

Congenital anemias due to ineffective erythropoiesis, such as sideroblastic anemia, congenital dyserythropoietic anemia and β-thalassemia intermedia (TI), are frequently associated with increased dietary iron absorption and progressive iron loading, and are therefore defined as “iron loading anemias”.1,2 Finch introduced the concept of the “erythroid regulator” of iron balance to illustrate the physiological mechanism by which the erythroid marrow expansion combined with ineffective erythropoiesis induces a positive iron balance.3
The identification of novel molecules that regulate iron metabolism has provided a deeper insight into the pathophysiology of iron loading anemias. In a mouse model of TI, increased iron absorption was found to be mediated by downregulation of hepcidin and upregulation of ferroportin.4 In patients with TI, an inverse relationship was observed between urinary hepcidin levels and both serum erythropoietin and soluble transferrin receptor (sTfR), taken as markers of erythropoietic activity.5 A number of studies investigated the hypothesis that release of the transforming growth factor β (TGF-β) superfamily members by the erythroblasts during the process of ineffective erythropoiesis might interfere with hepcidin production. The findings of these studies suggest that excess release of growth differentiation factor 15 (GFD15) and twisted gastrulation (TWSG1) may suppress hepcidin production dysregulating iron homeostasis in thalassemia syndromes.6,7 More recently, the hormone erythroferrone (ERFE), has been identified as a new erythroid regulator of hepcidin synthesis.8
β-thalassemia intermedia, hemoglobin E/β-thalassemia (mild and moderate forms), and hemoglobin H (HbH) disease are three clinically distinct forms of the so-called non-transfusion-dependent-thalassemias (NTDTs), a term used to label patients with hemoglobinopathies who do not require regular transfusion for survival. In patients with TI, the broad diversity of β-globin gene mutations, the co-inheritance of β-thalassemia, and the presence of genetic determinants associated with increased production of γ-globin chains in adult life are the main determinants for milder α/non-α-globin chain imbalance and transfusion independence. These factors are of particular relevance in Sardinia, where the most common type of TI as well as of β-thalassemia major is due to a nonsense mutation at codon 39 (c.118C>T) of the β-globin gene. HbH disease is the most severe non-fatal form of the α-thalassemia syndrome, mostly caused by molecular defects of the α-globin genes in which α-globin expression is decreased. HbH is a tetramer of β chains, which is unstable and causes a phenotype of mild to moderate chronic hemolytic anemia characterized by readily detectable HbH inclusion bodies in the peripheral blood cells.9
TI and HbH disease may have similar degrees of anemia, but hemolysis rather than ineffective erythropoiesis (IE) is the primary mechanism in HbH disease.9 Indeed, iron loading is much more common in TI than in HbH disease.9
In this study, we investigated the relationship between erythroid activity, body iron status, and hepcidin levels in 38 subjects with TI and 36 with HbH disease, all followed at the Ospedale Regionale per le Microcitemie, Cagliari, Sardinia, Italy. Patients with TI and HbH disease had either never been transfused or had received only sporadic transfusions during infections, pregnancy or surgery (less than 10 blood units in total, 5 years or more before the evaluation). Ferritin, hemoglobin and hematologic values were measured at the same time as sTfR, plasma hepcidin-25 and GDF15. One patient with HbH disease had received desferrioxamine before the study, while iron chelation therapy had been prescribed to 23 patients with TI, who, however, had shown suboptimal compliance. Eighteen patients with TI and 12 with HbH disease underwent liver iron concentration (LIC) measurement through magnetic resonance imaging within one year from the above-mentioned laboratory analysis (Ferriscan®-Resonance Health, Australia). Molecular diagnosis was performed with standard methods that have been previously described. Patients gave informed consent for genetic analysis. Iron parameters were analyzed as part of patient care in order to ameliorate their clinical management. Hemoglobin and red cell indices were determined with an automated red blood cell counter (Beckman Coulter LH 750), while serum ferritin and erythropoietin was measured with an automated chemiluminescence immunoassay analyzer (IMMULITE®2000, Siemens-DPC). sTfR was quantified using an immunonephelometric method (Dade Behring) on a BN II System analyzer, the quantification of serum GDF15 was performed with DuoSet ELISA for human GDF15 (R&D Systems) following the manufacturer’s protocol. Plasma hepcidin-25 measurements were performed by a combination of weak cation exchange chromatography and time-of-flight mass spectrometry (WCX-TOF MS).10 For statistical purposes, when hepcidin was below the lower limit of detection (i.e. <0.50 nM) it was considered as 0.25 nM; this may be considered a limitation of the study.
Characteristics of all individual patients with TI and HbH disease are given in the Online Supplementary Table S1A and B, and are summarized in Table 1. Reticulocyte percentage was lower in TI and reflects: i) the decreased number of differentiated cells despite the huge proliferation of medullary erythroid precursors; and ii) relative increased apoptosis in this disorder. Iron and erythropoietic parameters of both groups are given in Table 2 and in Online Supplementary Figure S2. Erythropoietin was increased in TI and in 25% of the HbH patients. sTfR was increased in both disorders. Elevations in both erythropoietin and sTfR indicate increased erythropoietic drive in both diseases, corroborating previous studies.5,11,12 Erythropoietin and sTfR in TI were 4 and 2 times more than in HbH disease, respectively. In agreement with these observations, in TI GDF15 levels were greatly elevated and correlated with sTfR (r2=14.5, P=0.01). In HbH disease, despite clearly elevated sTfR levels, the median GDF15 level was within reference range, with only 28% of the patients having levels above the upper limit of the reference range. Moreover, the hepcidin/GDF15 ratio was approximately 100 times lower for TI than for HbH (Table 2 and Online Supplementary Figure S1), suggesting that, in comparison to HbH, hepcidin levels in TI are also more sensitive to downregulation by GDF15. Considering both groups together, GDF15 was higher in males than in females (median value 4488 vs. 1499 pg/mL; P=0.049). Consistent with this increased erythropoietic drive in TI, hepcidin levels were significantly lower and (despite the iron chelation treatment) ferritin in this syndrome was significantly higher compared to HbH. LIC was also higher in TI, but this difference was not significant, probably due to treatment of most TI patients with desferrioxamine in combination with the relatively low number of patients for whom LIC was assessed (Table 2). Serum hepcidin was below the limit of detection in 30 subjects with TI and strongly reduced in the 8 others, while it was less than 0.5 nM only in one of the 36 patients with HbH disease: there was a significant difference in the proportion of hepcidin values below 0.50 nM between the two groups (P<0.001), while median values groups were 0.50 nM (range 0.50–0.50) and 2.4 nM (range 1.3–4.1), respectively (P<0.001).
Table 1.
Characteristics of patients with β-thalassemia intermedia and hemoglobin H disease.
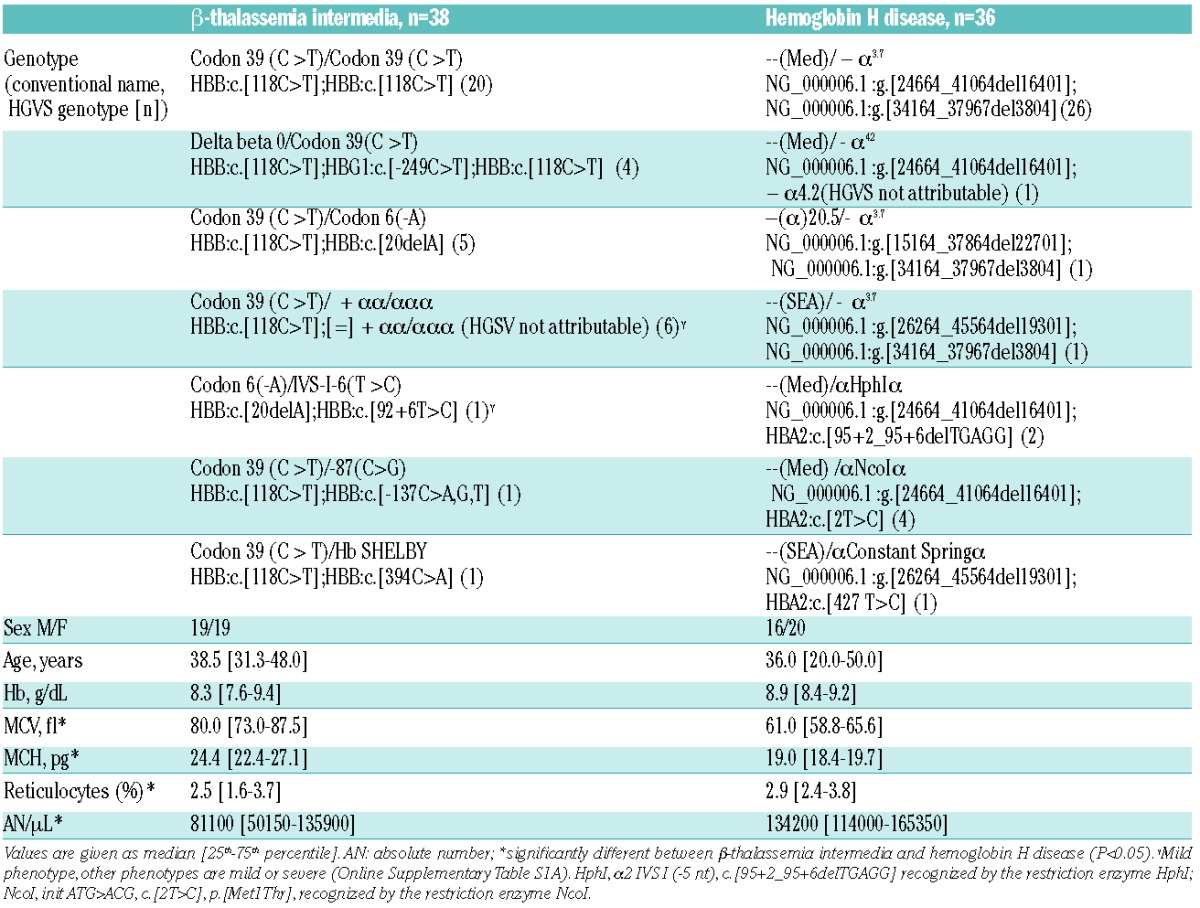
Table 2.
Iron and erythropoietic parameters of β-thalassemia intermedia and hemoglobin H disease.
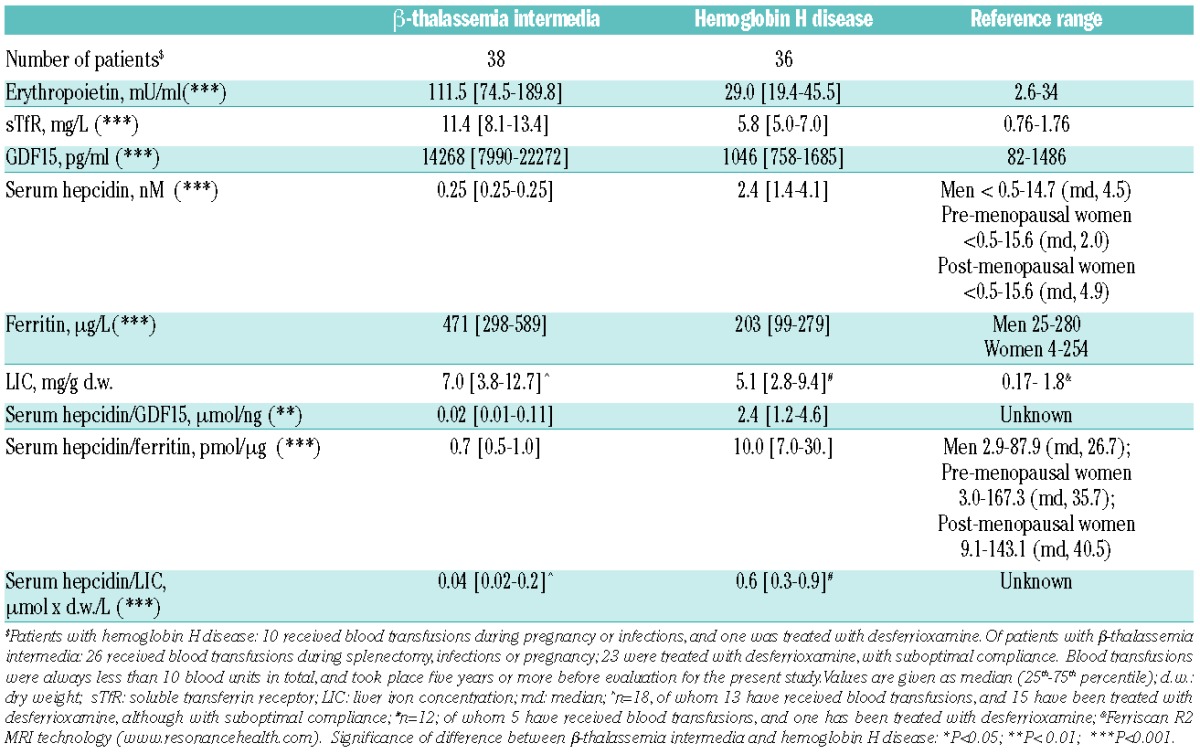
A binary logistic regression model (Cox & Snell: R2=0.624) for the occurrence of unmeasurable values of hepcidin (≤0.50 nM vs. >0.50 nM) considering the disease type and severity, GDF15, erythropoietin, sTfR, serum ferritin and sex, showed three variables significantly associated with the occurrence of very low values of hepcidin: disease (Odds Ratio (OR)=46.0 for TI vs. HbH disease; P=0.004), levels of GDF15 (OR=1.5 per 1000 pg/mL increase; P=0.033) and levels of serum ferritin (OR=12.0 per 100 ng/ml decrease; P=0.040). These data illustrate that low values of hepcidin are strongly driven by erythropoietic factors and iron stores, and in a different manner for TI and HbH disease. These observations are consistent with GDF15 as the factor contributing most to inhibition of hepcidin levels, in agreement with its previously proposed causal role in the IE-hepcidin-ferritin cascade.6
In addition, both the hepcidin/ferritin ratio and the hepcidin/LIC ratio were roughly 10-fold lower for TI than for HbH (Table 2 and Online Supplementary Figure S2). When compared to hepcidin/ferritin ratio of adults from a sample of the Dutch general population, patients with HbH have a median hepcidin/ferritin ratio that was substantially lower but still within the reference range (www.hepcidinanalysis.com).13 These observations of hepcidin levels that are inappropriately low for the body iron levels in TI, and to a lesser extent in HbH, corroborate the presence of a suppressive erythropoietic signal associated with IE that is strong in TI and only slightly increased in HbH.4,14,15 GDF15, erythropoietin and sTfR were significantly lower in the subjects with mild TI genotypes (n=7) than in the moderate and severe genotypes (n=31) (P<0.005), confirming the role of GDF15 as an IE response marker, whereas only sTfR was significantly lower in subjects with mild deletional HbH (n=29) compared to the more severe non-deletional HbH defects (n=7; P=0.003). It is interesting to note that, for these analyses, the number of subjects might be too low to allow definitive conclusions to be made.
This study demonstrates that the different extent of IE and chronic hemolysis in TI and HbH disease is well reflected by the diverse patterns of the GDF15-hepcidin-ferritin axis in the two populations, even at similar levels of anemia. In TI, the erythropoietic signal more strongly suppresses iron loading-induced signaling to hepcidin, and for this reason iron overload is more common in TI disease than in HbH disease. Larger studies are needed to clarify whether for both TI and HbH the various molecular arrangements in the respective β-and α-globin chains associated with different degrees of IE affect the above-mentioned axis and related tendency to develop iron overload.
Acknowledgments
Erwin Wiegerinck performed hepcidin-25 measurements and Geert Remy and Stan Verweij GDF15 measurements.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Cazzola M, Barosi G, Bergamaschi G, et al. Iron loading in congenital dyserythropoietic anaemias and congenital sideroblastic anaemias. Br J Haematol. 1983;54(4):649–654. [DOI] [PubMed] [Google Scholar]
- 2.Cazzola M, Beguin Y, Bergamaschi G, et al. Soluble transferrin receptor as a potential determinant of iron loading in congenital anaemias due to ineffective erythropoiesis. Br J Haematol. 1999;106(3):752–755. [DOI] [PubMed] [Google Scholar]
- 3.Finch C. Regulators of iron balance in humans. Blood. 1994;84(6):1697–1702. [PubMed] [Google Scholar]
- 4.Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109(11):5027–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–588. [DOI] [PubMed] [Google Scholar]
- 6.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13(9):1096–1101. [DOI] [PubMed] [Google Scholar]
- 7.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin in murine and human cells. Blood. 2009;114(1):181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fucharoen S, Viprakasit V. Hb H disease: clinical course and disease modifiers. Hematology Am Soc Hematol Educ Program. 2009:26–34. [DOI] [PubMed] [Google Scholar]
- 10.Kroot JJ, Laarakkers CM, Geurts-Moespot AJ, et al. Immunochemical and mass-spectrometry-based serum hepcidin assays for iron metabolism disorders. Clin Chem. 2010;56(10):1570–1579. [DOI] [PubMed] [Google Scholar]
- 11.Rees DC, Williams TN, Maitland K, Clegg JB, Weatherall DJ. Alpha thalassemia is associated with increased soluble transferrin receptor levels. Br J Haematol. 1998;103:365–369. [DOI] [PubMed] [Google Scholar]
- 12.Papassotiriou I, Traeger-Synodinos J, Kanavakis E, Karagiorga M, Stamoulakatou A, Kattamis C. Erythroid marrow activity and hemoglobin H levels in hemoglobin H disease. J Pediatr Hematol Oncol. 1998;20(6):539–544. [PubMed] [Google Scholar]
- 13.Galesloot TE, Vermeulen SH, Geurts-Moespot AJ, et al. Serum hepcidin: reference ranges and biochemical correlates in the general population. Blood. 2011;117(25):e218–225. [DOI] [PubMed] [Google Scholar]
- 14.Kroot JJ, Tjalsma H, Fleming RE, Swinkels DW. Hepcidin in human iron disorders: diagnostic implications. Clin Chem. 2011; 57(12):1650–1669. [DOI] [PubMed] [Google Scholar]
- 15.Papanikolaou G1, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]