

Genetic alterations of the mixed-lineage leukemia (MLL) gene are commonly implicated in the development of acute leukemias. In acute myeloid leukemia (AML) a partial tandem duplication (PTD) of MLL occurs in about 5%–11%1–4 of patients and has been linked to poor treatment outcome.1,5 Although recent studies show that MLL-PTD does not have a prognostic impact in CN-AML patients treated with intensive therapy,2,3 many patients still succumb to their disease,2,3,5–7 and the course of disease for patients not eligible for intensive chemotherapy is even more dismal. Thus, novel and innovative treatment approaches are needed. In acute leukemias, the MLL gene is also commonly affected by chromosomal rearrangements. Several MLL-rearranged leukemias were recently shown to be dependent on the only known histone 3 lysine 79 (H3K79) methyltransferase DOT1L.8,9 Genetic deletion of DOT1L impairs initiation and maintenance of MLL-rearranged leukemias in mouse models8–12 and pharmacological inhibition of DOT1L with the specific small molecules EPZ004777 or EPZ-5676 selectively kills MLL-rearranged leukemia cells in vitro13 and in vivo.14 Furthermore, DOT1L inhibition leads to a reversal of a characteristic gene expression signature, including downregulation of HOXA-cluster genes in MLL-rearranged leukemias.8–10 Based on these pre-clinical findings, EPZ-5676 is currently under investigation in a clinical phase I trial (clinicaltrials.gov identifier: 01684150). In the current study, we assessed whether MLL-PTD-positive leukemias that share critical biological features with MLL-rearranged leukemias, such as high expression of HOXA-cluster genes, similarly require DOT1L.
To investigate the effects of pharmacological DOT1L inhibition on cell growth of MLL-PTD positive leukemias, we first treated two human MLL-PTD-containing leukemia cell lines, EOL-1 and KOPM-88, with increasing doses of EPZ004777. MOLM-13 and HL-60 were also treated as positive and negative controls, respectively, and cell numbers were analyzed over a 14-day period. DOT1L inhibition reduced the cell number as compared to control for both MLL-PTD-positive cell lines and MOLM-13 in a dose-dependent manner, while HL-60 cells were unaffected (Figure 1A). IC50 values following 14 days of exposure to the compound indicate that EOL-1, KOPM-88 and MOLM-13 are all highly sensitive to DOT1L inhibition (Figure 1B). To validate the results from this first set of experiments, we further tested whether MUTZ-11, a third MLL-PTD positive leukemia cell line, was also sensitive to pharmacological DOT1L inhibition. As observed in the other MLL-PTD positive cell lines, exposure to 10 μM EPZ004777 led to profound impairment of cell proliferation in MUTZ-11 cells as compared to vehicle control (Figure 1A). Sensitivity to DOT1L-inhibition was also demonstrated in co-culture assays in 2 primary samples from patients with MLL-PTD-positive AML, and was shown to inhibit cell growth and induce differentiation (Online Supplemental Figure S2).
Figure 1.
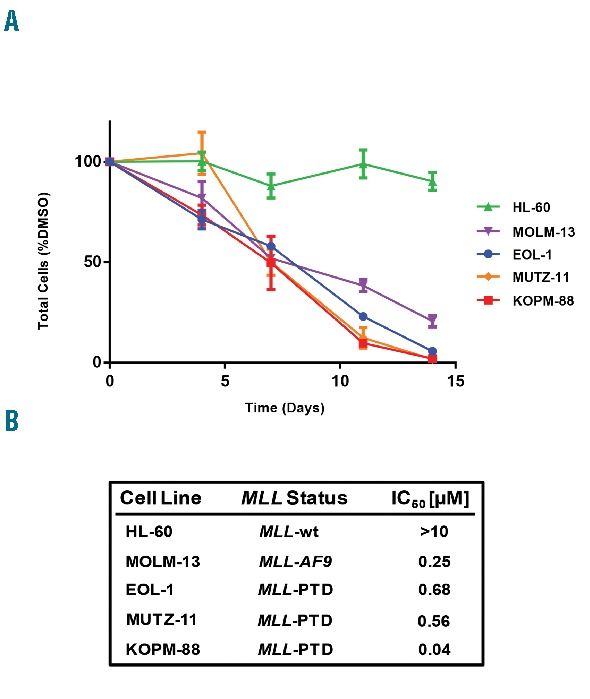
DOT1L inhibition impairs proliferation of MLL-PTD leukemia cell lines. (A) Growth of KOPM-88, EOL-1, MUTZ-11, MOLM-13 and HL-60 cells exposed to 10 μM EPZ004777. Viable cells were counted and re-plated at equal cell numbers in fresh media with fresh compound every 3–4 days. Results were plotted as percentage of split-adjusted viable cells in the presence of 10 μM EPZ004777 compared with DMSO vehicle control. (B) IC50 values for each cell line treated were determined graphically. Presence of MLL-PTD, MLL-AF9 translocation (MLL-AF9), or wild-type for MLL (MLL-wt) is indicated.
Next, we characterized the genome-wide distribution of H3K79me2 in MLL-PTD-positive leukemia cells using chromatin immunoprecipitation followed by next generation sequencing (ChIP-seq) in EOL-1 cells. We observed profound enrichment of H3K79me2 marks at the highly expressed HOXA-cluster locus (Figure 2A), resembling the pattern found in MLL-rearranged leukemias.8–11 ChIP-PCR confirmed H3K79me2 enrichment at HOXA9 and HOXA10 in EOL-1 and in KOPM-88 cells (data not shown). We then assessed the ability of EPZ004777 to inhibit global H3K79me2 marks in the MLL-PTD positive leukemias EOL-1 and KOPM-88, as well as MOLM-13 and HL-60 cells. After four days of treatment, immunoblot analysis confirmed the loss of global H3K79me2 in a dose-dependent manner (Figure 2B).
Figure 2.
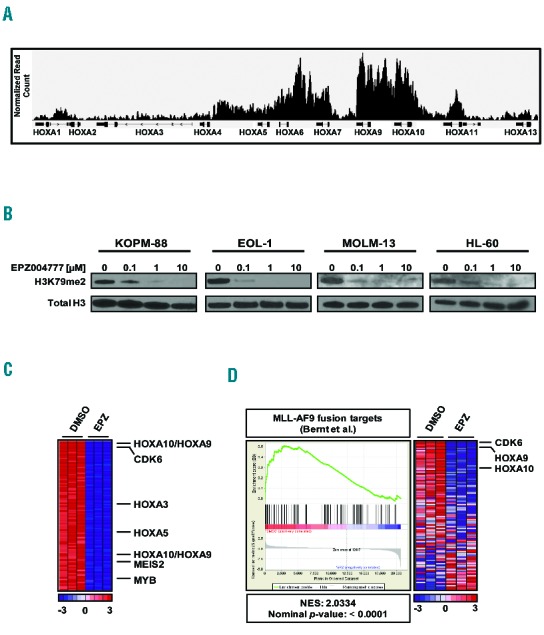
DOT1L inhibition abrogates H3K79me2 and leads to downregulation of MLL targets in MLL-PTD leukemia cell lines. (A) H3K79me2 profile of the HOXA-locus in the MLL-PTD positive EOL-1 cell line as assessed by ChIP-seq. (B) Western blot analysis of global H3K79me2 levels in KOPM-88, EOL-1, MOLM-13 and HL-60 human leukemia cell lines following four days of treatment with EPZ004777. (C) Heatmap of the top 100 down-regulated genes in biological triplicates of EOL-1 cells following 10 μM EPZ004777 treatment (right) for seven days as compared to DMSO vehicle control (left). (D) GSEA of genes down-regulated by 10 μM EPZ004777 treatment of EOL-1 cells for seven days as compared with genes bound by MLL-AF9 in MLL-AF9 transformed murine hematopoietic progenitors. The heatmap indicates differential expression of MLL-AF9 fusion genes between EOL-1 cells treated with either DMSO vehicle control (DMSO, left) or 10 μM EPZ004777 (EPZ, right).
In order to determine the effects of DOT1L-inhibition on gene expression, Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays were performed in EOL-1 cells. HOXA-cluster genes were found among the genes most significantly down-regulated after seven days exposure to EPZ004777 compared to DMSO vehicle control (Figure 2C and Online Supplementary Table S1). In fact, HOXA10 was the second most significantly down-regulated gene (Figure 2C and Online Supplementary Table S1). Quantitative PCR of HOXA9 and HOXA10 was used to validate the results from gene expression arrays and also revealed similar downregulation of these genes in KOPM-88 cells after seven days of DOT1L-inhibition (data not shown). Gene Set Enrichment Analysis (GSEA) of the genes down-regulated following EPZ004777 treatment demonstrated enrichment for common MLL-target genes and genes previously reported to be down-regulated after DOT1L-inhibition in MLL-AF9 leukemia cells (Figure 2D). These results indicate that MLL-PTD cells require DOT1L methyltransferase activity to maintain expression of select MLL-target genes and much of the MLL-associated gene expression program.
To assess the effect of EPZ004777 on the MLL-PTD positive cell lines in more detail, we analyzed changes in cell differentiation and apoptosis upon exposure to EPZ004777 by flow cytometry for the differentiation marker CD11b and for Annexin V and Sytox Blue staining, respectively. Both cell lines showed a progressive increase in CD11b expression over a 14-day period, with changes in expression levels apparent after seven days of DOT1L inhibition in both cell lines (Figure 3A). These changes in expression of CD11b were accompanied by a reduction of Sytox Blue negative viable cells and an increased Annexin V positivity in Sytox Blue negative cells (Figure 3B). In an independent validation experiment, we also observed a substantial increase in Annexin V binding in MUTZ-11 cells (data not shown). These data suggest that small molecule inhibition of DOT1L causes MLL-PTD positive leukemias to differentiate and undergo apoptosis.
Figure 3.
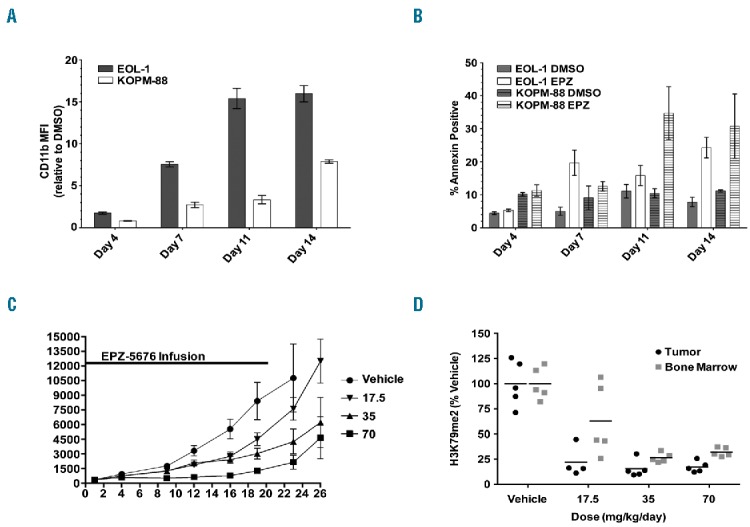
DOT1L inhibition causes differentiation and apoptosis in MLL-PTD leukemia cell lines and impairs MLL-PTD leukemia cell proliferation in vivo. (A) Mean fluorescent intensity (MFI) measured by flow cytometry for EOL-1 and KOPM-88 cells treated with 10 μM EPZ004777 for up to 14 days stained with CD11b antibody. Average MFI of EPZ004777 treated biological triplicates are plotted as the fold-change relative to DMSO vehicle control (EPZ/DMSO). Error bars: standard deviation. (B) EOL-1 and KOPM-88 cells were treated with 10 μM EPZ004777 (EPZ) or DMSO vehicle control for up to 14 days and analyzed by flow cytometry every 3–4 days for apoptosis by Annexin V binding. Error bars: standard deviation. (C) Effect of EPZ-5676 administration on the growth of EOL-1 xenograft tumors implanted subcutaneously (SC) in immunocompromised rats. Rats (n=10 per group) were dosed continuously by IV infusion with vehicle, with EPZ-5676 at 17.5 mg/kg/day, 35 mg/kg/day, or 70 mg/kg per day for 21 days. Mean tumor sizes are plotted for each group. Error bars represent the standard deviation. Tumor growth inhibition values on day 23 were 80, 60 and 29% for the 70, 35 and 17.5 mg/kg/day dose groups, respectively. EPZ-5676 blood plasma levels (mean±SD) were 1.31±0.68, 0.68±0.47 and 0.42±0.2 μM in the 70, 35 and 17.5 mg/kg/day dose groups, respectively. (D) Abrogation of H3K79me2 in a nude rat EOL-1 SC xenograft following seven days of continous EPZ-5676 infusion as assessed by quantitative ELISA. H3K79me2 levels were normalized to those of total histone H3 in the same sample and are plotted as a percent of the mean H3K79me2 level in tissue from the vehicle-treated group which is set at 100%. Horizontal lines represent the mean percent H3K79me2 values for each group.
Given the sensitivity of MLL-PTD cell lines to DOT1L inhibition in vitro, we wanted to determine whether a DOT1L inhibitor displayed anti-tumor activity in an in vivo model of MLL-PTD leukemia. For these experiments we used EPZ-5676, a DOT1L inhibitor molecule closely related to EPZ004777, but with improved potency and pharmacokinetic properties. We first confirmed the sensitivity of MLL-PTD cell lines to EPZ-5676 in 14-day cell proliferation assays (data not shown) and then developed a subcutaneous EOL-1 xenograft model in immunocompromised rats. The animals bearing established EOL-1 xenografts were treated with EPZ-5676 by continuous IV administration for 21 days. This led to robust dose-dependent tumor growth inhibition ranging from 80% in the highest dose group (70 mg/kg/day), to 29% in the lowest dose group (17.5 mg/kg/day) (Figure 3C). All doses were well tolerated and EPZ-5676 plasma levels averaged 1.31, 0.68, and 0.42 μM in the 70, 35 and 17.5 mg/kg/day groups, respectively. In a parallel experiment, H3K79 methylation was assessed in EOL-1 xenograft tissue and bone marrow tissue harvested from rats after seven days of treatment and revealed reduced H3K79me2 marks in all dose groups relative to vehicle-treated rats (Figure 3D). In addition, we found downregulation of HOXA9 mRNA levels in the highest dose group of 70 mg/kg/day after seven days of treatment (Online Supplementary Figure S1). At this time point, when cell proliferation was only mildly affected in vivo, we did not observe HOXA9 downregulation within the lower dose groups. The anti-proliferative effect in these dose groups might, therefore, be mediated by expression changes of other HOXA-cluster and MLL target genes, such as CDK6. Taken together, the results presented here demonstrate that pharmacological inhibition of DOT1L impairs the proliferation of leukemia cells bearing an MLL-PTD. Moreover, we show that inhibiting DOT1L in these leukemias abrogates global H3K79me2 in vitro and in vivo, and causes changes in gene expression that include downregulation of HOXA-cluster and other MLL target genes. In addition, MLL-PTD positive leukemia cells undergo differentiation, apoptosis, and ultimately cell death upon small molecule inhibition of DOT1L. Our data, therefore, suggest an important role of DOT1L in MLL-PTD positive leukemias.
The initial rationale for therapeutic targeting of DOT1L came from studies on MLL-rearranged leukemias.9 In these leukemias, several fusion partners such as AF9, ENL, and AF10, which normally interact with DOT1L, lead to dysregulation of DOT1L and elevated H3K79me2 at MLL-fusion target genes.9,15 This ultimately results in the activation of MLL-fusion-driven transcriptional programs, and thus, leukemogenesis.8,9,13 In MLL-PTD-positive leukemia, there is no evidence that the mutant MLL protein interacts directly with DOT1L. However, we observe a response pattern to the DOT1L inhibitor that is, in many ways, similar to what has been described for MLL-rearranged leukemias, including downregulation of HOXA-cluster and other MLL target genes followed by cell differentiation, apoptosis and cell death. Our data, therefore, support the hypothesis of an alternative mechanism by which DOT1L is involved in the development of MLL-PTD-positive leukemia. Future studies are required to elucidate the exact mechanism by which DOT1L might interact with the MLL-PTD protein and how it specifically contributes to MLL target gene activation. From a clinical perspective, the findings presented here have profound implications. MLL-PTD is the most common type of MLL alteration in adult AML.1 Although the status of MLL-PTD as an independent prognostic marker has recently been challenged in multivariate analyses, long-term survival rates are unsatisfactory, ranging between 15% in elderly to 50% in young adult patients.2,3 Targeting DOT1L is a highly specific and innovative approach to therapeutically address the epigenetic abnormalities found in MLL-rearranged leukemias. Our data provide a rationale to extend this approach to the patient population carrying a MLL-PTD. Due to the compound’s modest toxicity profile, estimated from pre-clinical data,13,14 this drug might be of particular value for the increasing population of elderly AML and MDS patients with co-morbid conditions that are not eligible for intensive chemotherapy. Further studies will shed light on the exact mechanism by which DOT1L ultimately contributes to MLL-PTD leukemogenesis.
Footnotes
Funding: supported by grants (to S.A.A.) from the National Institute of Health (CA66996 and CA140575), and the Leukemia and Lymphoma Society. M.W.M.K. was supported by the German Research Foundation (DFG; KU 2688/1-1).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Döhner K, Döhner H. Molecular characterization of acute myeloid leukemia. Haematologica. 2008;93(7):976–982. [DOI] [PubMed] [Google Scholar]
- 2.Whitman SP, Caligiuri MA, Maharry K, Radmacher MD, Kohlschmidt J, Becker H, et al. The MLL partial tandem duplication in adults aged 60 years and older with de novo cytogenetically normal acute myeloid leukemia. Leukemia. 2012;26(7):1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitman SP, Ruppert AS, Marcucci G, Mrozek K, Paschka P, Langer C, et al. Long-term disease-free survivors with cytogenetically normal acute myeloid leukemia and MLL partial tandem duplication: a Cancer and Leukemia Group B study. Blood. 2007;109(12):5164–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica. 2007;92(6):744–752. [DOI] [PubMed] [Google Scholar]
- 5.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Döhner K, Tobis K, Ulrich R, Frohling S, Benner A, Schlenk RF, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol. 2002;20(15):3254–3261. [DOI] [PubMed] [Google Scholar]
- 7.Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109(2):431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neff T, Armstrong SA. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood. 2013;121(24):4847–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121(13):2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia. 2013;27(4):813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122(6):1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernt KM, Armstrong SA. A role for DOT1L in MLL-rearranged leukemias. Epigenomics. 2011;3(6):667–670. [DOI] [PubMed] [Google Scholar]