

Abstract
By applying [1-13C]- and [2-13C]-glucose labeling schemes to the folded globular protein ubiquitin, a strong reduction of spectral crowding and increase in resolution in solid-state NMR (ssNMR) spectra could be achieved. This allowed spectral resonance assignment in a straightforward manner and the collection of a wealth of long-range distance information. A high precision solid-state NMR structure of microcrystalline ubiquitin was calculated with a backbone rmsd of 1.57 to the X-ray structure and 1.32 Å to the solution NMR structure. Interestingly, we can resolve structural heterogeneity as the presence of three slightly different conformations. Structural heterogeneity is most significant for the loop region β1-β2 but also for β-strands β1, β2, β3, and β5 as well as for the loop connecting α1 and β3. This structural polymorphism observed in the solid-state NMR spectra coincides with regions that showed dynamics in solution NMR experiments on different timescales.
Keywords: ubiquitin, 13C sparse labeling, heterogeneity, dynamics, solid-state NMR
Introduction
Despite recent progress in solid-state magic-angle spinning (MAS) technology, increased spectral resolution due to higher magnetic field strength and sophisticated isotope labeling strategies,1–3 assignment of solid-state NMR (ssNMR) backbone and side chain resonances remains a challenging task. Spectral overlap frequently limits the molecular weight of monomeric proteins or protein assemblies that can currently be studied by ssNMR.4–9
Recently we demonstrated the beneficial use of [1-13C]- and [2-13C]-glucose (glc) as bacterial 13C source during heterologous expression of α-synuclein10 and the type three secretion needles of Salmonella typhimurium11 and Shigella flexneri.12 A strong reduction of spectral crowding and enhancement in spectral resolution could be achieved, resulting in almost complete backbone assignment and a high number of long-range distance restraints, observable due to the concomitant reduction of dipolar truncation effects.10,13,14
Results and Discussion
Sparse labeling schemes enhance spectral resolution
Here, we applied the complementary [1-13C]- and [2-13C]-glucose labeling schemes to microcrystals of ubiquitin obtained by vapor diffusion with 40% 2-methyl-2,4-pentanediol (MPD) and 0.2M CdCl2 as crystallization solution in the reservoir.15 The crystallization condition used in our study differed slightly from previous studies.8,9,16 Using a suite of 2D ssNMR experiments (see Table SI, Supporting Information for further information) on sparsely labeled ubiquitin and additionally 3D experiments using band-selective homonuclear cross-polarization (BSH-CP) transfer on uniformly labeled ubiquitin as described previously,15 full backbone spectral resonance assignment of ubiquitin was achieved for residues M1 to V70, including the previously not assigned loop region T7-T12 connecting β-strands β1 and β2 (Table SII, Supporting Information). For the collection of interresidue long-range distance information, 2D 13C PDSD spectra were recorded on [1-13C]- and [2-13C]-glc-labeled samples using long mixing times of 400–900 ms. Using [1-13C]- and [2-13C]-glucose as sole 13C source has the advantage that only 1 out of 6 carbon atoms is 13C labeled,10,13 thus further reducing the level of labeling to 16.6% as compared to 66.6% or 33.3% for [1,3-13C]- and [2-13C]-glycerol.1,9
By the increased 13C spin-dilution, spectral crowding can be further reduced and 13C-13C dipolar couplings are strongly reduced, leading to reduced line width and improved resolution (compare Figs. S1 and S2, Supporting Information). This allowed for a high degree of completeness of manual spectral assignments both for backbone and side-chains where 91% of all resonances could be assigned (including residues M1 to V70). The assignment strategy is illustrated in Supporting Information Figure S3. Also the number of manually assignable cross-peak intensities in the PDSD spectra could be improved compared to previous studies on uniformly 15N 13C-labeled ubiquitin,8 resulting in 518 inter-residue distance restraints which includes 277 long range distance restraints. The advantage of combining three complementary labeling schemes (uniform-13C, [1-13C]-, and [2-13C]-glc) is illustrated in Figure 1(a–d) for V26 and T55 (see also Supporting Information Figs. S4 and S5).
Figure 1.

Distance restraints collected for V26 Cα (a,c) and T55 Cβ (b,d). (a, b) Excerpts of 2D PDSD spectra of [U-13C]-labeled ubiquitin with a mixing time of 50 ms (black) and of [1-13C]- and [2-13C]-glc-labeled ubiquitin with mixing times of 900 ms (green and magenta, respectively). Intraresidue and sequential correlations are labeled in black, medium- and long- range contacts are labeled in a residue-specific color: I3 (green), L15 (olive green), S20 (green), D21 (violet), T22 (cyan), I23 (orange), V26 (red), A28, K29 and I30 (all blue), T55 (red), S57 (blue), and D58 (pink). The correlation V26 Cα-Cγ2 highlighted by a red rectangle in the [2-13C]-glc spectrum (magenta) illustrates stereospecific information due to biosynthetic scrambling effects.17 (c,d) Illustration of the distance restraint collection for residues V26 (c) and T55 (d) on the X-ray structure (PDB ID: 3ONS) using the same color code as in (a,b). For the sake of clarity, no side-chains are shown for residues I23, A28, K29 and I30 in panel c).
Structure calculation
The structure of microcrystalline ubiquitin was determined using XPLOR-NIH.18 In total, 518 interresidue distance restraints, including 59 unambiguous, 126 network-anchored, and 333 ambiguous distance restraints were used (for further information see Supporting Information and Fig. S6). (The definition of network-anchored distance correlations is illustrated in Supporting Information Fig. S4). Each 13C-13C correlation in the PDSD spectra was assigned to a distance restraint range between 1 and 7 Å, where 7 Å was found to be the upper-distance limit leading to the best convergence (cf. Table SIII and Fig. S7, Supporting Information), which is consistent with previous protocols used in protein structure determination by ssNMR.1,8 (The reason for the large range of distances allowed during structure calculation is that in solid-state NMR, different from solution NMR, cross-peak intensities currently cannot be directly correlated to distances due to effects such as dipolar truncation or labeling statistics that skew the distance information). For sites which belong to flexible regions of ubiquitin (V5-K11, Q41, L67, L69, and V70),19,20 no medium- or long-range 13C-13C correlations could be detected [Fig. 2(c)]. Especially the reduced number of long range correlations for the hairpin β1-β2 is consistent with previous studies8,9 (cf. Fig. S8 and Table SVII, Supporting Information).
Figure 2.

(a) Solid-state NMR-ensemble of the 10 lowest-energy conformers calculated with 518 distance restraints. (b) Comparison between the X-ray structure of ubiquitin (PDB ID: 3ONS) (black) and the lowest energy structure (red). (c) Contact plot of distance restraints identified from 2D PDSD spectra of [1-13C]- and [2-13C]-glc-labeled samples with mixing times of 400 ms up to 900 ms. (blue: side chain-side chain, red: backbone-backbone and in black: backbone-side chain contacts). (d) Distance distribution of 59 unambiguous distance restraints as collected from the spectra; the corresponding distances as extracted from the X-ray structure are shown.
Figure 2(d) illustrates the distance distribution of the 59 unambiguous distance restraints as back-calculated from the X-ray structure (PDB ID: 3ONS). The majority of the observed correlations were found to correspond to distances between 4.5 and 7 Å (by comparison to the X-ray structure). Dihedral angle restraints were calculated from backbone chemical shift information using the program TALOS+ (Supporting Information Fig. S9).21 Further details on the structure calculation can be found in the Supporting Information.
Figure 2(a) shows the 10 lowest-energy structures. Small site-specific deviations between the ten lowest-energy structures could be observed in the first loop region β1-β2 (residues V5-G10), in the loop region α1-β3 (residues Q34-A46), in β-strand β4 (residues K48-Q49) and in the C-terminal β-strand β5 (residues H68-V70) (cf. Fig. S10, Supporting Information). An accuracy of 1.57 Å RMSD for the backbone (residues 1–70) was calculated by comparison to the X-ray structure of ubiquitin crystallized in MPD (PDB ID: 3ONS) [Fig. 2(b), see also Fig. S11, Supporting Information). Comparison to the solution NMR structure (PDB ID: 1D3Z)22 and the ssNMR structure crystallized in MPD (PDB ID: 2JZZ)9 resulted in accuracies of 1.32 Å and 1.98 Å RMSD for the backbone (residue 1–70) respectively (cf. Supporting Information Figs. S12, S13, S14, and S15). Site specific differences larger than 0.3 Å are observed mainly for flexible loop regions and the N-terminus of the protein. It should be noted that of all structures compared to, our structure agrees best with the residual dipolar coupling (RDC) refined solution NMR structure (PDB code: 1D3Z).22
Structural heterogeneity relates to internal dynamics
Interestingly, the careful analysis of the 2D and 3D correlation spectra revealed the presence of three slightly different conformations of ubiquitin, visible as separate peaks for several residues (Table SIV, Supporting Information). As the second and third conformation apparently overlap with the main conformation for the majority of residues and only the main conformation, which gave the strongest resonance intensities and most intense cross-correlations and highest number of unambiguous distance restraints could be unambiguously traced during the assignment process, a structure calculation was only possible for the main conformation (see above). Regions of structural heterogeneity (Met 1 to Leu 15, Glu 34 to Lys 48, Ser 65 to Val 70) show a strongly reduced number of distance restraints and thus appear less defined in the calculated ensemble of lowest energy structures, also for the solution NMR structural ensemble 1d3z22 and for the previous solid-state structure by Zech et al.9 (cf. Fig. S8, Supporting Information). Those regions of structural heterogeneity also show increased 15N chemical shift differences of the main conformation to the solution NMR structure (PDB code 1d3z) and the solid state NMR structure (PDB code 2JZZ) indicating structural differences of the backbone in those regions, while 13Cα and 13C′ appear very similar (cf. Tables SV and SVI, Supporting Information). Previous work8,9 did not report on multiple conformations. We explain the occurrence of multiple conformations by slightly different crystallization conditions compared to Zech et al.9
The observed structural polymorphism coincides with regions that are known to be dynamic, as investigated by solution NMR 15N relaxation23 and RDC-based measurements20,24 as well as recent high-resolution NMR relaxometry.25 In particular we were able to resolve the structural heterogeneity of the first two β-strands, β1 and β2, which form a β-hairpin, and the connecting loop region β1-β2 [Fig. 3(b)]. This loop region shows dynamics in solution both on the fast ps-ns time scale as investigated by 15N relaxation measurements as well as on the ns-µs time scale as investigated by RDC-based measurements20,24 [Fig. 3(b)]. Further we observed structural heterogeneity in the loop region α1-β3 (including the C-terminal tip of the α-helix α1, residues K33-Q40), the third β-strand (Q41-F45) and the adjacent residues up to Lys 48, as well as for the fifth β-strand β5 (residues T66-V70) [cf. Fig. 3(a)]. Those regions showed increased dynamics on the ns-µs time scales as revealed by RDC-based studies on ubiquitin.20,24,26 In particular loop β1-β2 and loop α1-β3, including the C-terminal tip of the α-helix, have been identified previously to be involved in a large amplitude collective motion, resembling a “pincer like” motion which was related to conformational sampling of ubiquitin during molecular recognition.20
Figure 3.
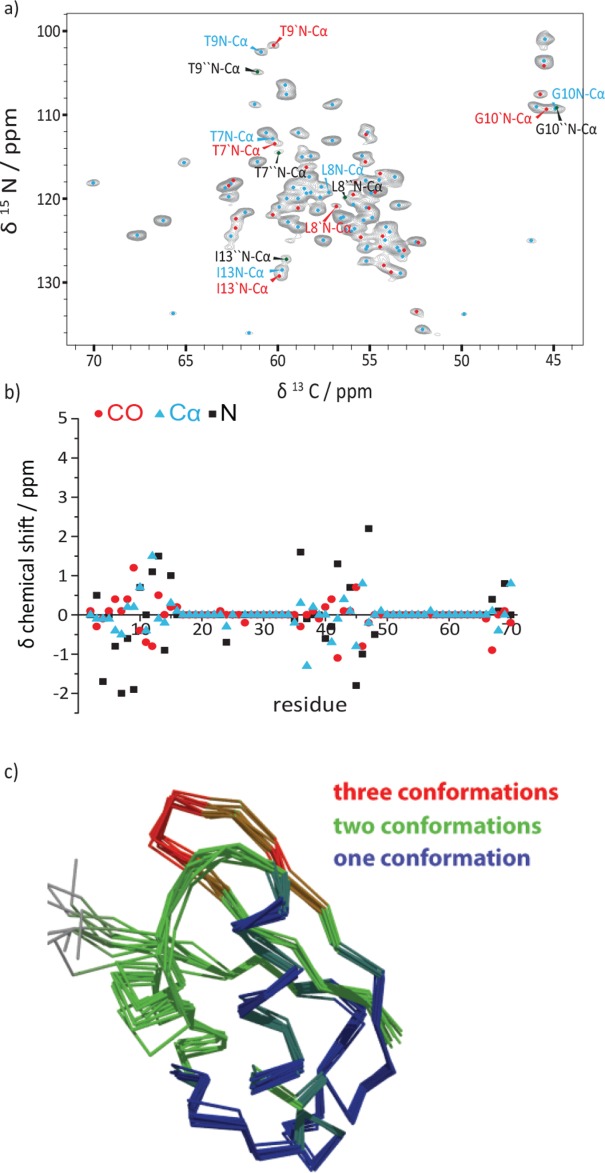
(a) 2D NCA ssNMR spectrum of uniformly 13C labeled ubiquitin sample. The correlations are labeled as follows: main (blue), second (red), and third conformation (black). The population distribution is calculated by integration of the T9 N-Cα cross peaks. (b) Illustration of chemical shift differences of the backbone atoms between the first- and the second conformation. (c) Solid-state NMR-ensemble of the 10 lowest-energy conformers (residues with three different conformations are colored in red, with two conformations in green and with one conformation in blue).
It is conceivable that the conformational sampling of ubiquitin in solution will be “frozen out” during the crystallization process of ubiquitin in MPD, leading to slightly different conformations, which can be identified based on their chemical shift differences in the solid state. It should be noted that Ala46, close to Lys48 whose side chain is the major recognition site of ubiquitin during poly-ubiquitination, shows the biggest difference in 15N chemical shifts [Fig. 3(b)]. More recent studies by Fenwick et al.27 suggested that β-strands β1, β2, β3, and β5 are involved in a concerted motion across the β-sheet mediated by the hydrogen bond network. The structural heterogeneity observed in the solid-state involves this β-strand network and the described loop regions but is, for example, largely absent for the α-helix, consistent with dynamics observed in solution. High B-factors for the first loop region of the X-ray structure of ubiquitin crystallized in MPD confirm the presence of heterogeneity in the loop region β1-β2.16 No significant temperature dependence of the structural polymorphism was observed, ruling out a temperature-dependent conformational exchange (Fig. S16, Supporting Information). Conformational differences rather seem to manifest during the crystallization process.
Microsecond dynamics in the 50 µs range has been observed for ubiquitin for I23, N25, T55, and V70 as investigated by 15N relaxation dispersion methods.28,29 With exception of T55 all those residues show structural heterogeneity in the present solid state NMR study. As however several additional residues show multiple conformations, which have been identified by RDC measurements to be dynamic (see above), we suggest that structural polymorphism/sample heterogeneity observed in our solid-state NMR studies are the result of conformational dynamics of the (soluble) protein on an even faster ns-µs time scale, accessible to RDCs [slower than the overall tumbling correlation time, 4 ns for ubiquitin at room temperature, but faster than the lower limit of 15N relaxation dispersion experiments (about 50 µs)]. Recently, the rate of structural inter-conversion between different ubiquitin conformations in the ground-state ensemble could be estimated to be in the order of a few microseconds.26
More recently, Michielssens et al.30 have identified several point mutations that affect the conformational ground state ensemble of ubiquitin in solution and shift the population towards a closed sub-state. We plan on crystallizing these ubiquitin mutants under identical conditions as described herein and will examine how those point mutations affect the occurrence of multiple conformations and if a different conformation becomes the dominant one.
Conclusions
In summary, the application of [1-13C]- and [2-13C] glc labeling schemes and BSH-CP based 3D spectra15 allowed for an almost complete spectral assignment of backbone and side-chain resonances of ubiquitin for the well-ordered residues 1–70. Using a wealth of long-range inter-residue distance information, an ubiquitin structure with a backbone RMSD of 1.57 to the X-ray structure solved in MPD9 and 1.32 Å to the solution NMR structure22 was calculated. The high spectral resolution and almost complete resonance assignment enabled the identification of structural polymorphism with up to three different conformations for the β-strands β1, β2, β3, and β5 as well as for the loop regions β1-β2 and α1-β3. Those regions of structural heterogeneity coincide with regions showing increased dynamics on the ns-µs time scale as previously revealed by solution NMR 15N relaxation and RDC-based experiments and were inferred to be involved in a conformational sampling process. This suggests that conformational dynamics in solution is “frozen out” during the crystallization process and manifests itself as a structural polymorphism observed by solid-state NMR.
Material and Methods
The recombinant expression and purification of ubiquitin was carried out as described in reference.31 The crystallization condition (40% MPD and 0.2M CdCl2) was identified by systematic screening using a commercial crystallization screen (Nextal MPD Suite, Qiagen). The solid-state NMR experiments were conducted on 850, 800, and 600 MHz spectrometers (Bruker Biospin, Germany) equipped with triple-resonance (1H, 13C, 15N) MAS probes. The MAS frequency was set in the range of 11–21 kHz using 4 and 3.2 mm ZrO2 rotors. Spectra were recorded at a sample temperature of 273 K, as determined from the 1H chemical shift of water in reference to DSS. 90° pulses employed RF fields of 83 kHz for 1H, 50 kHz for 13C, and 35 kHz for 15N. High-power 1H-13C decoupling (SPINAL-64)32 was applied with an RF field strength of about 83 kHz. The cross-polarization transfer (CP) from 1H to 13C or 15N was implemented with contact times of 200–1400 µs and a ramped shape (100–80%) on 1H. The homonuclear 2D 13C-13C correlation spectra were recorded using proton-driven spin diffusion (PDSD).33 For 2D 15N-13C correlation experiments, the contact time of the SPECIFIC 15N-13C CP transfer34 was set to 3–5 ms and a continuous wave decoupling of 83 kHz was applied. The 13C-13C transfer in the NCACX and NCOCX experiments was mediated by DARR mixing.35 The 2D H-C INEPT-spectrum was recorded with RF-pulse strengths as indicated above. All spectra were processed using Bruker Topspin 3.1 and analyzed using Sparky (version 3.100, T. D. Goddard and D. G. Kneller, University of California). In addition, also 3D spectra from reference15 were used. All 2D spectra are listed in Table SI, Supporting Information.
Data deposition
Chemical shifts are deposited in the BMRB (ID: 25123) and the determined structure in the PDB (ID: 2MSG).
References
- Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- Hong M. Determination of multiple phi-torsion angles in proteins by selective and extensive C-13 labeling and two-dimensional solid-state NMR. J Magn Reson. 1999;139:389–401. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- Lundstroem P, Vallurupalli P, Hansen DF, Kay LE. Isotope labeling methods for studies of excited protein states by relaxation dispersion NMR spectroscopy. Nature Protoc. 2009;4:1641–1648. doi: 10.1038/nprot.2009.118. [DOI] [PubMed] [Google Scholar]
- Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, Becker S, Baldus M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- Han Y, Ahn J, Concel J, Byeon I-JL, Gronenborn AM, Yang J, Polenova T. Solid-state NMR studies of HIV-1 capsid protein assemblies. J Am Chem Soc. 2010;132:1976–1987. doi: 10.1021/ja908687k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loquet A, Bardiaux B, Gardiennet C, Blanchet C, Baldus M, Nilges M, Malliavin T, Boeckmann A. 3D structure determination of the Crh protein from highly ambiguous solid-state NMR restraints. J Am Chem Soc. 2008;130:3579–3589. doi: 10.1021/ja078014t. [DOI] [PubMed] [Google Scholar]
- Manolikas T, Herrmann T, Meier BH. Protein structure determination from C-13 spin-diffusion solid-state NMR spectroscopy. J Am Chem Soc. 2008;130:3959–3966. doi: 10.1021/ja078039s. [DOI] [PubMed] [Google Scholar]
- Zech SG, Wand AJ, McDermott AE. Protein structure determination by high-resolution solid-state NMR spectroscopy: application to microcrystalline ubiquitin. J Am Chem Soc. 2005;127:8618–8626. doi: 10.1021/ja0503128. [DOI] [PubMed] [Google Scholar]
- Loquet A, Giller K, Becker S, Lange A. Supramolecular interactions probed by 13C-13C solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:15164–15166. doi: 10.1021/ja107460j. [DOI] [PubMed] [Google Scholar]
- Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A. Atomic model of the type III secretion system needle. Nature. 2012;488:276. doi: 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers J-P, Sgourakis NG, Gupta R, Loquet A, Giller K, Riedel D, Laube B, Kolbe M, Baker D, Becker S, Lange A. The common structural architecture of Shigella flexneri and Salmonella typhimurium type three secretion needles. PLoS Pathog. 2013;9:e1003245. doi: 10.1371/journal.ppat.1003245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loquet A, Lv G, Giller K, Becker S, Lange A. C-13 spin dilution for simplified and complete solid-state NMR resonance assignment of insoluble biological assemblies. J Am Chem Soc. 2011;133:4722–4725. doi: 10.1021/ja200066s. [DOI] [PubMed] [Google Scholar]
- Habenstein B, Loquet A, Giller K, Becker S, Lange A. Structural characterization of supramolecular assemblies by C-13 spin dilution and 3D solid-state NMR. J Biol NMR. 2013;55:1–9. doi: 10.1007/s10858-012-9691-9. [DOI] [PubMed] [Google Scholar]
- Shi C, Fasshuber HK, Chevelkov V, Xiang S, Habenstein B, Vasa SK, Becker S, Lange A. BSH-CP based 3D solid-state NMR experiments for protein resonance assignment. J Biomol NMR. 2014;59:15–22. doi: 10.1007/s10858-014-9820-8. [DOI] [PubMed] [Google Scholar]
- Huang KY, Amodeo GA, Tong L, McDermott A. The structure of human ubiquitin in 2-methyl-2,4-pentanediol: A new conformational switch. Protein Sci. 2011;20:630–639. doi: 10.1002/pro.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv G, Fasshuber HK, Loquet A, Demers JP, Vijayan V, Giller K, Becker S, Lange A. A straightforward method for stereospecific assignment of val and leu prochiral methyl groups by solid-state NMR: Scrambling in the [2–13C]Glucose labeling scheme. J Magn Reson. 2013;228:45–49. doi: 10.1016/j.jmr.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Haller JD, Schanda P. Amplitudes and time scales of picosecond-to-microsecond motion in proteins studied by solid-state NMR: a critical evaluation of experimental approaches and application to crystalline ubiquitin. J Biomol NMR. 2013;57:263–280. doi: 10.1007/s10858-013-9787-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange OF, Lakomek N-A, Fares C, Schroeder GF, Walter KFA, Becker S, Meiler J, Grubmueller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS plus: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from N-15 NMR relaxation. J Am Chem Soc. 1995;117:12562–12566. [Google Scholar]
- Lakomek N-A, Walter KFA, Fares C, Lange OF, de Groot BL, Grubmueller H, Brueschweiler R, Munk A, Becker S, Meiler J, Griesinger C. Self-consistent residual dipolar coupling based model-free analysis for the robust determination of nanosecond to microsecond protein dynamics. J Biomol NMR. 2008;41:139–155. doi: 10.1007/s10858-008-9244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Khan SN, Marquardsen T, Pelupessy P, Reiss V, Sakellariou D, Bodenhausen G, Engelke F, Ferrage F. Nanosecond time scale motions in proteins revealed by high-resolution NMR relaxometry. J Am Chem Soc. 2013;135:18665–18672. doi: 10.1021/ja409820g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban D, Funk M, Gulich R, Egger D, Sabo TM, Walter KFA, Fenwick RB, Giller K, Pichierri F, de Groot BL, Lange OF, Grubmueller H, Salvatella X, Wolf M, Loidl A, Kree R, Becker S, Lakomek N-A, Lee D, Lunkenheimer P, Griesinger C. Kinetics of conformational sampling in ubiquitin. Angew Chem Int Edit. 2011;50:11437–11440. doi: 10.1002/anie.201105086. [DOI] [PubMed] [Google Scholar]
- Bryn Fenwick R, Esteban-Martin S, Richter B, Lee D, Walter KFA, Milovanovic D, Becker S, Lakomek NA, Griesinger C, Salvatella X. Weak long-range correlated motions in a surface patch of ubiquitin involved in molecular recognition. J Am Chem Soc. 2011;133:10336–10339. doi: 10.1021/ja200461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JL, Szyperski T. Protein dynamics in supercooled water: The search for slow motional modes. J Biomol NMR. 2002;23:63–67. doi: 10.1023/a:1015397305148. [DOI] [PubMed] [Google Scholar]
- Massi F, Grey MJ, Palmer AG. Microsecond timescale backbone conformational dynamics in ubiquitin studied with NMR R-1p relaxation experiments. Protein Sci. 2005;14:735–742. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielssens S, Peters JH, Ban D, Pratihar S, Seeliger D, Sharma M, Giller K, Sabo TM, Becker S, Lee D, Griesinger C, de Groot BL. A designed conformational shift to control protein binding specificity. Angew Chem Int Ed. 2014;53:10367–10371. doi: 10.1002/anie.201403102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Etzkorn M, Heise H, Becker S, Baldus M. High-resolution solid-state NMR studies on uniformly C-13,N-15-labeled ubiquitin. Chembiochem. 2005;6:1638–1647. doi: 10.1002/cbic.200500085. [DOI] [PubMed] [Google Scholar]
- Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- Suter D, Ernst R. Spin diffusion in resolved solid-state NMR spectra. Phys Rev B. 1985;32:5608–5627. doi: 10.1103/physrevb.32.5608. [DOI] [PubMed] [Google Scholar]
- Baldus M, Petkova AT, Herzfeld J, Griffin RG. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol Phys. 1998;95:1197–1207. [Google Scholar]
- Takegoshi K, Nakamura S, Terao T. 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]