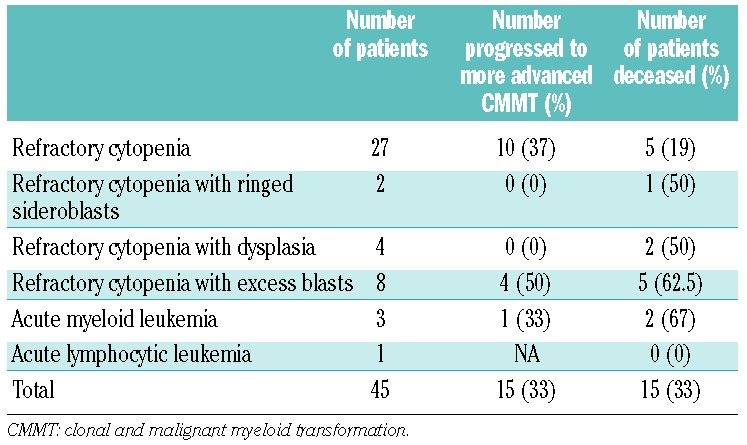
The majority of patients with CMMT had RC (60%), followed by RCEB (18%), RCD (9%), leukemia (AML, 7% and B-precursor acute lymphoblastic leukemia, 2%) and RCRS (4%) (Table 3).
Table 3.
Cytopathology of IBMFS-associated CMMT at presentation.
The risk of CMMT progression to more advanced CMMT tended to be higher in the RCEB group, being 70% within 3 years of the diagnosis of CMMT (Figure 3A). The rate of progression of RC was 37% and 65% at 3 and 10 years, respectively, after the diagnosis of CMMT (Figure 3B). Patients with RCRS and RCD had the lowest rates of progression (Figure 3C,D).
Figure 3.
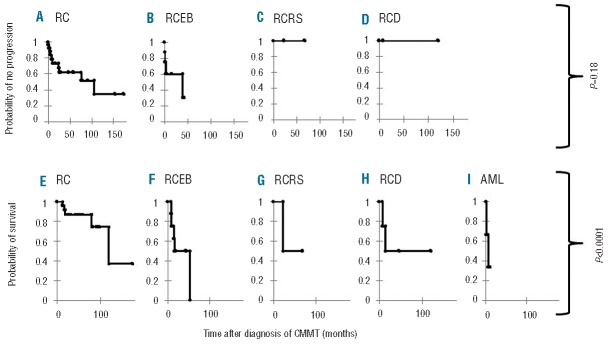
Impact of cytopathology on outcome of patients with clonal or malignant myeloid transformation (CMMT). Actuarial risk of progression after CMMT diagnosis among the various classes of cytopathology at presentation: (A) refractory cytopenia (RC), (B) refractory cytopenia with excess blasts (RCEB), (C) refractory cytopenia with ringed sideroblasts (RCRS), (D) refractory cytopenia with dysplasia (RCD). Comparative analysis between groups was not significant (P=0.18). Overall survival after CMMT diagnosis among the various classes of cytopathology at presentation: (E) refractory cytopenia (RC), (F) refractory cytopenia with excess blasts (RCEB), (G) refractory cytopenia with ringed sideroblasts (RCRS), (H) refractory cytopenia with dysplasia (RCD), (I) acute myeloid leukemia (AML). Comparative analysis showed a statistically significant difference (P<0.0001).
The overall survival after diagnosis of CMMT was significantly different between the various cytopathologic groups (Figure 3). Patients with RCEB and AML had the poorest overall survival (P<0.0001). Importantly, patients with RC did poorly with an overall survival of 37% at long-term follow-up of 10 years.
There was no correlation between cytopathology and bone marrow failure severity or specific cytogenetic abnormalities at presentation with CMMT (data not shown).
Cytogenetics of clonal and malignant myeloid transformation associated with inherited bone marrow failure syndromes and impact on outcome
CMCA were present in 93% of the patients at presentation with CMMT (Table 4). One patient developed a CMCA at follow-up. Monosomy 7, complex cytogenetics and i(7q) were the most common CMCA, and were seen in 36%, 21% and 11%, respectively, of the patients with CMCA. These abnormalities were analyzed for their prognostic values. Other cytogenetic groups were too small for statistical analysis.
Table 4.
Cytogenetics of bone marrow samples at diagnosis of IBMFS-associated CMMT.
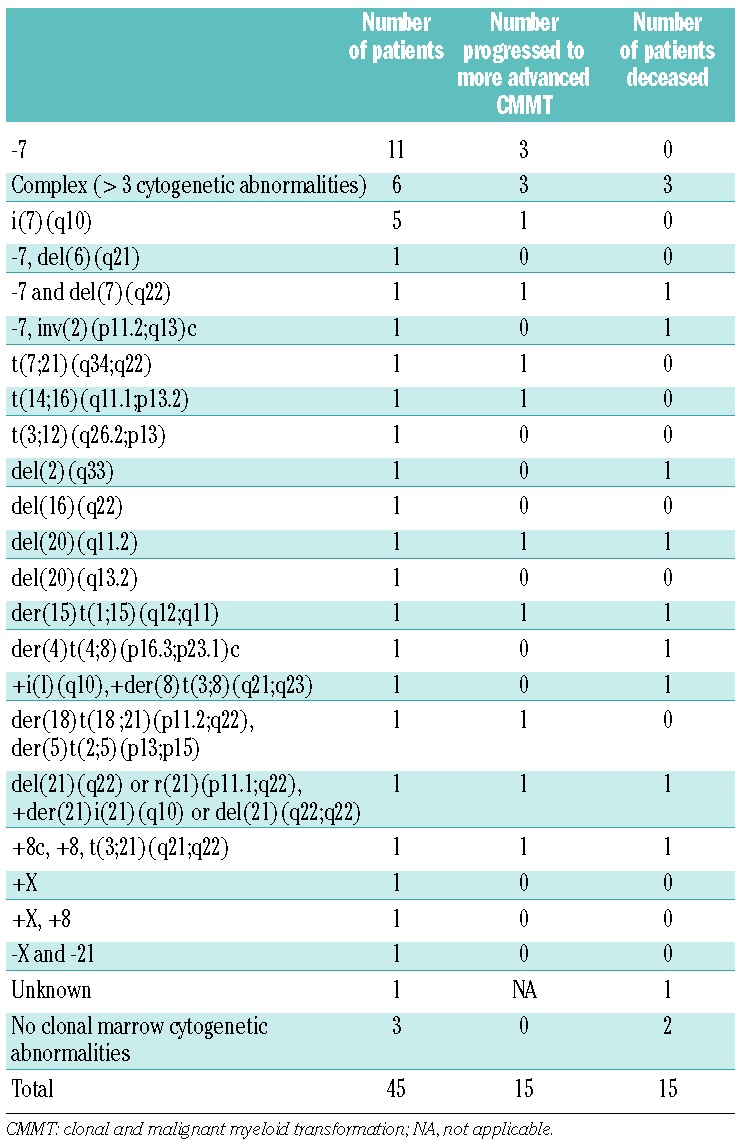
Rates of progression between the cytogenetic groups were significantly different. Patients with monosomy 7 and complex cytogenetics had the highest risks of progression of 54% and 60%, respectively, at 5 years after the diagnosis of CMMT (P=0.02; Figure 4A). There was no statistically significant difference in overall survival between the various cytogenetic groups (Figure 4B).
Figure 4.

Impact of cytogenetics on outcome of patients with clonal or malignant myeloid transformation (CMMT). (A) Actuarial risk of progression of CMMT; (B) overall survival of patients with CMMT.
All patients with i(7q) were alive at last follow-up; however, this anomaly was associated with significant morbidity. Among the four patients with SDS and i(7q), one patient suffered from severe multilineage cytopenia and one from severe neutropenia at presentation. A third patient with i(7q) developed an additional CMCA consisting of trisomy 8, as well as severe neutropenia at follow-up. One of the two patients with del(20q) developed RCEB and subsequently AML 11 years after the first appearance of this CMCA in the bone marrow.
There was no correlation between cytogenetics and the severity of bone marrow failure or specific cytopathology at presentation with CMMT (data not shown).
Discussion
The present study provides, for the first time, prognostic data related to the category, cytopathology and cytogenetics of IBMFS-associated CMMT in a large group of patients from one population-based registry. Our data showed an estimated 2284-fold higher risk of CMMT in patients with IBMFS than in the general population. Furthermore, the development of CMMT in patients with IMBFS signaled a high likelihood of malignant progression and significantly increased mortality.
The risk of CMMT among the whole group of IBMFS has never been studied. In our cohort of 320 patients, which included patients diagnosed with the most common categories of IBMFS, the estimated risk of CMMT by the age of 18 years was 37%. It is known that the cumulative risk of IBMFS-associated CMMT further increases in adulthood. However, similar to all other studies that have been conducted on IBMFS, our registry may also suffer from referral bias of patients with CMMT who are older than 18 years of age and who are not treated at pediatric centers. We did not, therefore, analyze certain factors (e.g. risk of CMMT) for patients older than 18 years of age.
Few population-based data are available about the differential risk of developing CMMT among patients with various categories of IBMFS.21 Although the various IBMFS share many clinical and morphological phenotypes, their respective IBMFS genes play roles in several different biochemical pathways. It is, therefore, reasonable to hypothesize that mutations in different IBMFS genes may have different impacts on the malignant potential and behavior of bone marrow cells. Our data indicate a significantly different CMMT risk by the age of 18 years between DBA, FA and SDS. CMMT was most common among FA patients, who had a 75% risk by this time, which is much higher than that described in other studies,5,8,22–24 and is even higher than that reported in studies in which patients with isolated clones were included.22 This may reflect our comprehensive registry inclusion criteria and meticulous collection of data on all bone marrow testing. SDS patients had a high risk of 20% of developing CMMT by the age of 18 years. This information provides the rationale for routine leukemia surveillance in children with FA and SDS.25 In contrast, no patient with DBA developed CMMT in our cohort, suggesting that cancer surveillance with annual bone marrow testing during childhood is not indicated in DBA. The need for surveillance for patients with DBA after the age of 18 years is not yet clear. The development of CMMT in children with K/SCN and dyskeratosis congenita is consistent with the literature and reinforces the need for surveillance in these populations.
Patients with unclassified-IBMFS manifest bone marrow failure and heterogeneous clinical and genetic characteristics. We found a substantially, and previously unreported, high risk of CMMT (24%) before 18 years of age in this population. This underlies the critical need to initiate cancer surveillance even when the precise syndrome and genetic group is unknown. Although the majority of patients with unclassified-IBMFS underwent extensive genetic testing that was negative, not all of them were tested for mutations in all known IBMFS genes. Some of the unclassified cases with CMMT in this group might, therefore, have mutations in genes such as RUNX1 and GATA2. Importantly, patients who present with idiopathic MDS are often not comprehensively tested for mutations in IBMFS. Hence, a proportion of the IBMFS patients who have neither a positive family history nor physical malformations and present with idiopathic MDS, would not be captured by our registry. The magnitude of this population of patients remains to be determined.
Our results indicate for the first time that CMMT is a progressive risk and significantly affects survival, which indicates a major need to develop strategies for prevention and early detection. It is noteworthy that the high rate of progression was identified at a median follow-up of 66 months, and that the prognosis may be even worse with longer follow-up. The impact of underlying category of IBMFS on the outcome of CMMT is unknown. The results of our study indicate no differences in disease progression and survival between patients with the common IBMFS after they develop CMMT. This might be due to the small numbers of patients in each category. Alternatively, it may be that the long-term outcome is substantially affected regardless of the specific IBMFS category. Interestingly, the pattern of progression and mortality was different between FA and SDS patients (Figure 3). FA patients had initially high progression and mortality rates and then a plateau, whereas SDS patients had a persistent risk of progression and drop in survival over time. The early and fast drop in survival in FA patients is likely due to early intervention with HSCT due to concomitant signs of severe cytopenia. Indeed, 11 of 14 patients with FA and CMMT underwent HSCT and the majority survived. Only one patient with SDS and CMMT underwent HSCT. The data therefore likely reflect the natural history of SDS patients with CMMT, and although past studies suggested that progression of these patients from CMCA to advanced MDS and AML might be slow,1,26,27 our data demonstrate that the risk of progression and mortality continue without a plateau from the time the CMMT is diagnosed. Although the number of SDS patients with CMMT is small, these results indicate a crucial need for further confirmation of these findings and for research into whether early intervention with HSCT in certain SDS patients could improve outcome.
In our study, RC was the most common cytopathology in IBMFS-associated CMMT.12,28 Patients with RC fared significantly better than those with RCEB with respect to progression and survival. The predicted risk of progression in those with RCEB was high early after diagnosis, in line with the findings of previous studies of MDS.29,30 Surprisingly, the risk of progression in patients with RC persisted and continued to increase over time with no apparent plateau. Clearly, the impact of early intervention with HSCT on overall survival in this group should be studied.
In contrast to de novo MDS, in which 30–50% of patients have a CMCA,31–33 CMCA were seen in 93% of our patients with IBMFS-associated CMMT at the time of presentation. Based on the International Prognostic Scoring System for adult MDS, monosomy 7 and complex cytogenetics (≥3 chromosomal abnormalities) are classified as poor prognostic features.34 Monosomy 7 occurred in one-third of our patients and was the most common CMCA, as in children and adolescents with de novo MDS.7 The predicted risk of progression and estimated survival of patients with monosomy 7 at 5 years after diagnosis of CMMT were 54% and 85%, respectively. Over one third of the patients with monosomy 7 did not receive a transplant and were alive at last follow-up. These results suggest a novel concept that HSCT may not be automatically necessary for all patients with monosomy 7, but only offered to those who have either significant or progressive cytopenia or excess blasts. Discovery of co-existing genetic alterations and definition of their prognostic significance will help to refine the transplant indications in this group. Patients with complex cytogenetics had a poor outcome in our study with a 60% predicted risk of progression as early as 1 year after the diagnosis of CMMT, and a 50% predicted overall survival at 5 years. This is similar to reports of poor outcome in patients with de novo MDS and complex cytogenetics.31,34,35
Isochromosome 7q and del(20q) can be seen for many years in the bone marrow from patients with stable SDS and can even become periodically undetectable.1,36–38 Surprisingly, in the present study, three out of four patients with i(7q) and SDS had significant complications at presentation or at last follow-up. In addition, one of the two patients with del(20q) developed RCEB and AML 11 years after the first appearance of this CMCA. These data may suggest that newly appearing clones with i(7q) and del(20q) signal a slowly progressive bone marrow dyscrasia that will eventually require treatment. Larger cohorts with longer follow-up are needed to determine the true impact of these cytogenetic abnormalities on progression to more severe bone marrow failure and/or leukemia.
Two classifications of pediatric MDS have been proposed due to increasing awareness of the biological and clinical differences between pediatric and adult MDS. In addition to the 2002 CCC pediatric MDS classification, a World Health Organization (WHO) pediatric MDS classification was developed in 2003 and revised in 2008.12,39–41 This classification focuses on MDS cytopathology and includes three classes: RC of childhood (RCC), RAEB (myeloblasts of 5–19%) and RAEB in transformation (RAEB-t, myeloblasts of 20–29%). RCRS was omitted from the pediatric WHO MDS classification due to its rarity in childhood. RCD was combined with RCC due to yet unclear differences from RCC. The pediatric WHO classification has several limitations. First, although 40% of children with MDS have an underlying IBMFS,11 the classification is mainly based on experience from de novo MDS and designed for this MDS category. The application of this classification to IBMFS has never been studied. Second, the omission of RCRS from the pediatric WHO classification did not take into account that despite its rarity, this cytopathology exists in children, as evidenced by this series and other reports in the literature.42 Had we used the pediatric WHO MDS classification we could not have categorized either of our cases of RCRS. Third, the significance of RCD has not been systematically and thoroughly studied in IBMFS. Although our numbers are small, there is a suggestion that this category carries a different risk of progression and survival than RC (Online Supplementary Figures S2 and S3). Hence, further studies are required before RCD is omitted as a separate entity from the pediatric MDS classifications. The pediatric CCC classification was designed to include all categories of MDS (de novo, therapy-related and syndrome-associated). To our knowledge the present study tests for the first time the prognostic significance of a pediatric MDS classification in a large cohort of patients with IBMFS-associated MDS or CMMT.
One of the cases in the present series did not fit the MDS diagnostic criteria proposed by Hasle et al.12 However, the patient clearly had MDS, since he had an MDS/AML predisposition syndrome (constitutional trisomy 8), progressive cytopenia and a hypercellular bone marrow. He did not have prominent dysplasia, ringed sideroblasts, excess blasts, cytogenetic abnormalities or indication of any other dietary, metabolic or infectious disorder that could account for the blood dyscrasia. We propose modifying the diagnostic criteria of pediatric MDS (Online Supplementary Table S1) and including cytopenia with hypercellular bone marrow that cannot be explained by causes other than MDS.
The data in the present study help to define the impact of category, cytopathology and bone marrow cytogenetic abnormalities on the characteristics and prognosis of IBMFS-associated CMMT. These components of the CCC classification simplify the wide assortment of variables that constitute pediatric MDS and CMMT into one scheme, and hence can be used clinically to describe the disease at presentation and follow-up. Due to the rarity of the disorders, the numbers in certain categories, cytopathologies and cytogenetic groups were small; thus, enrolling further patients and longer follow-up will be important for replicating the data and defining additional cytogenetic and genetic variables as risk factors. Emerging genomic data can be incorporated and be used to further characterize and understand IBMFS-associated CMMT.
Acknowledgments
The study was supported by grants from the C17 Research Network and Candlelighters Canada.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dror Y, Durie P, Ginzberg H, et al. Clonal evolution in marrows of patients with Shwachman-Diamond syndrome: a prospective 5-year follow-up study. Exp Hematol. 2002;30(7):659–669. [DOI] [PubMed] [Google Scholar]
- 2.Göhring G, Karow A, Steinemann D, et al. Chromosomal aberrations in congenital bone marrow failure disorders-an early indicator for leukemogenesis? Ann Hematol. 2007;86(10):733–739. [DOI] [PubMed] [Google Scholar]
- 3.Tonnies H, Huber S, Kuhl JS, et al. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood. 2003;101(10):3872–3874. [DOI] [PubMed] [Google Scholar]
- 4.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg PS, Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. 2008;93(4):511–517. [DOI] [PubMed] [Google Scholar]
- 6.Dokal I, Vulliamy T. Inherited aplastic anaemias/bone marrow failure syndromes. Blood Rev. 2008;22(3):141–153. [DOI] [PubMed] [Google Scholar]
- 7.Niemeyer CM, Baumann I. Myelodysplastic syndrome in children and adolescents. Semin Hematol. 2008;45(1):60–70. [DOI] [PubMed] [Google Scholar]
- 8.Butturini A, Gale RP, Verlander PC, et al. Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study. Blood. 1994;84(5):1650–1655. [PubMed] [Google Scholar]
- 9.Aul C, Giagounidis A, Germing U, Ganser A. Evaluating the prognosis of patients with myelodysplastic syndromes. Ann Hematol. 2002;81(9):485–497. [DOI] [PubMed] [Google Scholar]
- 10.Locatelli F, Zecca M, Pession A, et al. Myelodysplastic syndromes: the pediatric point of view. Haematologica. 1995;80(3): 268–279. [PubMed] [Google Scholar]
- 11.Mandel K, Dror Y, Poon A, Freedman MH. A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol. 2002;24(7):596–605. [DOI] [PubMed] [Google Scholar]
- 12.Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17(2):277–282. [DOI] [PubMed] [Google Scholar]
- 13.Teo JT, Klaassen R, Fernandez CV, et al. Clinical and genetic analysis of unclassifiable inherited bone marrow failure syndromes. Pediatrics. 2008;122(1):e139–148. [DOI] [PubMed] [Google Scholar]
- 14.Tsangaris E, Klaassen R, Fernandez CV, et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J Med Genet. 2011;48(9):618–628. [DOI] [PubMed] [Google Scholar]
- 15.Germing U, Aul C, Niemeyer CM, Haas R, Bennett JM. Epidemiology, classification and prognosis of adults and children with myelodysplastic syndromes. Ann Hematol. 2008;87(9):691–699. [DOI] [PubMed] [Google Scholar]
- 16.Cantu Rajnoldi A, Fenu S, Kerndrup G, et al. Evaluation of dysplastic features in myelodysplastic syndromes: experience from the morphology group of the European Working Group of MDS in Childhood (EWOG-MDS). Ann Hematol. 2005;84(7): 429–433. [DOI] [PubMed] [Google Scholar]
- 17.Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: incidence and survival in the United States. Cancer. 2007;109(8):1536–1542. [DOI] [PubMed] [Google Scholar]
- 18.Aul C, Giagounidis A, Germing U. Epidemiological features of myelodysplastic syndromes: results from regional cancer surveys and hospital-based statistics. Int J Hematol. 2001;73(4):405–410. [DOI] [PubMed] [Google Scholar]
- 19.Gologan R, Georgescu D, Tatic A, Radulescu I, Vasilache D. Epidemiological data from the registry of patients with myelodysplastic syndrome in a single hospital center of Romania. Leuk Res. 2009;33(11):1556–1561. [DOI] [PubMed] [Google Scholar]
- 20.Kuendgen A, Matsuda A, Germing U. Differences in epidemiology of MDS between Western and Eastern countries: Ethnic differences or environmental influence? Leuk Res. 2007;31(1):103–104. [DOI] [PubMed] [Google Scholar]
- 21.Hashmi SK, Allen C, Klaassen R, et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation. Clin Genet. 2011;79(5):448–458. [DOI] [PubMed] [Google Scholar]
- 22.Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 2003;101(4):1249–1256. [DOI] [PubMed] [Google Scholar]
- 23.Alter BP, Caruso JP, Drachtman RA, et al. Fanconi anemia: myelodysplasia as a predictor of outcome. Cancer Genet Cytogenet. 2000;117(2):125–131. [DOI] [PubMed] [Google Scholar]
- 24.Alter BP, Giri N, Savage SA, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010;150(2):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann NY Acad Sci. 2011;1242:40–55. [DOI] [PubMed] [Google Scholar]
- 26.Maserati E, Minelli A, Pressato B, et al. Shwachman syndrome as mutator phenotype responsible for myeloid dysplasia/neoplasia through karyotype instability and chromosomes 7 and 20 anomalies. Genes Chromosomes Cancer. 2006;45(4):375–382. [DOI] [PubMed] [Google Scholar]
- 27.Valli R, Pressato B, Marletta C, et al. Different loss of material in recurrent chromosome 20 interstitial deletions in Shwachman-Diamond syndrome and in myeloid neoplasms. Mol Cytogenet. 2013;6(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasaki H, Manabe A, Kojima S, et al. Myelodysplastic syndrome in childhood: a retrospective study of 189 patients in Japan. Leukemia. 2001;15(11):1713–1720. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22(3):538–543. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–2088. [PubMed] [Google Scholar]
- 31.Wang H, Wang X, Xu X, Lin G. Cytogenetic features and prognosis analysis in Chinese patients with myelodysplastic syndrome: a multicenter study. Ann Hematol. 2010;89(6): 535–544. [DOI] [PubMed] [Google Scholar]
- 32.Parlier V, van Melle G, Beris P, et al. Hematologic, clinical, and cytogenetic analysis in 109 patients with primary myelodysplastic syndrome. Prognostic significance of morphology and chromosome findings. Cancer Genet Cytogenet. 1994;78(2):219–231. [DOI] [PubMed] [Google Scholar]
- 33.Billstrom R, Thiede T, Hansen S, et al. Bone marrow karyotype and prognosis in primary myelodysplastic syndromes. Eur J Haematol. 1988;41(4):341–346. [DOI] [PubMed] [Google Scholar]
- 34.Hasle H, Baumann I, Bergstrasser E, et al. The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia. 2004;18(12): 2008–2014. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Wang XQ, Xu XP, Lin GW. Cytogenetic evolution correlates with poor prognosis in myelodysplastic syndrome. Cancer Genet Cytogenet. 2010;196(2):159–166. [DOI] [PubMed] [Google Scholar]
- 36.Cunningham J, Sales M, Pearce A, et al. Does isochromosome 7q mandate bone marrow transplant in children with Shwachman-Diamond syndrome? Br J Haematol. 2002;119(4):1062–1069. [DOI] [PubMed] [Google Scholar]
- 37.Minelli A, Maserati E, Nicolis E, et al. The isochromosome i(7)(q10) carrying c.258+2t>c mutation of the SBDS gene does not promote development of myeloid malignancies in patients with Shwachman syndrome. Leukemia. 2009;23(4):708–711. [DOI] [PubMed] [Google Scholar]
- 38.Pressato B, Valli R, Marletta C, et al. Deletion of chromosome 20 in bone marrow of patients with Shwachman-Diamond syndrome, loss of the EIF6 gene and benign prognosis. Br J Haematol. 2012;157(4):503–505. [DOI] [PubMed] [Google Scholar]
- 39.Baumann I, Niemeyer CM, Bennett JM, International Agency for Research on Cancer. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue, Childhood myelodysplastic syndrome. World Health Organization, Geneva, Switzerland: Swerdlow S, Campo E, Harris NL. (eds), 2008. [Google Scholar]
- 40.Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program. 2011:84–89. [DOI] [PubMed] [Google Scholar]
- 41.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 42.Huijgens PC, van der Veen EA, Meijer S, Muntinghe OG. Syndrome of Shwachman and leukaemia. Scand J Haematol. 1977; 18(1):20–24. [DOI] [PubMed] [Google Scholar]