

Acquired aplastic anemia (AA), characterized by pancytopenia in peripheral blood (PB) and bone marrow (BM) hypoplasia, is a bone marrow failure syndrome. The late evolution to myelodysplastic syndromes (MDS)/acute myeloid leukemia (AML) is the most common clonal complication in refractory patients and in those who do not achieve a robust response.1,2 The reported rates of clonal evolution varied in some studies from 1.7%–57% during an observation period of 5–11 years.1–3 The evolution of chromosomal abnormalities including monosomy 7 has been associated with a poor prognosis, but some abnormal cytogenetics, for example, +8 and del13q, have been associated with a good response to immunosuppressive therapy (IST).4 Single nucleotide polymorphism array karyotype abnormalities could identify those AA patients who were at risk of clonal evolution.5 Mutations of DNMT3A and BCOR may be associated with a risk of transformation to MDS.6 However, no reliable biomarkers that predict prognosis and MDS evolution are currently known in AA. AA has genetic instability, and acquired somatic mutations of ASXL1, TET2, RUNX1, TP53, K-RAS and N-RAS typically occurred in MDS/AML.7–11 We postulated that these mutations might be an early event in AA evolution to MDS/AML, and could predict MDS/AML evolution and prognosis. In this study, we analyzed mutations in ASXL1, TET2, RUNX1, TP53, K-RAS and N-RAS in Chinese AA patients and showed that somatic mutations that were common in myeloid malignancies also existed in AA. Moreover, patients with different mutations showed distinct clinical and biological features.
Bone marrow aspirates were collected from 440 patients with pancytopenia between February 2012 and September 2014 at a single institution (Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Science & Peking Union Medical College). A total of 138 patients with AA diagnosed according to standard criteria12 had complete clinical data for this study; 16 iron deficiency anemia or megaloblastic anemia patients (age range 32–77 years) were analyzed as controls. Tables 1 and 2 list the clinical and biological characteristics of these AA patients. All patients were screened for PNH clone using the combination of FLAER with multicolor flow cytometry to detect expression of GPI-anchored proteins on peripheral blood red cells and granulocytes. Chromosome analyses were performed on unstimulated bone marrow cells after 24 h cultures using the G-and/or R-banding techniques. Somatic mutations of TET2, ASXL1, RUNX1, TP53, N-RAS and K-RAS genes were searched by direct sequencing exons and consensus splicing sites after PCR amplification of genomic DNA. Exons studied were: 1) TET2 (reference sequence: NM_001127208.2), exons 3 and 11; 2) ASXL1 (reference sequence: NM_015338.5), exon 12; 3) RUNX1 (reference sequence: NM_001754), exons 3–8; 4) TP53 (reference sequence: NM_000546.5), exons 5–8; 5) N-RAS (reference sequence: NM_002524.4), codon 12 and 13; 6) K-RAS (reference sequence: NM_004985.3), codon 12 and 13. The primers used for sequencing are listed in Online Supplementary Table S1. Previously annotated single nucleotide polymorphisms in the database (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes) were discarded.
Table 1.
Comparison of clinical and biological characteristics of 138 aplastic anemia patients according to ASXL1 mutation.
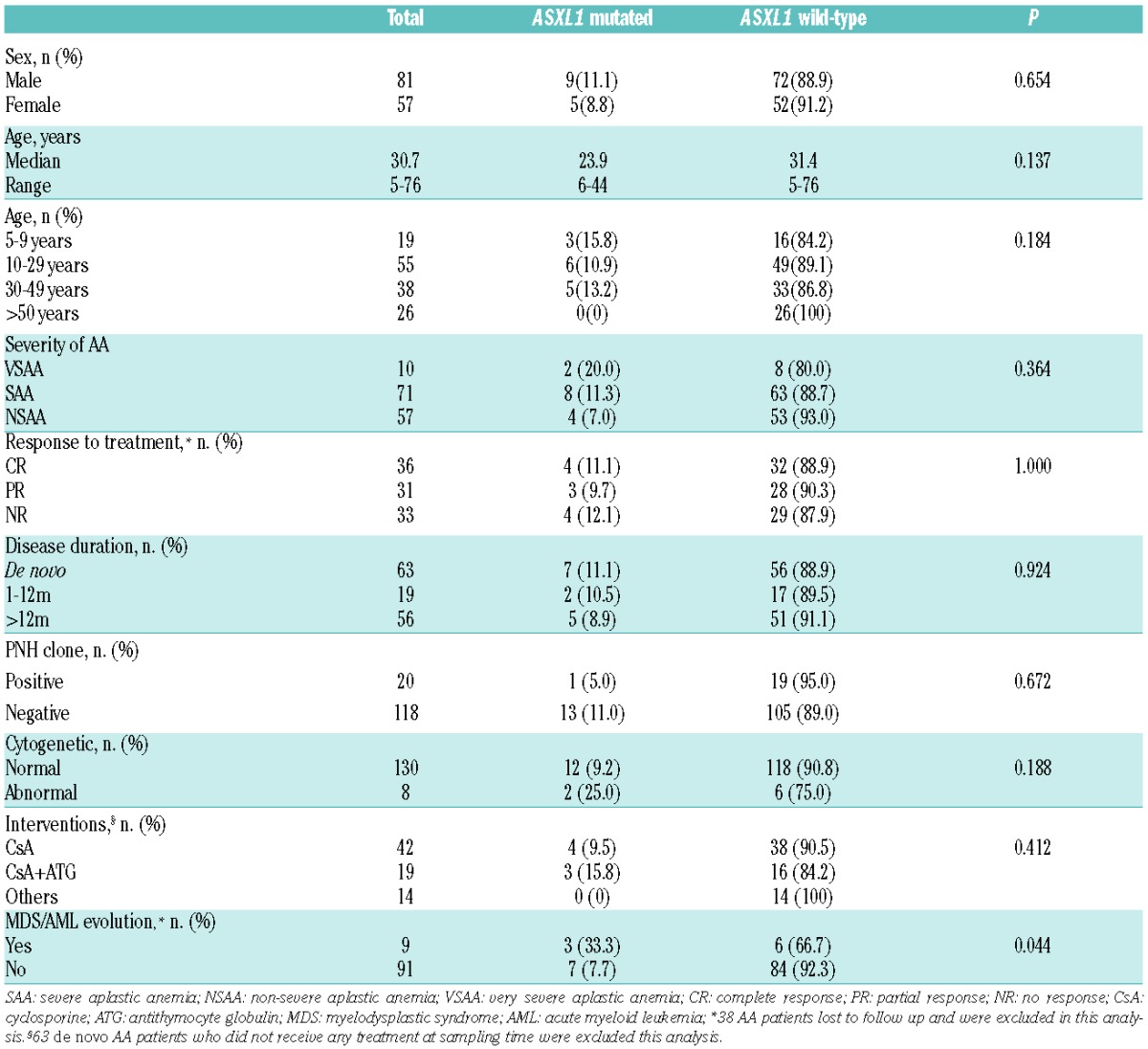
Table 2.
Comparison of clinical and biological characteristics of 138 aplastic anemia patients according to TET2 mutation.
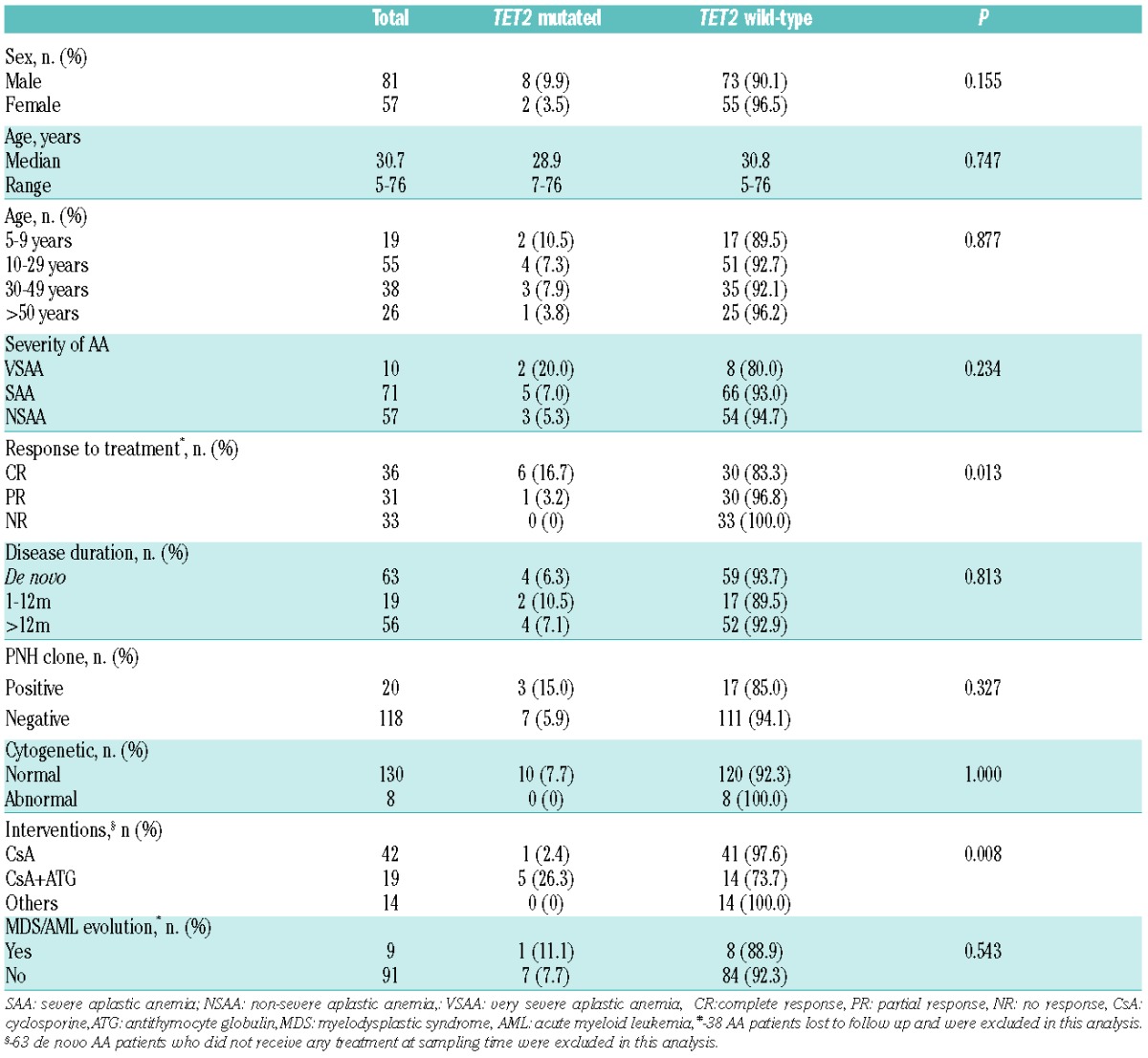
A total of 24 of 138 (17.4%) patients with AA were found to harbor mutations, including ASXL1 mutations in 14 patients, TET2 in 10 patients; no mutations were detected in RUNX1, TP53, K-RAS and N-RAS. All mutations were heterozygous, including missense (n=13), nonsense (n=8), frameshift (n=4), non-frameshift deletion (n=1), and splice site (n=1) changes (Figure 1 and Online Supplementary Tables S2 and S3). Comparisons of clinical and biological variables between patients with and without ASXL1/TET2 mutations are shown in Tables 1 and 2. All ASXL1 and TET2 mutations were isolated. ASXL1 mutations had different relationship with clinical features and biological characteristics compared with those of TET2 mutations. Somatic mutations of ASXL1 were the most frequent abnormality and were seen in 14 of 138 (10.1%) patients. Median age of patients with ASXL1 mutations was lower than those without ASXL1 mutations (23.9 years vs. 31.4 years). Patients under nine years of age had the highest incidence (3 of 19, 15.8%), but the difference was not significant (P=0.184). Also there were no significant differences between patients with or without ASXL1 mutations in terms of sex (P=0.654), severity of AA (P=0.364), duration of disease (P=0.924), interventions (P=0.412), or response to treatment (P=1.000) (Table 1). TET2 mutations were seen in 10 of 138 (7.3%) patients, and were closely associated with prior interventions. Mutations were detected in 5 of 19 (26.3%) patients who had received antithymocyte globulin (ATG)-based IST, compared with patients receiving cyclosporine (CsA)-based IST (1 of 42, 2.4%) and others (0 of 14, 0%); these differences were significant (P=0.008). TET2 mutations also were closely related with a good response. The frequency of TET2 mutations in patients with CR (6 of 36, 16.7%) was higher than those with PR (1 of 31, 3.2%) or NR (0%, 0 of 33); this difference was also significant (P=0.013). There was no significant difference between patients with or without TET2 mutations in terms of age (P=0.877), sex (P=0.155), severity of AA (P=0.234), or duration of disease (P=0.813) (Table 2).
Of 138 patients, the PNH clone was negative in 118 patients, positive in 20 (14.5%); 10 were detected at diagnosis, and after diagnosis in the other 10. The metaphase cytogenetic karyotype was normal in 130 patients, trisomy 8 (n=3), del (13) (q12–q21) (n=2), monosomy 7 (n=1), 16qh+ (n=1) and complex karyotype (n=1) were detected in the remaining 8 patients (Online Supplementary Table S4). ASXL1 and TET2 mutations had no relationship with the PNH clone (P=0.672 and P=0.327, respectively) (Tables 1 and 2). ASXL1 mutation were detected in 2 of 8 (25%) patients with abnormal cytogenetics, compared with patients with normal cytogenetics (12 of 130, 9.2%), but the difference was not significant (P=0.188) (Table 1), possibly because of the limited number of cases. Surprisingly, TET2 mutation had no relationship with abnormal cytogenetics (P=1.000), and was not detected in any patients with an abnormal cytogenetic profile (Table 2).
Of the 100 AA patients who had complete clinical data available for analysis in evolution to MDS, progression to MDS was seen in 9 patients (Online Supplementary Table S5). The median time of progression to MDS was 36.3 months (range 7–86 months), and in 6 of 9 patients occurred less than three years from diagnosis. Seven of 9 patients received CsA-based IST, the others ATG-based IST; 2 patients who evolved to MDS subsequently progressed to AML. The cytogenetic karyotype at the time of evolution was normal (n=5), monosomy 7 (n=1), trisomy 8 (n=1), del(13)(q12–q22) (n=1), and complex karyotype (n=1), respectively. The frequency of ASXL1 mutations was higher in patients who progressed to MDS than those who did not (3 of 9, 33.3% vs. 7 of 91, 7.7%; P=0.044) (Table 1). AA patients with ASXL1 mutations had a greater risk of transformation to MDS in univariate analysis (P=0.014) (Online Supplementary Figure S1A). Surprisingly, TET2 mutations had no relationship with evolution to MDS in univariate analysis (P=0.464) (Online Supplementary Figure S1B); 1 of 9 (11.1%) patients with TET2 mutation evolved to MDS, compared to 7 of 91 (7.7%) patients without TET2 mutation, although this difference was not significant (P=0.543) (Table 2).
ASXL1 and TET2 mutations have been reported in various myeloid malignancies,7–11 especially MDS/AML. Evolution of AA to MDS/AML is a serious and common long-term complication.1–2,13 Little has been known about somatic mutations in AA until now. In this study, we found mutations in epigenetic regulator genes including TET2 and ASXL1 in 17.4% patients with AA. Mutations were detected at any stage of disease. The rate of mutation was similar to those reported in AA patients in the UK.6 ASXL1 mutations were the most common mutation in AA, and were associated with a transformation to MDS. Meanwhile, ASXL1 mutations were relatively more frequently found in patients with abnormal cytogenetics compared with patients with normal cytogenetics. Kulasekararaj et al.6 also found that 7 of 12 AA patients with ASXL1 mutations showed progression to MDS and were associated with 40% risk of transformation to MDS. In their report, ASXL1 mutation occurred more frequently in older patients, but we found ASXL1 mutation was more common in younger Chinese patients and those under nine years of age had the highest incidence. For the first time, TET2 mutations were found in AA. Surprisingly we found that AA patients with TET2 mutations had a better response to IST than those without mutations. The advantage resulted at least in part from the fact that TET2 mutations occurred more frequently in patients who had received ATG-based IST, and that TET2 mutations were not associated with abnormal cytogenetics and had no adverse effect on transformation to MDS. In addition, TET2 mutations were also associated with longer survival, lower risk of transformation to AML, and a molecular marker for good prognosis in patients with MDS.14,15 TET2 mutations may have a good prognostic implication in AA, but this result needs to be further confirmed with larger series of patients.
In summary, we identified TET2 and ASXL1 mutations in Chinese patients with AA. These important data may predict disease outcomes in AA patients of diverse genetic backgrounds.
Acknowledgments
The authors would like to thank all of the doctors and nurses in the therapeutic Centre of Anemic Diseases and the research team of the Clinical Laboratory Centre for their professional assistance.
Footnotes
Funding: this work was supported by grants from the National Natural Science Foundation of China (No. 81330015, No. 81370606 and No. 81300388) and a grant of Tianjin Municipal Science and Technology Commission (No.14JCQNJC10700).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. JAMA. 2003; 289(9):1130–1135. [DOI] [PubMed] [Google Scholar]
- 2.Kojima S, Ohara A, Tsuchida M, et al. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood. 2002;100(3):786–790. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Li X, Ge M, et al. Long-term follow-up of clonal evolutions in 802 aplastic anemia patients: a single-center experience. Ann Hematol. 2011;90(5):529–537. [DOI] [PubMed] [Google Scholar]
- 4.Maciejewski JP, Risitano A, Sloand EM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002;99(9):3129–3135. [DOI] [PubMed] [Google Scholar]
- 5.Afable MG, Wlodarski M, Makishima H, et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011; 117(25):6876–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a sub-group aplastic anemia patients that progress to myelodysplastic syndrome. Blood. 2014;124(17):2698–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. [DOI] [PubMed] [Google Scholar]
- 9.Dicker F, Haferlach C, Sundermann J, et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia, 2010;24(8):1528–1532. [DOI] [PubMed] [Google Scholar]
- 10.Boultwood J, Perry J, Pellagatti A, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24(5):1062–1065. [DOI] [PubMed] [Google Scholar]
- 11.Fidler C, Watkins F, Bowen DT, Littlewood TJ, Wainscoat JS, Boultwood J. NRAS, FLT3 and TP53 mutations in patients with myelodysplastic syndrome and a del(5q). Haematologica. 2004; 89(7):865–866. [PubMed] [Google Scholar]
- 12.Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70. [DOI] [PubMed] [Google Scholar]
- 13.Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16(1 Suppl): S119–S125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Ai X, Gale RP, et al. TET2, ASXL1 and EZH2 mutations in Chinese with myelodysplastic syndromes. Leuk Res. 2013;37(3):305–311. [DOI] [PubMed] [Google Scholar]
- 15.Olivier Kosmider, Véronique, Gelsi-Boyer, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood. 2009;114(15):3285–3291. [DOI] [PubMed] [Google Scholar]