

Abstract
Preventing untoward immune responses against a specific antigen is a major challenge in different clinical settings such as gene therapy, transplantation, or autoimmunity. Following intramuscular delivery of recombinant adeno-associated virus (rAAV)-derived vectors, transgene rejection can be a roadblock to successful clinical translation. Specific immunomodulation strategies potentially leading to sustained transgene expression while minimizing pharmacological immunosuppression are desirable. Tolerogenic dendritic cells (TolDC) are potential candidates but have not yet been evaluated in the context of gene therapy, to our knowledge. Following intramuscular delivery of rAAV-derived vectors expressing an immunogenic protein in the nonhuman primate model, we assessed the immunomodulating potential of autologous bone marrow-derived TolDC generated in the presence of IL10 and pulsed with the transgene product. TolDC administered either intradermally or intravenously were safe and well tolerated. While the intravenous route showed a modest ability to modulate host immunity against the transgene product, intradermally delivery resulted in a robust vaccination of the macaques when associated to intramuscular rAAV-derived vectors-based gene transfer. These findings demonstrate the critical role of TolDC mode of injection in modulating host immunity. This study also provides the first evidence of the potential of TolDC-based immunomodulation in gene therapy.
Introduction
Recombinant adeno-associated virus-derived vectors (rAAV) have emerged in the last decade as a promising vector platform for gene therapy. Indeed, one single in vivo administration of rAAV can result in efficient and stable transgene expression.1–5 However, immune responses against the transgene product and/or the capsid emerged as a potential roadblock before treating patients.6 Targeting therapeutic genes to the skeletal muscle is likely to be problematic using the intramuscular (IM) route, despite being exploited currently in many clinical conditions, including orphan muscular genetic diseases or affections necessitating the secretion of therapeutic factors. Indeed, after IM administration of rAAV, immune rejection of heterologous as well as autologous transgene-products has often been observed in large species,3,7–11 making clinical translation difficult. Even if the use of immunosuppressive drugs have shown some success to promote long-term transgene expression after in vivo rAAV delivery in animal models,9,10,12 such unspecific treatment is not desirable because it is potentially associated with important toxicity and increased risk of infections and cancer, in particular when gene therapy protocols are addressed to infants affected with inherited diseases. Aside from strategies targeting the vector construct itself, cell-based immunomodulation aiming to induce antigen-specific immune tolerance, is an attractive potential alternative. Dendritic cells (DCs), with their inherent ability to direct antigen-specific presentation towards immunity or tolerance pathways emerged recently as promising cell candidates. In the last decade, a considerable number of reports indicated that the bone-marrow (BM) or monocyte-derived DC generated in vitro in the presence of suppressive cytokines or inhibitory pharmacological agents, such as IL-10,13–15 rapamycin,16,17 vitamin D3 alone or in combination with dexamethasone18,19 have in vitro tolerogenic properties. In vivo, we and others have shown that the administration of such tolerogenic DCs (TolDC) in rodents was able to regulate autoreactive or alloreactive T-cell responses and promote or restore antigen-specific tolerance in autoimmune diseases20–22 and transplantation.23–26 A pioneer clinical study has reported that antigen-loaded autologous TolDC injected intradermally (ID) in healthy volunteers are well tolerated. Moreover, this study provided a first proof of principle in humans as it demonstrated in vivo specific modulation of effector T-cell responses.27,28 More recently, the first phase 1 clinical trial using TolDC in type 1 diabetes also reported that ID injection of TolDC is safe and well tolerated.29 Since then, two TolDC-based clinical trials in rheumathoid arthritis have begun21 and one will soon be performed by us in kidney transplant patients.26 To our knowledge, TolDC have not yet been evaluated in the context of transgene rejection following rAAV-based gene transfer. Furthermore, there is still an evident lack of translational studies demonstrating the efficacy of TolDC-based immunotherapy in large animal models. Indeed, only one recent study reported the generation of nonhuman primate (NHP) TolDC and their safety after their intravenous (IV) administration.15 Recently, the same group demonstrated the ability of donor TolDC in association with CTLA4Ig+/− rapamycin to prolong MHC mismatched renal allograft survival in rhesus monkeys.30
We have previously described the generation of cynomolgus macaque immature BM-derived DC (iBMDC) with in vitro tolerogenic properties31 and reported that these cells are able to inhibit T lymphocytes via heme oxygenase-1 immunosuppressive molecule.32 Here, we propose an improved generation protocol in the presence of IL10 (iBMDC10) that has been shown to increase DC tolerogenic properties and resistance to maturation.14 We further evaluated the potential of iBMDC10 in the NHP model after IM injection of a rAAV that expresses the cynomolgus erythropoietin (cmEpo) and the doxycyclin (Dox)-sensitive transactivator rtTA. In our previous studies, more than 85% of the macaques injected IM with rAAV from various serotypes carrying this construct, developed antibodies and a T-cell response against the TetON transactivator rtTA, leading to the loss of regulated cmEpo expression (Refs. 3,11,33 and our unpublished data). We investigated here whether autologous iBMDC10 pulsed with rtTA protein and injected either via ID or IV routes were able to promote long-term gene transfer.
Results
In vitro generation of macaque immature BM-derived DC in the presence of IL10 leads to higher tolerogenic properties and resistance to maturation after rtTA pulse
A recent study has described the generation of human monocyte-derived tolerogenic DC in the presence of IL10 (called DC10).14 In order to increase the tolerogenic capability of macaque iBMDC that we described earlier,31 we generated them in the presence of recombinant IL-10, in addition to low dose of granulocyte-macrophage colony-stimulating factor (GM-CSF). The phenotype of DC populations generated in the absence of IL10 (iBMDC) and in its presence (iBMDC10) was assessed by flow cytometry (Figure 1a). Neither of these DC populations expressed CD34, T, B, or natural killer (NK) cell markers (Figure 1a and data not shown). iBMDC and iBMDC10 defined homogeneous populations that express CD11c, CD11b, and HLA-DR DC markers. Interestingly and in accordance with the phenotype of human DC10,14 macaque iBMDC10 displayed higher expression of CD25 than their counterparts generated with GM-CSF alone, and the proportion of CD14/CD16 positive cells in iBMDC10 appeared also higher. Furthermore, the marker of costimulation CD86 was much less expressed in iBMDC10 condition than iBMDC (9 versus 30% HLA-DR/CD86 double positive cells, respectively). Thus, iBMDC and iBMDC10 populations displayed different phenotypes with less costimulation molecules in the presence of IL10.
Figure 1.

Phenotypical and functional characterization of macaque iBMDC versus iBMDC10 cells. Bone marrow cells were used to generate iBMDC in the presence of GM-CSF and iBMDC10 in the presence of GM-CSF and IL-10. Results are representative of DC generated from two different BM macaque donors. (a) iBMDC and iBMDC10 phenotyping using flow cytometry. Cells were stained with anti-CD20, anti-CD3, anti-CD11b, anti-CD11c, anti-HLA-DR, anti-CD86, anti-CD34, anti-CD25, anti-CD14, and anti-CD16 antibodies. The numbers within FACS dot plots quadrants indicate the percentages of DC cells expressing relevant markers (as representative of three different experiments). (b) Assessment of suppressive activity of irradiated iBMDC versus iBMDC10 cells added to a mixed lymphocyte reaction (MLR) using 3H-thymidine uptake assay where proliferation levels are expressed in cpm. The ratio 1 DC for 16 MLR is shown as a representative result of two independent experiments. (c) Assessment of stimulative ability of iBMDC versus iBMDC10 on allogeneic PBMC. Proliferation was measured by 3H-thymidine uptake (cpm). A range of DC: PBMC ratio from 1:6 to 1:126 was realized. (d) Expression of HLA-DR and CD86 costimulatory molecule on the surface of iBMDC and iBMDC10 cells using flow cytometry. Marker expression was assessed after overnight incubation in medium alone or with rtTA protein, in the absence or in the presence of IL10. The percentages of double positive HLA-DR/CD86 cells are indicated in the corresponding dot plot quadrants. (e) Detection of IL10 transcripts (expressed in relative quantity (RQ)) in iBMDC versus iBMDC10 using a quantitative reverse-transcription PCR assay. IL10 transcripts were detected in DC cultured overnight in medium alone or with rtTA protein. cpm, counts per minute; iBMDC, immature bone-marrow-derived dendritic cells; PBMC, peripheral blood mononuclear cells.
We next compared the tolerogenic properties of iBMDC and iBMDC10 cell populations in vitro. We already demonstrated the ability of iBMDC to inhibit a mixed lymphocyte reaction.32 In the same setting of a lymphocyte inhibition assay, iBMDC10 were also able to suppress T-cell proliferation to the same degree than iBMDC as shown in Figure 1b. Interestingly, when both DC types were evaluated for their capacity to stimulate proliferation of allogeneic T cells, iBMDC10 induced significant less T-cell proliferation than iBMDC (Figure 1c), confirming that iBMDC generated in the presence of IL10 were poor stimulators of T lymphocytes, and better candidate for in vivo evaluation.
To specifically modulate the immune response against the rtTA transgene product following rAAV-based gene transfer, iBMDC and iBMDC10 were pulsed with rtTA protein. As antigen loading can induce in vitro maturation of immature DC, we assessed the expression of CD86 marker on iBMDC and iBMDC10 pulsed with rtTA (Figure 1d). iBMDC showed an increase of CD86 expression after rtTA pulse (43 versus 24% of HLA-DR/CD86 double positive cells in unpulsed cells). In contrast, iBMDC10 displayed a very low increase of CD86 expression despite antigen pulse (21 versus 17% HLA-DR/CD86 double positive cells for unpulsed cells), indicating that iBMDC10 were more resistant to maturation. When IL10 was added to the culture during rtTA antigen pulse, even lower expression of CD86 was observed in both iBMDC and iBMDC10 (15 versus only 4%, respectively, in HLA-DR/CD86 double positive cells). As the tolerogenic effect of DC10 was previously shown to be IL10-dependent,14 we next assessed by quantitative RT-PCR whether our DCs were able to produce IL10 per se (Figure 1e). A higher expression of IL-10 transcript was detected in iBMDC10 as compared to iBMDC, be it in the absence or the presence of rtTA protein. After antigen pulse, the level of IL-10 expression did not increase in iBMDC. In the opposite, a twofold to threefold increase of IL-10 transcript levels was observed in iBMDC10 after rtTA pulse. These results further confirm that iBMDC10 are better candidates for in vivo evaluation as they displayed higher tolerogenic properties than iBMDC. Finally and before moving on toward in vivo assessment, the efficiency of iBMDC10 pulse was demonstrated by the intracellular presence of rtTA as detected using anti-rtTA immunohistochemistry as shown in Supplementary Figure S1. We then moved forward and evaluated their in vivo potential in a macaque model of rAAV-based IM gene transfer.
cmEpo gene expression after rAAV and iBMDC10 administration
In order to evaluate the tolerogenic potential of autologous rtTA-pulsed iBMDC10, we used an immunogenic rAAV-based gene transfer model that we have previously reported.3,11,33 Briefly, it consists of IM delivery of rAAV1 vector that expresses the cmEpo under the control of the Dox-sensitive transactivator rtTA. In a previous study, when this vector was injected via the IM route at a dose of 1 × 1011 viral genomes/kg, three out of four macaques developed antibodies as well as T-cell responses against the tetON transactivator rtTA, leading to the loss of cmEpo regulated expression.11 Here, we investigated whether the injection of autologous iBMDC10 pulsed with rtTA protein in combination with rAAV1-based gene transfer, was able to modulate the host immune response against the transactivator rtTA and to promote persisting cmEpo expression. The experimental strategy is schematically described in Figure 2a. We compared ID (group iBMDC10-ID, Mac 4 and Mac 5) and IV (iBMDC10-IV, Mac 6 to Mac 10) modes of DC injection, as both have been previously described in TolDC-based immunotherapy protocols. A group of three animals (Mac 1 to Mac 3) injected with the rAAV1 vector alone was monitored in parallel as the control group. All these macaque groups are described in Table 1. Because it was shown that the vector dose delivered per IM injection site as well as the local level of transgene expression are determinant factors for antitransgene immunity,34,35 we injected comparable doses comprised between 0.8 × 1011 and 1 × 1011 vg in each site for all the animals, except Mac 5 who received 2 × 1011 vg per site (Table 1). After repetitive Dox inductions, the expression of the cmEpo reporter gene was monitored by enzyme-linked immunosorbent assay in animal serum samples until more than 1 year postinjection and is shown for the three groups of animals (Control, iBMDC10-ID, and iBMDC10-IV) in Figure 2b and Supplementary Figure S2. The iBMDC10-IV group showed the highest proportion of animals with persisting cmEpo gene transfer. Indeed, two out of five animals never showed a loss of cmEpo regulation and were able to respond to Dox induction during at least 1-year postgene transfer. In contrast, all animals from iBMDC10-ID and control groups showed a loss of cmEpo expression within maximum 138 days postinjection (Figure 2b and Supplementary Figure S2 for individual Epo graphs).
Figure 2.

In vivo evaluation of iBMDC10 in an AAV-based gene transfer model. (a) The experimental strategy of macaque in vivo study model is schematically represented. rAAV1 vector expressing the reporter Epo gene under the control of the tetracyclin-dependant Tet-ON rtTA transactivator was injected via multiple intramuscular (IM) sites in the anterior tibialis muscle. The total vector dose was 1 × 1011 viral genome/kg. Autologous iBMDC10 cells pulsed with rtTA protein in the presence of IL10 were injected 1 day prior to rAAV administration and at 2 months postgene transfer either via intravenous (IV) or intradermal (ID) routes at a dose of ≈ 1 × 106 cells/kg. iBMDC10-ID group received an additional cell injection at 3 months postgene transfer. Follow-up of macaques was realized during more than 18 months postgene transfer and consisted in measuring Epo transgene expression in macaque sera by ELISA upon regular doxycyclin administration. Anti-rtTA immune responses and PBMC phenotype were also monitored. (b) Epo gene expression persistence in control, iBMDC10-ID, and iBMDC10-IV macaque groups was monitored by ELISA in macaque sera and is shown until 1 year postgene transfer in a survival graph type. Control (n = 3), iBMDC10-ID (n = 2), and iBMDC10-IV (n = 5) macaque groups are compared. Control and iBMDC10-IV groups are not statistically different: P value = 0.66. Statistics were not performed for iBMDC10-ID group (only two individuals). Epo levels for each macaque are shown in Supplementary Figure S2. (c) Quantification of vector transgene transcripts by quantitative RT-PCR in the tibialis muscle at one site of vector injection for macaques from control, iBMDC10-ID, and iBMDC10-IV groups. Transgene transcript levels are expressed in relative quantity (RQ) after normalization with endogenous HPRT gene. The threshold of PCR sensitivity is represented in grey in the graph and determined at 8 × 10-3. Control and iBMDC10-IV groups are not statistically different: P value = 0.57. Statistics were not performed for iBMDC10-ID group (only two individuals). ELISA, enzyme-linked immunosorbent assay; iBMDC, immature bone-marrow-derived dendritic cells; PBMC, peripheral blood mononuclear cells; rAAV, recombinant adeno-associated virus-derived vectors.
Table 1. Summary of macaque groups and treatments.
| Group | Primate | rAAV1 | Vector injection number and vector dose per site | iBMDC10 |
|---|---|---|---|---|
| Control (n = 3) | Mac 1 | Intramuscular (1 × 1011vg/kg) | 3 (0.9 × 1011 vg) | No injection |
| Mac 2 | 3 (1 × 1011 vg) | No injection | ||
| Mac 3 | 4 (0.9 × 1011 vg) | No injection | ||
| iBMDC10-ID (n = 2) | Mac 4 | Intramuscular (1 × 1011vg/kg) | 6 (0.8 × 1011 vg) | Intradermal (≈1 × 106/kg) |
| Mac 5 | 4 (2 × 1011 vg) | |||
| iBMDC10-IV (n = 5) | Mac 6 | Intramuscular (1 × 1011 vg/kg) | 3 (0.9 × 1011 vg) | Intravenous (≈1 × 106/kg) |
| Mac 7 | 3 (1 × 1011 vg) | |||
| Mac 8 | 4 (1 × 1011 vg) | |||
| Mac 9 | 4 (1 × 1011 vg) | |||
| Mac 10 | 4 (1 × 1011 vg) |
iBMDC, immature bone-marrow-derived dendritic cells.
In order to further assess whether the loss of inducible cmEpo gene expression is correlated to an immune elimination of transduced cells, we quantified gene transcripts in muscle samples obtained at the sites of IM vector injection, at more than 18 months postinjection. RNA transgene levels were quantified in noninjected contralateral tibialis muscle for each macaque and were all negative (data not shown). In contrast, in the site of IM injection, iBMDC10-IV and iBMDC10-ID groups showed opposite transgene transcripts levels (Figure 2c). Indeed, Mac 4 and Mac 5 in iBMDC10-ID group displayed very low transgene transcripts (with Mac 4 nearly at the limit of qPCR sensitivity determined at 8 × 10−3) in accordance with the loss of cmEpo regulation within the first 5 months post-AAVr injection. In contrast, all animals in the iBMDC10-IV group showed significant higher transgene transcript levels at long term time points. Strikingly, we were also able to detect transgene transcripts in animals from control group despite the loss of Epo expression within the first 5 months post-AAVr injection and this finding is currently under investigation. We next investigated whether irreversible cmEpo loss in the iBMDC10-ID group was correlated to humoral and cellular immune responses against the transactivator rtTA, as reported in our previous studies.3,11
Monitoring of humoral and cellular immune responses against the rtTA transgene product
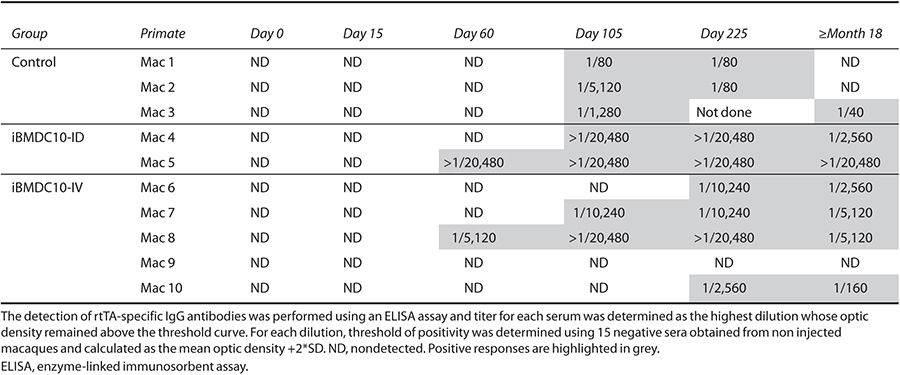
The presence of circulating anti-rtTA IgG antibodies was monitored by enzyme-linked immunosorbent assay at days 0, 15, 60, 105, 225, and more than 18 months postinjection. Kinetics of IgG appearance as well as antibody titers are presented in Table 2. No significant difference was observed between the three groups. Indeed, the majority of animals exhibited anti-rtTA IgG antibodies, except Mac 9 from DC-IV group. IgG were detectable from day 60 to 105 postinjection and titers appeared variable between animals regardless of their experimental group, and were comprised between 1/80 and >1/20,480. Nevertheless, both Mac 4 and Mac 5 from iBMDC10-ID groups showed high titers (>1/20,480). Furthermore, no decrease of antibody levels until more than 18 months postinjection was observed for Mac 5 (Table 2), in contrast to all other animals that showed a decrease in IgG levels over time.
Table 2. Detection of anti-rtTA IgG antibodies in macaque sera obtained before treatment (day 0) and at days 15, 60, 105, 225 and at ≥ than 18 months postgene transfer.
We next assessed anti-rtTA T-cell responses using an IFNγ ELISpot assay. Macaque peripheral blood mononuclear cells (PBMCs) were obtained at more than 18 months postinjection, and restimulated with an overlapping peptide library covering the rtTA protein and divided in five peptide pools. Results are summarized in Table 3 and Supplementary Table S1, and representative graphs for two animals from each group are represented in Figure 3. In the control group, two out of three macaques (Mac 2 and Mac 3) did not show detectable IFNγ secretion at 18 months but only a transitory response around 3 months postinjection (Supplementary Figure S3), while Mac 1 was still positive at 18 months postinjection (Figure 3, Mac 1 and 2, top left panel and Table 3). Both Mac 4 and Mac 5 from iBMDC10-ID group showed a persisting anti-rtTA T-cell response at 18 months postinjection as demonstrated by a significant IFNγ secretion in response to peptide pools 1 and 2 for Mac 4 (Figure 3 central panel and Table 3). Animals from iBMDC10-IV group did not show detectable or significant responses (Figure 3, Mac 6 and Mac 8, bottom panel and Table 3). In the iBMDC10-ID group, PBMC from Mac 4 showed the highest IFNγ secretion in response to rtTA peptide pool 1 (1051.67 spot-forming colonies/106 cells, 8.4 time fold as compared to the threshold of positivity). In this macaque, and using a CD4 versus CD8 magnetic depleting strategy, we were able to show that this IFNγ response was predominantly mediated by CD8 T lymphocytes (Supplementary Figure S4).
Table 3. Summary of anti-rtTA IFNγ T cell responses using an ELISpot assay.
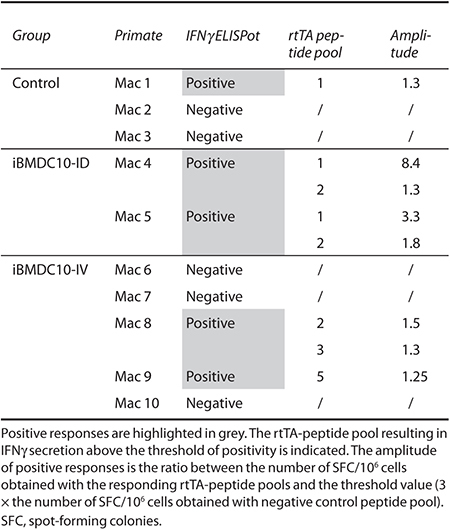
Figure 3.

Anti-rtTA T cell responses using an IFNγ ELISpot assay. Representative results of two macaques from control, iBMDC10-ID, and iBMDC10-IV groups are shown. Macaque PBMC obtained at ≥18 months postgene transfer were stimulated using an overlapping 15 per 10 amino acid peptide library covering the rtTA protein and divided in five pools (p1–p5). Negative controls consisted in PBMC cultured with either medium alone (nonactivated cells, NA) or an unrelated peptide pool (C−). Positive control (C+) consisted in PMA/ionomycin activation. IFNγ secretion was measured as spot forming cells (SFC) per 106 PBMC. Threshold of positivity (dotted line) consists in SFC > 50 spots and three times the number of SFC obtained with negative peptide pool control (C−). For positive responses, statistical analysis was performed using a DFR test, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05. Results for all the macaques are summarized in Table 3 and Supplementary Table S1. PBMC, peripheral blood mononuclear cells.
It was previously shown that human and mouse DC10 were able to modulate the immune system via the induction of IL-10-producing Tr1 cells.14 Using an ELISpot assay, we further investigated whether IL10 is secreted by rtTA-stimulated PBMC in iBMDC10-IV group where effector IFNγ responses were negative in two out of five animals or only weak/transitory in the rest of the cohort. In all animals, we were not able to detect IL10 secretion similarly to iBMDC10-ID group and control animals (Supplementary Figure S5, two representative macaques are shown for each group).
PBMC phenotyping of macaque groups by flow cytometry
In order to investigate whether iBMDC10 administration has an impact on blood immune cell populations, we next analyzed the frequency of relevant circulating immune cells using flow cytometry. Macaque PBMC were obtained at more than 18 months postinjection, at a time when T cells from iBMDC10-ID macaques were still responding to rtTA, in contrast to iBMDC10-IV group. PBMC from four untreated macaques constituted the mock control group. Gating strategy of CD3+ T cells, CD20+ B cells, CD56+ NK cells as well as T-cell subpopulations, is represented in Figure 4a. The percentage means of each cell population among our primate groups have been compared. In the first Tol-DC-based phase 1 clinical trial for diabetes type 1, patient who received Tol-DC ID exhibited an increase in peripheral B cells (B220+ CD11c−) as early as 1 week after DC delivery.29 The preliminary data of the trial suggest a potential involvement of these cells in the modulation of the immune system as demonstrated recently in NOD mouse model by the same group.36 In our hands, lower B cell percentages were observed in the iBMDC10-ID group as compared to other animal cohorts (5.65 versus 15.23, 17.35, and 8.65% in iBMDC10-IV, control and mock groups, respectively), while in contrast NK percentage appeared slightly augmented in these macaques (Figure 4b). While it is difficult to draw a conclusion because the small size of our cohorts, it is important to note that Mac 4 and Mac 5 from the iBMDC10-ID group showed homogeneous percentages. Regarding T cells, and while total CD3+ cells did not show a significant difference between the animal groups (Figure 4c, left panel), the ratio between CD3+/CD8+ and CD3+/CD4+ T cells appeared augmented in iBMDC10-ID group, suggesting a higher number of circulating CD8 T cells in these animals (Figure 4c, central panel). Even if this observation does not reflect antitransgene T-cell specific responses, it was interestingly correlated to the detection of a predominant CD8-mediated anti-rtTA T-cell response in iBMDC10-ID macaques in the ELISpot assay as reported above (Figure 3 and Supplementary Figure S4). Finally, no statistical difference was observed for regulatory T-cell percentages in our macaque groups (Figure 4c, right panel).
Figure 4.

PBMC macaque phenotyping using flow-cytometry. PBMCs were obtained at ≥18 months postgene transfer from macaques of control, iBMDC10-ID, and iBMDC10-IV groups. PBMCs from four untreated macaques of the same age range than our experimental individuals constituted the mock group. (a) Gating strategy. After lymphocyte cell gating using FSC and SSC parameters, doublets were excluded using FSC-H and FSC-A parameters and live cells were gated using a DAPI staining. CD3+ T cells, CD20+ B cells, CD56+ NK cells, CD8+, and CD4+ T cells among CD3+ populations were gated. Regulatory T cells (Treg) consisted in CD3+ CD4+ CD25Hi Fox P3+ cells. (b) Mean percentages of CD20+ B (left graph), CD56+ NK (central graph), and CD3+ T (right graph) cells among total PBMC live cells for mock, control, iBMDC10-ID, and iBMDC10-IV macaque groups. (c) Mean percentages of CD25Hi Fox P3+ regulatory T cells (Treg) among CD3+/CD4+ T cell population (left graph) and CD8+/CD4+ ratio among CD3+ T cell population (right graph) are shown for each macaque group. For b and c panels, mock, control and iBMDC10-IV groups are not statistically different whereas statistics were not performed for iBMDC10-ID group (n = 2). DAPI, 4’,6-diamidino-2-phenylindole; FSC, forward scatter; iBMDC, immature bone-marrow-derived dendritic cells; PBMC, peripheral blood mononuclear cells; SSC, side scatter.
Discussion
At a time where IM rAAV-based gene delivery to the skeletal muscle is already being assessed in phase 1 clinical trials for the treatment of a wide variety of genetic diseases affecting the skeletal muscle,37,38 or necessitating the secretion of therapeutic factors,39–41 host immunity against the transgene product emerged as a major limit in translational studies.3,7–11 Controlling these immune responses through cell-mediated antigen specific immunomodulation is an attractive alternative as compared to conventional immunosuppressive regimen that expose the patient to considerable side effects. Here, we evaluated whether autologous iBMDC10 are able to modulate antitransgene immunity in a NHP model of IM rAAV1-based gene transfer. We already described a protocol allowing the generation of macaque iBMDC with in vitro tolerogenic properties using only low doses of GM-CSF.31 IL10 cytokine was shown to increase the tolerogenic properties of DC when added to the culture.42 In this study, the generation of macaque iBMDC in the presence of IL10 in addition to GM-CSF also allowed to improve their ability to resist to maturation in particular upon antigen pulsing. An additional important feature of our iBMDC10 is their ability to secrete IL10 per se as described earlier for human and rat TolDC.43 Another study has described an IL10 secreting human DC population naturally occurring in vivo and inducible in vitro, that are able to induce suppressive Tr1 T cells via an IL10-dependent pathway.14
So far, very few studies have evaluated the impact of the route of TolDCs administration versus their tolerogenic potential in vivo. Migration of DC to draining lymph nodes was shown (mainly in tumor positive vaccination strategies) to be a limiting step. It is assumed that only 1–4% of injected DC are able to reach peripheral lymphoid organs upon their in vivo delivery44,45 and that cell migration to lymph nodes is more efficient when DC are injected through ID or intranodal routes, as compared to IV delivery. However, it is assumed that ID route is rather immunogenic and is more generally used for vaccination protocols, in contrast to IV route.46,47 In addition, in vivo migration of immature TolDC generated in the presence of IL10 could be decreased because of IL10-mediated CCR7 chemokine receptor downregulation,48 which is essential for cell homing to lymph nodes. For all these reasons and because local administration of TolDC was already shown to be safe in healthy humans27,28 and more recently in diabetic patients,29 we hypothesized that ID injection of iBMDC10 in the area of rAAV draining lymph nodes could be in favor of rtTA immunomodulation. As reported previously in humans,28,29 ID delivery of iBMDC10 was well tolerated by the animals. Nevertheless, the two injected-macaques showed a rapid and irreversible loss of gene transfer efficiency correlated to a robust and sustained IFNγ secretion upon T-cell stimulation with the transgene product. Surprisingly, viral genome copies at the site of injection were detectable at comparable levels than other groups at late time points (Supplementary Figure S6, 0.01 and 0.27 vg/dg for Mac 4 and Mac 5, respectively). Thus, it seems that the loss of transgene expression in iBMDC10-ID group is not due (at least partially) to a conventional CD8-mediated cytotoxic elimination of transduced cells as expected, but rather a result of other mechanisms which need further investigations in our model. One explanation for the results obtained after ID administration of rtTA-loaded iBMDC10 could be an inflammation-mediated maturation of the cells once injected in the area of rAAV draining. Indeed, IM injection of the vector could induce local inflammatory “danger signals” due to either the injection itself, or a component in the viral preparation. Subsequent rAAV-mediated expression of rtTA could result in the reactivation of the immune system already primed via the delivered DC. Moreover, rAAV serotype 1 used in the study could favor these pathways as compared to other AAV serotypes because of high vector concentration at the site of injection, which increases the risk of local inflammation and levels of rtTA expression.34,49 Finally, rAAV were shown to activate in vivo toll like receptor 9-Myd88 pathway and subsequent IFN type 1 secretion50–52 which may increase the likelihood of maturation of the injected iBMDC10. All this events could explain the difference we have with the first clinical studies where subcutaneous injection of antigen-loaded DC did result in effective T-cell immune modulation through CD8+ regulatory T cells.27,28 One observation that supports our hypothesis was the fact that autologous iBMDC10 loaded with rtTA and injected ID were not able to vaccinate per se three pilot macaques, in the absence of rAAV administration (Supplementary Figure S7). Only a weak and transitory antibody response was observed in one out of three macaques and no T-cell proliferation in response to rtTA stimulation was observed for the three pilot macaques as compared to an immunized macaque (Supplementary Figure S7).
As an alternative to ID delivery of autologous iBMDC10 pulsed with rtTA, we evaluated IV administration that has shown great potential in rodent models of allograft and autoimmune diseases.23,25 In our setting, while IV administration of iBMDC10 allowed long term gene transfer with no anti-rtTA transgene T-cell response in two out of five animals (Mac 6 and Mac 10), we were not able to formally demonstrate the efficiency of the strategy. Indeed, the other animals exhibited either transitory or weak anti-rtTA T-cell effector responses and IgG antibodies were detected in iBMDC10-IV group similarly to the other conditions. Furthermore, all the animals of the group (including Mac 6 and Mac 10) presented muscle infiltrates at the site of injection (>18 months pi) similarly to the other groups (Supplementary Figure S8), with no predominant necrosis observed even for iBMDC10-ID animals. At this stage, we cannot exclude that the observed muscle infiltrations were due to another component than the in situ expression of the immunogenic rtTA transgene. A potential involvement of the viral capsid itself is potentially possible as recently described in α anti-trypsin-deficient patients 1 year after AAV1 IM injection.53
TolDC were shown to modulate the immune system through the induction and expansion of different subsets of T-suppressive cells based on the method of TolDC generation. Neither the induction of IL10-secreting Tr1 cells was demonstrated after DC IV delivery, nor an increase in peripheral regulatory FoxP3+ T cells.29 Even if iBMDC10-ID and iBMDC10-IV groups received different doses of DC (three injections in ID group versus two in IV group) we don’t think that this could explain our results. Indeed, in diabetic patients, TolDC were administered ID once every 2 weeks for a total of four administrations without inducing such effect.29 In our study, strong anti-rTA immunity was observed as early as the two first ID iBMDC10 administrations suggesting that the active vaccination in our setting is rather due to a rapid immunization as far as the inflammatory signals were induced by IM vector injection. Moreover, the number of IV TolDC injections was shown to not be determinant in a rat heart allograft model with repeated cell injections not able to improve graft survival.29 Our strategy did not result in an efficient modulation of rtTA transgene immunity but this is likely not due to an insufficient DC cell dose because it was within the same range or even higher than the doses described in pioneer clinical studies where a total of 2 × 106 or four injections of 107 were administered locally to healthy28 or diabetic individuals,29 respectively.
Because anticapsid immunity that is induced following in vivo rAAV injection is also an important feature in rAAV-based protocols, we checked whether iBMDC10 treatment had an effect on the onset or amplitude of anti-AAV1 humoral response in our macaques that were all seronegative prior to the protocol. All the animals displayed anti-AAV1 IgG antibodies (data not shown) and neutralizing factors (Supplementary Figure S9) regardless of the experimental group, with Mac 4 showing the highest titer. Thus, iBMDC10 administration did not modulate anti-AAV responses.
A potential explanation for the limited efficiency of our rtTA-pulsed iBMDC10 is a possible uptake and presentation of rtTA antigen by endogenous macaque DC, resulting in the priming of the host immune system. This uptake can even be higher after ID administration because of the AAV-induced inflammatory environment and subsequent iBMDC10 maturation. Similarly, in a murine model of allogeneic skin graft using alloantigen-bearing TolDC, tolerance was not achieved because antigen uptake and presentation by recipent DC.54 Whether TolDC pulse with the antigen is necessary for specific tolerance is not clear. Indeed, we already showed that unpulsed recipient TolDC were able to specifically induce tolerance against donor alloantigen in rodent models of allotransplantation.23,25 Thus, the injection of unloaded iBMDC10 may be an alternative to evaluate in our gene transfer model. Finally, one way to increase the efficiency of IV administration of TolDC for future clinical application could be their association to a minimal short-term immunosuppressive regimen that will not interfere with T regulatory-induced expansion and function. Indeed, the potential of such strategy has already shown convincing data in NHP and rodent models.23,25,30
In conclusion, TolDC-based immunotherapy is currently considered as an attractive approach to minimize the use of immunosuppressive drugs in various clinical settings where host immunity is problematic such autoimmune diseases and transplantation. The first TolDC-based phase 1 clinical trial has been reported recently for type 1 diabetes29 and others are ongoing or imminent in rhumathoid arthritis patients (Queensland and New Castle Universities) or for kidney transplant in our team (Nantes University). Controlling host immune responses against the transgene product after rAAV-based gene therapy emerged recently as an additional potential application of TolDC cell therapy. Our study in NHP demonstrated that ID and IV DC administrations are both safe and well tolerated. IV DC delivery could be more appropriate but still needs additional improvements because it was not able to modulate anti-transgene immunity in our rAAV-based gene transfer model. In contrast, where rAAV are considered not to be vectors of choice for positive vaccination as compared to adenoviral reagents because of a limited ability to transduce antigen presenting cells,55,56 it appears that ID injection of DC pulsed with the transgene product associated to IM rAAV-based gene transfer is able to induce persisting and efficient active vaccination. All these findings highlight the critical role that the route of TolDC administration plays in shifting the in vivo balance from immunomodulation to immunogenicity, and provide important information for the design of future TolDC-based clinical trials in general, and host immune response immunodulation strategies in rAAV-based gene therapy protocols in particular.
Materials and Methods
A large part of this work was performed under the control of our quality management system that is approved by Lloyd’s Register Quality Assurance LRQA to meet requirements of international Management System Standards ISO 9001:2008. It has been implemented to cover all activities in the laboratory including research experiments and production of research-grade viral vectors.
Animals and in vivo experiments
Nonhuman primates (Macaca fascicularis) were purchased from BioPrim, (Baziège, France). All animals were used for experimentation in accordance with our institutional and national ethical guidelines. We selected male and female primates that had no detectable neutralizing antibody against AAV serotypes 1 in the serum. The weight of all animals was comprised between 3 and 5 kg except for Mac 5 (8 kg). For blood and bone marrow samples and vector or cell injections, primates were anesthetized with IM injection of 20 µg/kg medetomidine (Domitor; Pfizer, Paris, France) associated to 8 mg/kg of ketamine (Imalgène; Rhone Merieux, Toulouse, France). IM vector administration was performed as described previously49 in the absence of any immunosuppressive regimen, under ketamine/medetomidine/morphine-induced anesthesia maintained with 1.5% isoflurane. A total vector dose of 1 × 1011 vg/kg split over three to six pretattooed injection sites along one tibialis anterior muscle was administered in a volume per site of 320–420 µl. Long term muscle samples (injected and contralateral noninjected) were obtained at animal necropsy for iBMDC10-ID and iBMDC10-IV macaques, and via surgical biopsies for the control group. During the protocol, primates were monitored by clinical observations, respiratory rate measurements, temperature measurements, and when necessary electrocardiography, oxymetry, and capnography. Analgesia was performed with 0.1 mg/kg of morphin (Morphine, Cooper, France). Euthanasia was performed with IV injection of sodium pentobarbital (Dolethal, Vétoquinol, France) after 0.1mg/kg morphin-induced analgesia.
Recombinant AAV-1 vector
The rtTA/Epo vector plasmid consist in (i) the cynomolgus macaque Epo (cmEpo) complementary DNA under the control of the Dox-inducible TetO-cytomegalovirus promoter and (ii) an expression cassette encoding the reverse transactivator protein (rtTA-M2) under the control of the muscle-specific 1-kb human desmin promoter. Recombinant AAV-1 vectors were manufactured using 293 cell transfection method, and purified by cesium chloride density gradients followed by extensive dialysis against phosphate-buffered saline.
Generation and injection of iBMDC and iBMDC10
Macaque DCs were generated from bone marrow precursors as described earlier.32 Briefly, after red blood cell lysis, macaque cells (1 × 106cells/ml) were cultured in complete medium supplemented with 1% macaque serum (collected from our animals) and recombinant human GM-CSF (100 U/ml, Novartis, Basel, Switzerland). On day 3, the supernatant was replaced by fresh medium containing GM-CSF, macaque serum and with or without recombinant human IL-10 (20 ng/ml, R&D Systems, France). On day 7, the adherent cells were harvested. In some cases, the adherent iBMDC and iBMDC10 were cultured overnight with purified recombinant rtTA protein (10 µg/ml) with or without recombinant human IL-10 (20 ng/ml, R&D Systems). Three pilot animals received autologous iBMDC10 ID in the inguinal lymph nodes area. For Mac 4 and Mac 5, autologous iBMDC10 were injected ID in the inguinal and popliteal lymph nodes areas of the rAAV-1 vector injected leg. Mac 6 to 10 received autologous iBMDC10 by IV route. Each animal received the first injection the day before the AAVr injection and the second one, 2 months later, during the first Dox induction of the cmEpo system. Mac 4 and Mac 5 received a third injection of iBMDC10 three months post-AAVr injection. Cell dose was ~1 × 106 iBMDC10/kg.
Follow-up of cmEpo expression
Dox (Ratiopharm, Ulm, Germany) was given IV (20 mg/kg the first day, then 10 mg/kg). The induction protocol started 2 months after rAAV-rtTA/Epo delivery and consisted of 3-day Dox administration, as described previously.11 Regular Dox inductions were performed to assess Epo inducible expression. Serum cmEpo levels were measured by enzyme-linked immunosorbent assay (Quantikine IVD kit; R&D Systems) during up to 1 year postinjection. The loss of Epo expression was defined as the day corresponding to the last peak of Epo secretion obtained in injected animals.
Cell phenotyping by flow cytometry
The phenotypic analysis of either iBMDC/iBMDC10 cells or PBMC obtained from animals were analyzed using mouse antibodies (Abs) against human antigens cross-reacting with the cynomolgus macaque, or species-specific antibodies when available. Matching isotype controls mouse Abs was used. Cells were stained using DAPI (4’,6-diamidino-2-phenylindole), anti-CD20 (clone L27), anti-CD3 (clone SP34-2), anti-CD8 (clone RPA-T8), anti-CD4 (clone M-T477), anti-CD11b (clone ICRF44), anti-HLA-DR (clone L243), anti-CD86 (clone 2331), anti-CD34 (clone 564), anti CD16 (clone 3G8), anti-CD14 (clone M5E3), and anti-CD56 (clone MY31; BD Biosciences, Le Pont de Claix, France); anti-CD11c (clone 3.9), anti-CD25 (clone BC96), and anti-Foxp3 (clone 236A/E7; ebioscience). Foxp3 staining was performed on permeabilized cells, according to the manufacturer’s protocol (eBioscience, Paris, France). Cells were acquired using FACSCalibur and FACSCantoII cytometers (BD Biosciences) and results were analysed using Cell Quest (BD Biosciences) and FlowJo softwares (Treestar).
MLR and lymphocyte inhibition assays
Irradiated iBMDC and iBMDC10 were cocultured with 1 × 105 allogeneic PBMC isolated from peripheral blood by Ficoll-Hypaque density gradient. Coculture was performed in triplicates using graded doses of iBMDC/iBMDC10 versus PBMC (1:8, 1:16, 1:32, 1:64, and 1:128). After 5 days, PBMC proliferation was measured by 3H-thymidine uptake during the last 9 hours, and was expressed as counts per minute measured in a liquid scintillation counter (Betaplate, Perkin Elmer, Turku, Finland).
Allogeneic MLRs were performed between PBMCs (1 × 105) from two different macaques (one of them was irradiated). Graded doses of iBMDC or iBMDC10 provided from third party macaques were added to these MLRs and cultured for 3 days. Responder T-cell proliferation was measured by 3H-thymidine uptake as described above.
Quantitative RT-PCR assays
Total RNA from cells or muscle samples obtained from animals was isolated using TRIzol (Life Technologies, Saint Aubin, France) and purified by chloroforme extraction. Total RNA was then treated with RNAse-free TURBO DNAse from TURBO DNA-free kit (Life Technologies) according to manufactured instructions to eliminate DNA contamination. Reverse transcription was performed on RNA using an oligo dT primer (Life Technologies) and an M-MLV Reverse Transcriptase (Life Technologies). For IL10 transcripts, quantitative PCRs were performed using the GenAmp 7700 sequence detection system (Life Technologies) with SYBR Green core reagents (Life Technologies). Primer sequences were designed to amplify IL-10 cDNA (5′- CTGCCTAACATGCTTCGAGATC-3′ and 5′ AACCCTTAAAGTCCTCCAGCAA-3′) and hypoxanthine-guanine phosphoribosyl transferase (HPRT) cDNA (5′-CGTGATTAGCGATGATGAACC-3′ and 5′-ATCCAGCAGGTCAGCAAAGA-3′). Thermal cycling conditions comprised a 2-minute incubation at 50 °C, a 10-minute denaturation at 95 °C, followed by 40 cycles of denaturation (95 °C for 15 seconds), annealing (60°C for 1 minute), extension (Tm for 15 seconds), and 10 minutes at 72 °C. Tm was 77 °C for HPRT and 80°C for IL-10. The Ct results obtained for IL10 were normalized with HPRT values using RQ = 2−ΔCt where ΔCt = Ct IL10 − Ct HPRT.
For muscle tissues, the quantification of transgene mRNA was performed by targeting the polyA\BGH sequence with the following primers: forward primer 5′-TCTAGTTGCCAGCCATCTGTTGT-3′; reverse primer 5′-TGGGAGTGGCACCTTCCA-3′ and polyA\BGH probe 5′ (6 FAM)-TCCCCCGTGCCTTCCTTGACC-3′ TAMRA. Datas were normalized by quantifying the endogenous HPRT mRNA, using the following primers to target the HPRT sequence: forward primer 5′-GCTTTCCTTGGTCAGGCAGTA-3′; reverse primer 5′-TGGAGTCCTTTTCACCAGCA-3′; and HPRT probe 5′ (6 FAM) AATCCAAAGATGGTCAAGGTCGCAA-3′ TAMRA. Quantification was conducted using AB StepOne Plus (Life Technologies) machine with Taqman chemistry. The polyA\BGH PCR was done using the following program: initial denaturation 20 seconds at 95 °C followed by 45 cycles of 1 second at 95 °C and 20 seconds at 60 °C. The HPRT PCR was done using the following program: initial denaturation 20 seconds at 95 °C followed by 40 cycles of 3 seconds at 95 °C and 30 seconds at 62 °C. The Ct results obtained for the transgene were normalized with HPRT values using RQ = 2−ΔCt.
Follow-up of anti-TetR humoral immune responses in NHP
Detection of anti-rtTA antibodies was conducted using an enzyme-linked immunosorbent assay. Nunc MaxiSorp P96 plates (Sigma-Aldrich, France) were coated overnight at 4 °C with recombinant rtTA protein (at 5 µg/ml, Proteogenix, France). After 3 phosphate-buffered saline washings and 0.1% Tween 1% gelatin saturation for 1 hour at 37 °C, wells were incubated for 2 hours at 37 °C with 12 serial twofold dilutions of macaque sera (from 1/10 to 20,480) then for 1 hour at 37 °C with goat horseradish peroxidase-conjugated anti-rhesus IgG (Cliniscience, France). Positive controls consisted in a serum from an anti-rtTA immunized macaque and a commercial polyclonal anti-TetR antibody (MoBiTec, Germany). Revelation was performed using 2.2-3,3′,5,5′-Tetramethylbenzidine (TMB, BD OptEIA, BD Biosciences) and absorbance of duplicate samples were read at 450 nm with a correction at 570 nm on a Multiskan Go reader (Thermo Scientific, France). Threshold of positivity was determined using 15 negative sera obtained from unrelated naïve macaques as mean of optic density for each dilution +2*SD. IgG titers for experimental animals was defined as the last sera dilution with optic density remaining above the threshold curve.
Follow-up of anti-TetR cellular immune responses in NHP
Anti-TetR cellular immune responses were evaluated with an IFNγ ELISpot assay using frozen PBMC isolated at >18 months postinjection. The assay was performed using an overlapping peptide library covering the rtTA sequence (15 per 10 mers, Pepscreen, Sigma) split in five peptide pools. Briefly, the method consisted in plating 2 × 105 cells in human anti-IFNγ antibody (MabTech, France) precoated MultiScreenHTS filter plates with polyvinyldiene difluoride membrane (PVDF, Millipore, France). Cells were restimulated with rtTA-derived pool peptides at a final concentration of 10 μg/ml. Medium alone and an irrelevant pool of peptides served as negative controls, while cells stimulated with phorbol-12-myristate-13-acetate (PMA, 10 ng/ml, Sigma)/calcium ionophore A23187 Mi Calcium Salt (ionomycin, 250 ng/ml, Sigma) served as a positive control. After incubation with a biotinylated anti-IFN-γ antibody (clone 7-B6-1, MabTech, France) and ExtrAvidin alkalin phosphatase (Sigma-Aldrich), enzymatic reaction was revealed using NBT/BCIP (Pierce, Thermo Scientific). Spot number was determined using an ELISpot reader ELR07 (AID, Straßberg, Germany) and analyzed with AID ELISpot Reader Software V7.0 (AID, Germany). Responses were considered positive when the number of spot-forming colonies per million cells was >50 and at least threefold higher than the peptide pool negative control (C−).
Statistical analysis
A nonparametric Mann-Whitney statistical test was performed using GraphPad Prism version 4. Mean values were considered statistically different if the P value was below 0.05 (*P < 0.05; **P < 0.01; ***P < 0.001). For ELISpot results, statistical analysis was performed with the DFR(2×) test (distribution free resampling statistical test).
Acknowledgments
The authors thank the Boisbonne Center at ONIRIS (Nantes) for nonhuman primates housing and care, the Vector Core (http://www.atlantic-gene-therapies.fr/production-de-vecteurs/centre-de-production-de-vecteurs/) at the University Hospital of Nantes for providing the rAAV1 vector stock, the flow cytometry core facility of Nantes University (Cytocell), and the cellular and tissular imaging core facility of Nantes University (MicroPICell). The study was supported by the INSERM, the University Hospital of Nantes, The French Ministry of education, the “Fondation pour la Thérapie Génique en Pays de Loire,” the AFM-Telethon (Association Française contre les Myopathies), the National Research Agency (ANR-09-BLAN-0265 GENETOL program), the “Region Pays de La Loire” in the context of IMBIO-DC consortium, the “Fondation Centaure,” and the “Fondation Progreffe”. This work was also supported by the IHU-Cesti project that received French government financial support managed by the National Research Agency (ANR-10-IBHU-005 program). The IHU-Cesti project is also supported by Nantes Metropole and Région Pays de la Loire.
The authors declare no conflict of interest.
References
- Bohl D, Salvetti A, Moullier P, Heard JM. Control of erythropoietin delivery by doxycycline in mice after intramuscular injection of adeno-associated vector. Blood. 1998;92:1512–1517. [PubMed] [Google Scholar]
- Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA. 2009;106:16363–16368. doi: 10.1073/pnas.0904514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre D, Blouin V, Provost N, Spisek R, Porrot F, Bohl D. Lack of an immune response against the tetracycline-dependent transactivator correlates with long-term doxycycline-regulated transgene expression in nonhuman primates after intramuscular injection of recombinant adeno-associated virus. J Virol. 2002;76:11605–11611. doi: 10.1128/JVI.76.22.11605-11611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RW, Yang EY, Couto LB, Hagstrom JN, Elwell D, Fields PA. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- Rivera VM, Gao GP, Grant RL, Schnell MA, Zoltick PW, Rozamus LW. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood. 2005;105:1424–1430. doi: 10.1182/blood-2004-06-2501. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenuaud P, Larcher T, Rabinowitz JE, Provost N, Cherel Y, Casadevall N. Autoimmune anemia in macaques following erythropoietin gene therapy. Blood. 2004;103:3303–3304. doi: 10.1182/blood-2003-11-3845. [DOI] [PubMed] [Google Scholar]
- Gao G, Lebherz C, Weiner DJ, Grant R, Calcedo R, McCullough B. Erythropoietin gene therapy leads to autoimmune anemia in macaques. Blood. 2004;103:3300–3302. doi: 10.1182/blood-2003-11-3852. [DOI] [PubMed] [Google Scholar]
- Herzog RW, Mount JD, Arruda VR, High KA, Lothrop CD. Muscle-directed gene transfer and transient immune suppression result in sustained partial correction of canine hemophilia B caused by a null mutation. Mol Ther. 2001;4:192–200. doi: 10.1006/mthe.2001.0442. [DOI] [PubMed] [Google Scholar]
- Ross CJ, Twisk J, Bakker AC, Miao F, Verbart D, Rip J. Correction of feline lipoprotein lipase deficiency with adeno-associated virus serotype 1-mediated gene transfer of the lipoprotein lipase S447X beneficial mutation. Hum Gene Ther. 2006;17:487–499. doi: 10.1089/hum.2006.17.487. [DOI] [PubMed] [Google Scholar]
- Toromanoff A, Adjali O, Larcher T, Hill M, Guigand L, Chenuaud P. Lack of immunotoxicity after regional intravenous (RI) delivery of rAAV to nonhuman primate skeletal muscle. Mol Ther. 2010;18:151–160. doi: 10.1038/mt.2009.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields PA, Arruda VR, Armstrong E, Chu K, Mingozzi F, Hagstrom JN. Risk and prevention of anti-factor IX formation in AAV-mediated gene transfer in the context of a large deletion of F9. Mol Ther. 2001;4:201–210. doi: 10.1006/mthe.2001.0441. [DOI] [PubMed] [Google Scholar]
- Amodio G, Gregori S. Human tolerogenic DC-10: perspectives for clinical applications. Transplant Res. 2012;1:14. doi: 10.1186/2047-1440-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. 2010;116:935–944. doi: 10.1182/blood-2009-07-234872. [DOI] [PubMed] [Google Scholar]
- Zahorchak AF, Kean LS, Tokita D, Turnquist HR, Abe M, Finke J. Infusion of stably immature monocyte-derived dendritic cells plus CTLA4Ig modulates alloimmune reactivity in rhesus macaques. Transplantation. 2007;84:196–206. doi: 10.1097/01.tp.0000268582.21168.f6. [DOI] [PubMed] [Google Scholar]
- Naranjo-Gómez M, Raïch-Regué D, Oñate C, Grau-López L, Ramo-Tello C, Pujol-Borrell R. Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med. 2011;9:89. doi: 10.1186/1479-5876-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol. 2007;178:7018–7031. doi: 10.4049/jimmunol.178.11.7018. [DOI] [PubMed] [Google Scholar]
- Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis. 2010;69:2042–2050. doi: 10.1136/ard.2009.126383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7:610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- Thomas, DC, Wong, FS,, Zaccone, P, Green, EA, Wållberg, M. Protection of islet grafts through TGF beta induced tolerogenic DC. Diabetes. 2013;62:3132–3142. doi: 10.2337/db12-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilkens CM, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol. 2013;172:148–157. doi: 10.1111/cei.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llanos C, Mackern-Oberti JP, Vega F, Jacobelli SH, Kalergis AM. Tolerogenic dendritic cells as a therapy for treating lupus. Clin Immunol. 2013;148:237–245. doi: 10.1016/j.clim.2013.04.017. [DOI] [PubMed] [Google Scholar]
- Bériou G, Pêche H, Guillonneau C, Merieau E, Cuturi MC. Donor-specific allograft tolerance by administration of recipient-derived immature dendritic cells and suboptimal immunosuppression. Transplantation. 2005;79:969–972. doi: 10.1097/01.tp.0000158277.50073.35. [DOI] [PubMed] [Google Scholar]
- Hill M, Thebault P, Segovia M, Louvet C, Bériou G, Tilly G. Cell therapy with autologous tolerogenic dendritic cells induces allograft tolerance through interferon-gamma and epstein-barr virus-induced gene 3. Am J Transplant. 2011;11:2036–2045. doi: 10.1111/j.1600-6143.2011.03651.x. [DOI] [PubMed] [Google Scholar]
- Pêche H, Trinité B, Martinet B, Cuturi MC. Prolongation of heart allograft survival by immature dendritic cells generated from recipient type bone marrow progenitors. Am J Transplant. 2005;5:255–267. doi: 10.1111/j.1600-6143.2004.00683.x. [DOI] [PubMed] [Google Scholar]
- Moreau A, Varey E, Bériou G, Hill M, Bouchet-Delbos L, Segovia M. Tolerogenic dendritic cells and negative vaccination in transplantation: from rodents to clinical trials. Front Immunol. 2012;3:218. doi: 10.3389/fimmu.2012.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM. Antigen-bearing immature dendritic cells induce peptide-specific CD8(+) regulatory T cells in vivo in humans. Blood. 2002;100:174–177. doi: 10.1182/blood.v100.1.174. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34:2026–2032. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzelarab MB, Zahorchak AF, Lu L, Morelli AE, Chalasani G, Demetris AJ. Regulatory dendritic cell infusion prolongs kidney allograft survival in nonhuman primates. Am J Transplant. 2013;13:1989–2005. doi: 10.1111/ajt.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau A, Chiffoleau E, Beriou G, Deschamps JY, Heslan M, Ashton-Chess J. Superiority of bone marrow-derived dendritic cells over monocyte-derived ones for the expansion of regulatory T cells in the macaque. Transplantation. 2008;85:1351–1356. doi: 10.1097/TP.0b013e31816f22d6. [DOI] [PubMed] [Google Scholar]
- Moreau A, Hill M, Thébault P, Deschamps JY, Chiffoleau E, Chauveau C. Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase-1 in rodents and in nonhuman primates. FASEB J. 2009;23:3070–3077. doi: 10.1096/fj.08-128173. [DOI] [PubMed] [Google Scholar]
- Favre D, Provost N, Blouin V, Blancho G, Chérel Y, Salvetti A. Immediate and long-term safety of recombinant adeno-associated virus injection into the nonhuman primate muscle. Mol Ther. 2001;4:559–566. doi: 10.1006/mthe.2001.0494. [DOI] [PubMed] [Google Scholar]
- Arruda VR, Schuettrumpf J, Herzog RW, Nichols TC, Robinson N, Lotfi Y. Safety and efficacy of factor IX gene transfer to skeletal muscle in murine and canine hemophilia B models by adeno-associated viral vector serotype 1. Blood. 2004;103:85–92. doi: 10.1182/blood-2003-05-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cao O, Swalm B, Dobrzynski E, Mingozzi F, Herzog RW. Major role of local immune responses in antibody formation to factor IX in AAV gene transfer. Gene Ther. 2005;12:1453–1464. doi: 10.1038/sj.gt.3302539. [DOI] [PubMed] [Google Scholar]
- Di Caro V, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Involvement of suppressive B-lymphocytes in the mechanism of tolerogenic dendritic cell reversal of type 1 diabetes in NOD mice. PLoS One. 2014;9:e83575. doi: 10.1371/journal.pone.0083575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson S, Hentati F, Rigolet A, Behin A, Romero NB, Leturcq F. A phase I trial of adeno-associated virus serotype 1-γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain. 2012;135 Pt 2:483–492. doi: 10.1093/brain/awr342. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, Poirier A. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults. Hum Gene Ther. 2006;17:1177–1186. doi: 10.1089/hum.2006.17.1177. [DOI] [PubMed] [Google Scholar]
- Gaudet D, Méthot J, Déry S, Brisson D, Essiembre C, Tremblay G. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- Buelens C, Willems F, Delvaux A, Piérard G, Delville JP, Velu T. Interleukin-10 differentially regulates B7-1 (CD80) and B7-2 (CD86) expression on human peripheral blood dendritic cells. Eur J Immunol. 1995;25:2668–2672. doi: 10.1002/eji.1830250940. [DOI] [PubMed] [Google Scholar]
- Chauveau C, Rémy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106:1694–1702. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63:12–17. [PubMed] [Google Scholar]
- Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59:56–58. [PubMed] [Google Scholar]
- Herzog C. Influence of parenteral administration routes and additional factors on vaccine safety and immunogenicity: a review of recent literature. Expert Rev Vaccines. 2014;13:399–415. doi: 10.1586/14760584.2014.883285. [DOI] [PubMed] [Google Scholar]
- Shepard CC, Walker LL, Van Landingham RM, Ye SZ. Sensitization or tolerance to Mycobacterium leprae antigen by route of injection. Infect Immun. 1982;38:673–680. doi: 10.1128/iai.38.2.673-680.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama T, Morelli AE, Onai N, Hirao M, Matsushima K, Tahara H. Mammalian and viral IL-10 enhance C-C chemokine receptor 5 but down-regulate C-C chemokine receptor 7 expression by myeloid dendritic cells: impact on chemotactic responses and in vivo homing ability. J Immunol. 2001;166:7136–7143. doi: 10.4049/jimmunol.166.12.7136. [DOI] [PubMed] [Google Scholar]
- Toromanoff A, Chérel Y, Guilbaud M, Penaud-Budloo M, Snyder RO, Haskins ME. Safety and efficacy of regional intravenous (r.i.) versus intramuscular (i.m.) delivery of rAAV1 and rAAV8 to nonhuman primate skeletal muscle. Mol Ther. 2008;16:1291–1299. doi: 10.1038/mt.2008.87. [DOI] [PubMed] [Google Scholar]
- Sudres M, Ciré S, Vasseur V, Brault L, Da Rocha S, Boisgérault F. MyD88 signaling in B cells regulates the production of Th1-dependent antibodies to AAV. Mol Ther. 2012;20:1571–1581. doi: 10.1038/mt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Luo X, Bird A, Li S, Koeberl DD. Deficiency in MyD88 Signaling Results in Decreased Antibody Responses to an Adeno-Associated Virus Vector in Murine Pompe Disease. Biores Open Access. 2012;1:109–114. doi: 10.1089/biores.2012.0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huang X, Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest. 2009;119:2388–2398. doi: 10.1172/JCI37607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller C, Chulay JD, Trapnell BC, Humphries M, Carey B, Sandhaus RA. Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J Clin Invest. 2013;123:5310–5318. doi: 10.1172/JCI70314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth LA, Ratnasothy K, Moreau A, Alcock S, Sagoo P, Meader L. Tolerogenic Donor-Derived Dendritic Cells Risk Sensitization In Vivo owing to Processing and Presentation by Recipient APCs. J Immunol. 2013;190:4848–4860. doi: 10.4049/jimmunol.1200870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jooss K, Yang Y, Fisher KJ, Wilson JM. Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J Virol. 1998;72:4212–4223. doi: 10.1128/jvi.72.5.4212-4223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mays LE, Vandenberghe LH, Xiao R, Bell P, Nam HJ, Agbandje-McKenna M. Adeno-associated virus capsid structure drives CD4-dependent CD8+ T cell response to vector encoded proteins. J Immunol. 2009;182:6051–6060. doi: 10.4049/jimmunol.0803965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.