

Abstract
The structure of insulin, a glucose homeostasis-controlling hormone, is highly conserved in all vertebrates and stabilized by three disulfide bonds. Recently, we designed a novel insulin analogue containing a fourth disulfide bond located between positions A10-B4. The N-terminus of insulin's B-chain is flexible and can adapt multiple conformations. We examined how well disulfide bond predictions algorithms could identify disulfide bonds in this region of insulin. In order to identify stable insulin analogues with additional disulfide bonds, which could be expressed, the Cβ cut-off distance had to be increased in many instances and single X-ray structures as well as structures from MD simulations had to be used. The analogues that were identified by the algorithm without extensive adjustments of the prediction parameters were more thermally stable as assessed by DSC and CD and expressed in higher yields in comparison to analogues with additional disulfide bonds that were more difficult to predict. In contrast, addition of the fourth disulfide bond rendered all analogues resistant to fibrillation under stress conditions and all stable analogues bound to the insulin receptor with picomolar affinities. Thus activity and fibrillation propensity did not correlate with the results from the prediction algorithm.
Keywords: disulfide, prediction, insulin, protein design, stability
Statement: A fourth disulfide bond has recently been introduced into insulin, a small two-chain protein containing three native disulfide bonds. Here we show that a prediction algorithm predicts four additional four disulfide insulin analogues which could be expressed. Although the location of the additional disulfide bonds is only slightly shifted, this shift impacts both stability and activity of the resulting insulin analogues.
Introduction
Insulin is a key hormone regulating glucose homeostasis and an important drug for treatment of diabetes, a disease affecting more than 350 million people world wide.1 Diabetes has evolved into an epidemic and especially in the developing countries, where lack of cooling facilities poses an additional problem, the numbers of patients are increasing with a tremendous speed.1,2 The stability of insulin at elevated temperatures is therefore important for its shelf-life and the safety of the patients. Furthermore the use of continuous pumps as an alternative to multiple daily injections increases the necessity for high physical stability of insulin. Insulin is subjected to shear forces in the pumps and is known to readily form fibrils under such physical stress.3,4
The insulin molecule consists of two peptide chains, the A-chain and the B-chain, linked by two disulfide bonds. In addition the A-chain also contains an intramolecular disulfide bond. The A-chain consists of 21 amino acids containing two α-helices in A1-A8 and A12-A20 connected by a loop, whereas the B-chain consist of 30 amino acids containing a central α-helix in position B9-B19 flanked by two turns and flexible regions in both termini.5,6 Insulin forms higher oligomeric states, dimers at micromolar concentration and hexamers at millimolar concentration with addition of zinc. Binding of small aromatic alcohols, like phenol and m-cresol in the hexameric form, facilitates a conformational change of the N-terminal end of the B-chain extending the central helix to include positions B1-B8.7–9 The oligomeric states of insulin stabilize the molecule3,10,11 but it is the monomeric state of insulin which facilitates its binding to the insulin receptor.12
The three native disulfide bonds have been conserved in the insulin structure for more than half a billion years and are of major importance for the stability of the molecule.13–16 Recently, we introduced an insulin analogue containing a fourth disulfide bond between position A10 in the A-chain and B4 in the B-chain, (A10C-B4C)** which was not only more stable but also showed indications of being more potent than human insulin.17 Here we describe three additional insulin analogues where the position of one of the cysteine residues in the fourth disulfide bond is altered. The analogues were engineered using prediction tools and knowledge about the structure. Furthermore, we compared the binding affinity and stability of these new analogues with the published results for the A10C-B4C 4SS-insulin‡ analogue. We also discuss the use of disulfide bond prediction tools and the different factors effecting successful prediction of stable disulfide bonds.
Results
Engineering and expression
We have previously shown that an analogue containing an additional disulfide between position A10 in the A-chain and B4 in the B-chain could successfully be expressed, folded, and secreted from the yeast, Saccharomyces cerevisiae expression system. We therefore tested the ability of disulfide bond prediction algorithms to predict the locations of disulfide bonds in the 4SS-insulin analogue already constructed and also in adjacent positions. The Schrödinger software BioLuminate was used to identify new potential disulfide bonds in a crystal structure of human insulin (Protein data base (PDB) ID: 1MSO18).19–22 In order to investigate if the natural occurring disulfide bonds could be predicted, all three disulfide bonds in the crystal structure of human insulin were broken and the cysteins were substituted with alanine. The cysteine mutation method was applied on this structure with a 5 Å Cβ-Cβ cut-off distance. All three native disulfide bonds were identified as potential disulfide bonds. A weighted score for each of the bonds were calculated in the software where the lower the score the better the prediction. The weighted scores were 456 (A7C-B7C), 466 (A11C-A6C), and 348 (A20C-B19C) for the native disulfide bonds illustrating the scores found for highly plausible disulfide bonds. After protein preparation the cysteine mutation method was then applied on the crystal structure of human insulin (1MSO). We chose to focus mainly on the N-terminal part of insulin as this area does not contribute important receptor binding positions,23–26 thus giving higher possibilities of producing active analogues. Also as this area is highly flexible, restraining it by an additional disulfide bond could have a higher impact on stability.27 Different potential disulfide bonds were identified based on varying the cut-off values for the Cβ-Cβ distance (Table1). A10C-B3C was identified with default cut-off value (5 Å) while the cut-off values had to be increased in order to identify A10C-B2C, A10C-B4C, A10C-B5C, and A10C-B6C. A10C-B1C was not identified as a potential disulfide bond up to the maximal evaluated cut-off value (15 Å). The weighted scores were calculated after refinement and A10C-B3C and A10C-B5C had scores comparable to the scores obtained for the naturally occurring disulfide bonds.
Table 1.
Identification of Disulfide Bonds in Human Insulin
| Cβ-Cβ distance cut-off (Å) | Disulfide bond | Weighted score | |
|---|---|---|---|
| Based on one structure | 15 | A10-B1 | — |
| 10 | A10-B2 | 10,436 | |
| 5 | A10-B3 | 442 | |
| 7 | A10-B4 | 10,483 | |
| 6 | A10-B5 | 471 | |
| 9 | A10-B6 | 10,504 | |
| Based on trajectory | 8 | A10-B1 | 10,490 |
| 8 | A10-B2 | 10,545 | |
| 7 | A10-B3 | 10,418 | |
| 8 | A10-B4 | 498 | |
| 7 | A10-B5 | 412 | |
| 8 | A10-B6 | 518 |
As the X-ray crystal structures do not picture the full flexibility of the protein, it could be plausible to introduce a fourth disulfide bond in such an area of the molecule even though the distance, as determined from the X-ray structure, is not optimal for disulfide bond formation and therefore not predicted by the prediction tools in the initial screening method. The N-terminal end of the B-chain is very flexible as evident by the B-factors in the 1MSO structure (Fig. 1). This flexibility was also observed for the A10C-B4C insulin analogue where a small bend was formed in order to facilitate the formation of the fourth disulfide bond, [Fig. 2(B)].
Figure 1.
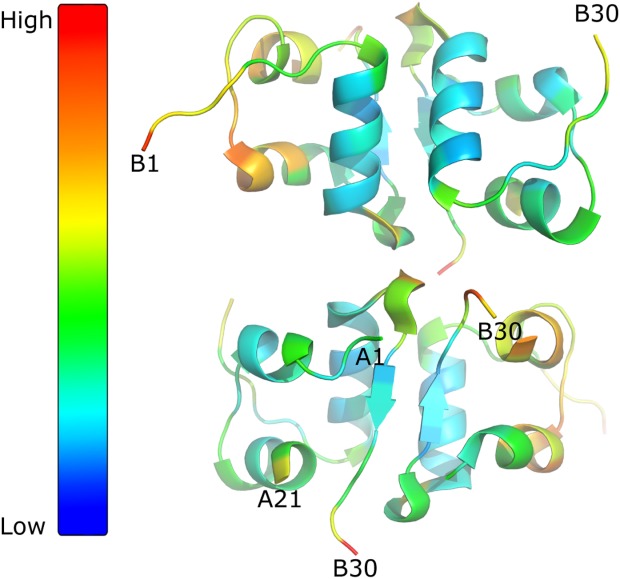
B-factors for insulin structure. Insulin structure (PDB code: 1MSO; one dimer from the hexameric form), seen in two different orientation and visualize by PyMOL. The B-factor increases from blue going to red as illustrated by color bar in the left site.
Figure 2.
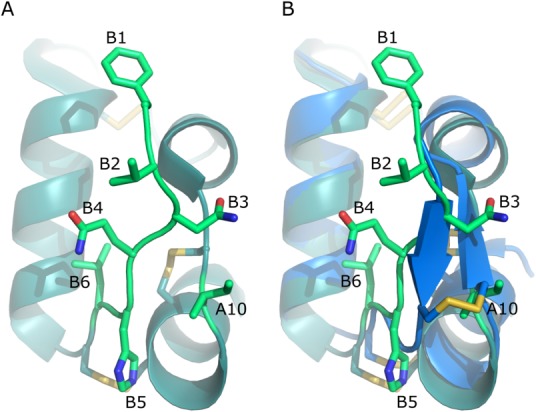
Selecting positions for the fourth disulfide bond. (A) Highlight of the side chains used in the 4SS-inslin analogue design and (B) how the disulfide bond is formed in the A10C-B4C analogue, PDB code 4EFX.17
To account for the flexibility, another strategy was employed. Sampling of different structures was performed by making a molecular dynamics simulations.28,29 The cysteine mutation method was applied on the trajectory from the simulation. With a cut-off distance of 8 Å between Cβ-Cβ all six disulfide bonds were identified as potential disulfide bonds. The weighted scores were calculated after refinement and A10C-B4C, A10C-B5C, and A10C-B6C had scores comparable to the scores obtained when the naturally occurring disulfide bonds were predicted (Table1).
Based on the positions identified by the BioLuminate the following analogues were constructed and investigated for expression in the yeast expression system: A10C-B1C, A10C-B2C, A10C-B3C, A10C-B5C, and A10C-B6C [Fig. 2(A)]. The analogues were expressed as insulin precursors (see Methods). The expression yield of the five analogue precursors were analyzed by RP-HPLC with human insulin (HI) as an external standard (Table2).
Table 2.
Data on Selected Position for Introduction of Addition Disulfide Bond
| Disulfide bond | Distance Cβ-Cβ (Å)a | Expression yield precursor (%) | Receptor bindingb (%) | Metabolic responseb (%) | Melting temperature (°C) | |
|---|---|---|---|---|---|---|
| Tm1 | Tm2 | |||||
| HI | 100 | 100 | 100 | 64.2 | NA | |
| A10C-B1C | 12.0 | ∼20 | 40 ± 1 | 31.8 | 59.8 | 87.8 |
| A10C-B2C | 9.2 | ∼40 | 70 ± 10 | 65 | 68.4 | 87.4 |
| A10C-B3C | 4.4 | ∼80 | 38 ± 1 | 22.5 | 97.4 | NA |
| A10C-B4C | 6.4 | ∼70c | 156 ± 16c | 132 | 98.8‡ | NA |
| A10C-B5C | 5.3 | <10 | NA | NA | NA | NA |
| A10C-B6C | 8.4 | <1 | NA | NA | NA | NA |
PDB 1MSO chain A and B.
Relative to HI, ±STD, n = 3.
Ref. 17.
The precursor (i.e. single chain insulin with a mini C-peptide and an N-terminal spacer peptide) with a proposed fourth disulfide between A10C-B6C was expressed with a minute expression yield, only identified by MALDI-MS and no corresponding peak was detected by HPLC. The precursor with a disulfide bond between A10C-B5C was expressed with yield <10% relative to the HI precursor. During conversion to the mature analogue which occurs at pH ∼9, this analogue was unstable and lost during the process (likely due to disulfide scrambling). The analogue A10C-B3C, which had been predicted with a low score based on the prepared crystal structure was expressed with the highest yield, ∼80% relative to the HI precursor, which was higher than observed for the previous A10C-B4C 4SS-insulin analogue. The A10C-B2C precursor was expressed with a yield of ∼40% relative to the HI precursor and the A10C-B1C precursor with a yield of ∼20% relative to the HI precursor. The three precursors (A10C-B1C, A10C-B2C, and A10C-B3C), which were expressed and stable during enzymatic conversion to mature insulin analogues, were purified as previously described.17 The precursors were converted to desB30 insulin analogues (analogues lacking the threonine in position 30 in the B-chain) using lysine specific Achromobacter lyticus protease.
Receptor binding
Receptor binding affinities for the three purified analogues were measured using the A isoform of the receptor in a scintillation proximity assay (SPA) as previously described.23 The binding receptor affinities were determined by competition of the 4SS-insulin analogue and [125I]TyrA14-labeled insulin (Novo Nordisk A/S) in the SPA assay. The results were analyzed according to a four-parameter logistic model and the affinities were expressed relative to the HI standard [IC50(insulin)/IC50(analogue) × 100%], see Table2. Representative binding curves are shown in Figures 3.
Figure 3.
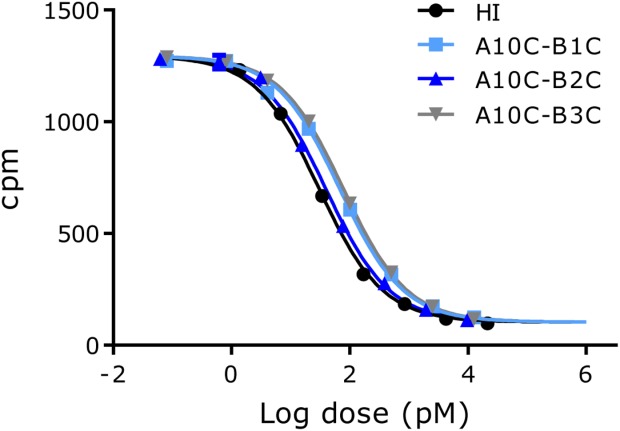
Representative insulin receptor binding curves for human insulin and the 4SS-insulin analogues. Each point on the graph represents the mean ± SD, n = 4 within one assay.
All three analogues bound to the insulin receptor with high affinities; A10C-B1C and A10C-B3C both bound close to 50% relative to HI whereas A10C-B2C bound slightly better at 70%. Their ability not only to bind but also to activate the receptor and elicit a metabolic response was confirmed in a lipogenesis assay. The results reflected the receptor binding affinities with a tendency of A10C-B3C having a lower response compared with its receptor binding affinity (Table2).
Stability
Amyloid fibril formation
The propensity of the three analogues to form amyloid fibrils was tested in a thioflavin T (ThT) assay. Fibrillation was observed within the first few hours of incubation for HI (Fig. 4). Addition of Zn and phenol to HI known to induce the formation of R6-hexamers,9 increased the lag time before fibril formation to ∼5 h. The 4SS-insulin analogues were tested without addition of Zn and phenol and no signs of fibril formation were observed after vigorous shaking for 45 h at 37 °C.
Figure 4.
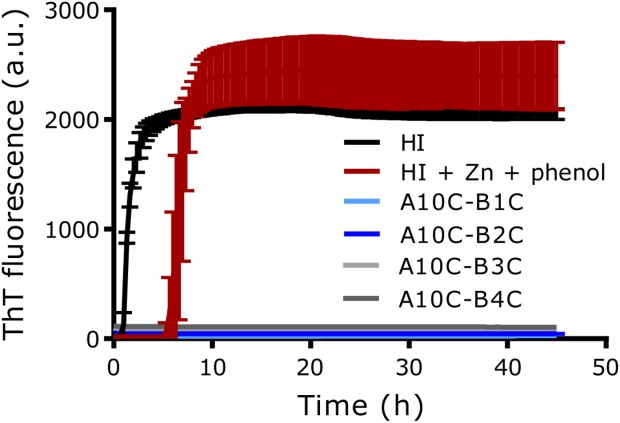
Thioflavin-T (ThT) assay. Time course of ThT fluorescence is shown for human insulin, human insulin with 3Zn/hexamer and 30 mM phenol, and 4SS-insulin analogues during 45 h shaking (960 rpm) at 37 °C. SD is indicated for each point, n = 4.
Thermal stability
The thermal stabilities of the analogues were assessed by differential scanning calorimetry (DSC) (Fig. 5) and circular dichroism (CD) (Fig. 6). The melting temperatures (Tm) for the three analogues were determined by DSC as the maximum of the endothermic peak, see Table1. Unfortunately, as the endothermic unfolding transition had not been completed for any of the 4SS-insulin analogues, before a sharp exotherm occurred due to precipitation/aggregation at ∼100 °C, the unfolding of the analogues was irreversible and it was therefore not possible to calculate thermodynamic parameters.
Figure 5.
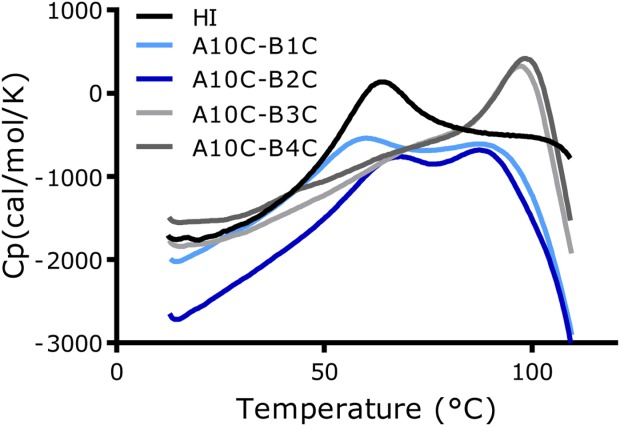
Differential scanning calorimetry (DSC) Thermographs for human insulin and 4SS-insulin analogues recorded from 10 to 110 °C with a scan rate of 1 °C/min are shown. Insulin analogues (0.2 mM) were formulated in a 0.2 mM phosphate buffer pH 7.5 which was also used as reference buffer.
Figure 6.
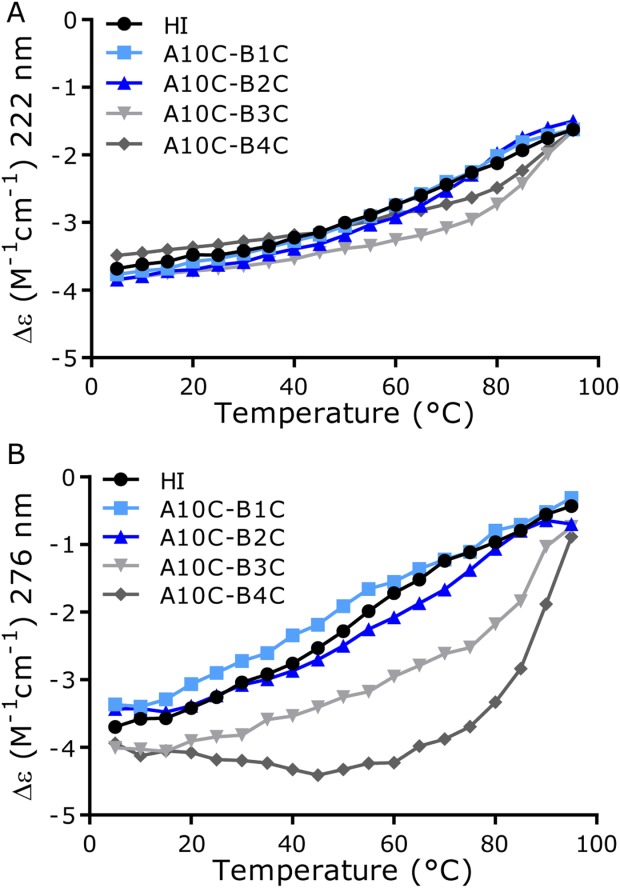
CD measurement of thermal unfolding. (A) Far-UV spectra at 222 nm. (B) Near-UV spectra at 276 nm.
The thermograph for A10C-B3C looked very similar to previously reported thermograph for A10C-B4C analogue. This was also reflected in the melting temperature which was only ∼1 °C lower. The thermographs for A10C-B1C and A10C-B2C also resembled each other but were different from the thermographs for A10C-B3C and A10C-B4C as both contained two peaks with the first melting temperature close to that of HI and the second ∼10 °C lower than that observed for the A10C-B3C and A10C-B4C analogues.
The temperature effect on secondary structure of the three new 4SS-insulin analogues together with the A10C-B4C analogue was analyzed by circular dichroism (CD). Two different spectral regions were analyzed; 222 nm, [Fig. 6(A)], characteristic for α-helix content and 276nm, [Fig. 6(B)], showing Tyr exposure to the solvent, i.e. side-chain exposure by either changed self-association state or side-chain unfolding.
The fourth disulfide bonds did not have a large effect on unfolding of the backbone helixes. A10C-B1C and A10C-B2C were close to identical and were very similar to HI. A10C-B3C and A10C-B4C were also very similar to each other. Most noticeably, they diverged from HI and the other two 4SS-insulin analogues after ∼50/60 °C, where they appeared to maintain the helix up to around 85 °C after which they again merged with HI and the two other 4SS-insulin analogues. The spectra at 276 nm showed that the A10C-B1C and A10C-B2C analogues were again very similar to that of HI. There was a tendency that the side chains were generally slightly more exposed for A10C-B1C and for the A10C-B2C slightly less exposed compared with HI. In contrast, a divergence between the HI curve and the curves for A10C-B3C and A10C-B4C was seen. For A10C-B3C, there was the same tendency of divergence from the HI curve observed also at this wavelength suggesting lower solvent exposure relative to HI until ∼85 °C. This again indicated the higher stability of this analogue assuming the same oligomerization state as HI. In contrast, the curve for the A10C-B4C analogue had a unique profile with a decrease in exposure from 5 to ∼40 °C with a slight increase up to ∼85 °C following by a steep increase with the curve ending close to the others. This was a strong indication of a change in oligomerization state in the initial phase combined with unfolding occurring at elevated temperature.
Discussion
Introduction of an additional disulfide bond (both inter- or intramolecular) to stabilize a protein has been investigated for many different proteins.30–38 The difficult aspect of disulfide bond engineering is selecting the best positions for introduction of the cysteine residues involved in formation of the new disulfide bond. It is important that the new disulfide bond does not hinder access and flexibility of the areas essential for protein's function and does not interfere with folding of the protein by disulfide scrambling or interference with hydrophobic core formation. It is also important that the positions of the two cysteine residues favor the formation of a disulfide bond. Several different algorithms were developed to predict possible positions of new disulfide bonds in proteins29,39,40 based on distances and angles between atoms of the disulfide bond identified from the known structures in the PDB database. Additional factors such as flexibility and solvent accessibility have also been incorporated in the upgraded versions of the softwares19,22,27,29,39,41 to achieve higher prediction success rates.
In the area of interest new four disulfide bonds containing insulin analogues were discovered by using the disulfide bond prediction tool in the Schrödinger BioLuminate software. In order to identify new possible disulfide bonds in the expressed insulin precursors, changes to the structure (allowing flexibility) and bond criteria (changing default settings) had to be allowed. The usefulness of the prediction tools is illustrated as some correlation is seen between the extent of changes that had to be made for the predicted disulfide bonds and the expression yields of the precursor. It was not possible to express all of the predicted precursors, as the A10C-B6C was not expressed in a measurable yield. This disulfide was identified using the X-ray structure method by increasing the cut-off for the Cβ-Cβ distance to 9 Å and resulted in a high weighted score. It was also identified in the trajectory method with a score comparable to the naturally occurring disulfide positions. This illustrates that using the trajectory method could lead to false positives. The precursors for A10C-B3C and A10C-B4C were expressed with the highest yield. According to the melting temperatures, these were also the most stable insulin analogues tested. A10C-B3C was identified using the crystal structure with default cut-off settings and had a weighted score comparable to the values obtained for the naturally occurring disulfide bonds. In order to identify A10C-B4C, the prediction using the trajectory method had to be used with an increased cut-off value for the Cβ-Cβ distance but resulting in a low weighted score. In this case the trajectory method reveals one of the two analogues achieving the highest stability, which has not been identified as a good candidate when using only the crystal structure method. In order to identify A10C-B2C, the cut-off value had to be increased and the weighted scores were high using both the crystal structure and the trajectory methods. The disulfide bond in the expressed precursor A10C-B1C could not be identified when the crystal structure method was used. Based on the trajectory method with increased cut-off for the Cβ-Cβ distance, it was identified but with a high weighted score. The A10C-B5C bond was identified from the crystal structure using an increased cut-off value for the Cβ-Cβ distance with weighted score comparable to the naturally occurring disulfide bonds. Compared with the A10C-B4C disulfide bond, the A10C-B5C precursor had a much lower yield and was shown to be unstable presumably due to disulfide bond scrambling.
Explanations of protein stabilization by disulfide bonds have generally been attributed to both entropic and enthalpic effects. One theory claims that a decrease in entropy of the unfolded state causing destabilization of this state compared with the folded state.42 Another theory claims it is a result of an increase in enthalpy for the folded state compared with the unfolded state as a result of burial of non-polar groups.43 No clear evidence of either of the theories has been found and the answer to the question is more likely a combination of the two depending on each protein sequence.44,45 Prior attempts to stabilize proteins have indicated that the highest degree of stabilization is achieved when disulfide bonds were introduced in regions with high flexibility.27 For all four 4SS-insulin analogues restriction of the otherwise very flexible N-terminal end of the B-chain was achieved. The degree of structural flexibility is increasing from the B4 to the B1 position in native HI so the degree of constrains introduced is increasing in the same order. It could therefore be speculated that the analogue with the highest stability was A10C-B1C. As evident by both DSC and CD this is not the case as stabilization appears to be going in the opposite direction increasing from B1 to B4. It could be speculated that formation of the disulfide bonds in A10C-B1C and A10C-B2C resulted in greater strain on the structure, as it had to be arranged in a dis-favorable conformation in order to facilitate bond formation thereby reducing the stability of the molecules. The identification of the A10C-B2C disulfide bond was only made after using a cut-off value of the Cβ-Cβ distance that were much higher than the optimal 5 Å and the A10-B1 disulfide bond was only identified when the trajectory method was used. In all cases the weighted scores for the prediction were high indicating that this position is not optimal. Our results contradict the theories that the highest stability is gained when the disulfide bond is introduced in the most flexible area indicating that this rule is protein-dependent.26
Insulin amyloid fibril formation has been thought to occur through an unfolding step of the two flexible terminals of the B-chain with emphasis on the C-terminus.4 Already with the first 4SS-insulin analogue we showed that reducing the flexibility of the N-terminus completely hindered fibril formation and these results were further supported by our current results as none of the 4SS-insulin analogues formed fibrils when vigorously shaken for 45 h.
In general the N-terminal part of the B-chain is not believed to be important for receptor binding as illustrated with desB1, desB1-2, and desB1-3 analogues (analogues lacking one, two or three amino acids in the B-chain N-terminus) retaining 89%, 86%, and 68% biological activity, respectively.46 It has been speculated that a conformational change from T- to R-state occurs upon binding.47 The three new 4SS-insulin analogues still retain between 38 and 70% of receptor binding affinity, i.e. picomolar affinity. This reduced binding is likely a result of the restricted flexibility and not their inability to form the R-state.17 The restriction of flexibility and changes in structure compared with HI is not equal for all four analogues even though their positions are located in close proximity to each other. Thus, albeit the new disulfide bonds are located away from positions known to be involved in receptor binding, the changes in structure can still affect the activity, which, in this case, result in lower binding affinities of the three new analogues compared with the first A10C-B4C analogue. This also provides evidence of the strong effect of the location of the bond not only on stability but also on activity.
The two methods, one predicting possible positions for introduction of disulfide bonds directly from the crystal structure and the other taking flexibility into account using a trajectory from MD simulations gave varying results. Based on these results, we propose the following strategy for disulfide bond prediction: (1) the method based on available crystal structures should indicate the best possible positions that are independent of flexibility in the local area of the protein structure. In our case this was the A10C-B3C analogue, which was identified with a low score and gave the highest yield and best stability. In this case A10C-B5C was also identified but appeared to have problems with disulfide scrambling possibly due to the proximity of the native disulfide bond in position A7-B7. These kinds of problems are not taken into account in the prediction tools; (2) the method based on MD simulations could be an advantage for disulfide bonds located in a flexible areas. The disulfide bond discovered before this work, A10C-B4C, was not found by the first method indicating that it was not a good position. It was clear from the crystal structure of this analogue that flexibility around the B4 position was necessary for its formation. Using the MD simulation this is taken into account and the A10C-B4C analogue got a good weighted score. This method also indicates that A10C-B6C disulfide bond should be formed, however, this analogue was not expressed. Thus introducing the flexibility could also give rise to false positives. A10C-B1C and A10C-B2C were expressed even though both were only predicted with high weighted scores despite changing default setting for distances and using the MD simulation. However, it was clear that these disulfide bonds did not result in the same high stabilization of the molecule as was seen for the other expressed and purified analogues. This is possibly due to a high amount of strain needed to form these disulfide bonds and also indicates that not all disulfide bonds lead to structure stabilization even if predicted by the algorithms. At last the prediction tool does not take activity into account which was also evident with the 4SS-insulin analogue and here it is necessary to utilize prior knowledge about function and activity when choosing position for disulfide bond formation.
Introduction of disulfide bonds into proteins with the goal of obtaining stabilized active analogues of a protein of interest is not straightforward. Small changes in location may result in large effect on both stability and activity as illustrated with the 4SS-insulin analogues. Although algorithms helping in selecting the best positions for novel disulfide bonds exist, in-depth knowledge about the molecule is essential for obtaining the best functional version of the protein.
Materials and Methods
Modeling
Protein preparation was done with the Protein Preparation Wizard in Maestro version 9.7 and 10.021,22 and disulfide predictions were carried out using the cysteine mutation panel within BioLuminate version 1.4 and 1.732 In order to identify possible disulfide bonds, different distance cut-off values between the beta carbons was used (5–15 Å). The selected mutants were refined with gas phase minimization. In order to identify possible disulfide bonds, both the crystal structure of human insulin (1MSO) and the trajectory from the molecular dynamics simulation were used. In the Protein Preparation Wizard the bond orders were assigned, hydrogen atoms added, disulfide bonds created, protonation state assigned with PROPKA at pH 7.0 and finally a restrained minimization was performed. If not stated otherwise the default cut-off of Cβ-Cβ distance was used. The molecular dynamics simulations were performed with Desmond version 4.0 and with OPLS2005 force field.22,28 The simulations were performed with the protein in a water box of 10 Å as buffer and sodium ions in order to neutralize the system. The simulations were performed at constant temperature (300 K) and pressure (1 bar). The thermostat method used was Nose-Hoover chain and the barostat method used was Martyna-Tobias-Klein. The cut-off for Coulombic interactions was 9.0 Å. The far time step size was 0.006 ps. The simulation was run for 100 ns.
Plasmids construction and expression
Material, vectors, strains, and construction were as previously described.24,48,49 Shortly, the two cysteine residues were introduced in selected positions in the coding sequence by overlapping PCRs. The insulin analogues were expressed as precursors in Saccharomyces cerevisiae consisting of a spacer GluGluAlaGluAlaGluAlaProLys (EEAEAEAPK) peptide followed by the B-chain (B1-B29), a mini C-peptide Ala Ala Lys (AAK) and lastly the A-chain (A1-A21). The expression yields of the insulin precursors were determined by reversed-phase HPLC (RP-HPLC) based on peak area with human insulin as external standard. The mass determined by MALDI as previous described.50
Purification
The precursors were fermented in continuous fermenters. The cells were removed from the culture supernatant by centrifugation and were then acidified. The precursor was partially purified and concentrated by cation exchange chromatography. The precursors were converted to mature insulin analogues lacking the B30 threonine by treatment with A. lyticus protease (Novo Nordisk A/S) as previously described.17 The analogues were further purified by RP-HPLC using a formic acid buffer system and lyophilised.
Circular dichroism spectroscopy
Far-UV and Near-UV CD spectra were recorded with a Jasco J-715 spectropolarimeter (Jasco Scandinavia AB) calibrated with (1S)-(þ)-camphor-10-sulfonic acid.
All samples were prepared in a concentration of 50 μM in a 10 mM phosphate buffer, pH 7.6. The samples were measured in the near-UV range (245–350 nm) using a 10 mm light path and in the far-UV range (200–260 nm) using a 1 mm light path. The samples were analyzed with a spectra recorded every 5 min starting with a temperature of 5 °C and increasing the temperature 1 °C per minute until 95 °C.
Receptor binding assay
Receptor binding was measured on the A isoform of the receptor in a scintillation proximity assay (SPA) as previously described.23 Briefly, the binding receptor affinities were determined by competition of the 4SS-insulin analogue and [125I]TyrA14-labelled insulin (Novo Nordisk A/S) in the SPA assay. The 4SS-insulin analogue (n=4) was tested together with a human standard (n = 4) in one plate. The data was analyzed according to a four-parameter logistic model51 and the affinities were expressed relative to the human insulin standard [IC50(insulin)/IC50(analogue) × 100%].
Metabolic potency determination
The metabolic potency determinations were done by lipogenesis essentially as described before.12 Shortly, primary rat adipocytes from SPRD rats were isolated and placed in a degradation buffer containing Hepes buffer, Krebs buffer, 0.1% HSA, collagenase, and glucose. The cells were shaken vigorously for 1 h at 37 °C and the cell suspensions were filtered, washed twice and resuspended in incubation buffer containing Hepes buffer, Krebs buffer and 0.1% HSA. 100 µL aliquots were distributed in 96-well PicoPlates and incubated 2 h at 37 °C with gentle shaking together with 10 µL glucose solution containing d-[3-3H]glucose and glucose and of increasing concentration of 10 µL insulin analogue. The incubation was stopped by adding 100 µL MicroScint E (Packard) and the plates were counted in a TopCount NXT (PerkinElmer Life Science). The data were analyzed according to a four-parameter logistic model45 and the metabolic potency were expressed relative to a HI standard [EC50(insulin)/EC50(analogue) × 100%].
Differential scanning calorimetry
The differential scanning calorimetry (DSC) measurements were performed essentially as described before.11 The insulin analogues were formulated at 0.2 mM in a 0.2 mM phosphate buffer pH 7.5 which were also used as reference buffer. The samples were heated from 10 °C to 110 °C with a scan rate of 60 °C per hour.
Thioflavin T fibrillation assay
The Thioflavin T (ThT) assay was performed as previously described.12 Briefly, the samples were prepared freshly. The experiment was performed at 37 °C and the plate with the samples was incubated for 10 min before the first measurement and then measured every 20 min for up to 45 h. Between each measurement, the plate was continuously shaken (960 rpm) and heated. Each shown time point is the mean of the four replicas with standard deviation error bars.
Acknowledgments
Berit Bergerud Hansen, Kirsten Vestergaard, Anne Ahrensberg Jensen, Louise Qvistgaard, Rikke Lill Rønhede, provided excellent technical assistance. All authors employed at Novo Nordisk own Novo Nordisk stocks.
Footnotes
The analogues will be named by the location of the 4th disulfide bond e.g. A10C-B4C for a disulfide bond between position ten in the A-chain and four in the B-chain for the rest of the article.
An insulin analogue with four disulfide bonds will be abbreviated an 4SS-insulin analogue.
References
- Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, Rao M, Ali MK, Riley LM, Robinson CA, Ezzati M. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Brange J, Langkjær L. Insulin structure and stability. Pharm Biotechnol. 1993;5:315–350. doi: 10.1007/978-1-4899-1236-7_11. [DOI] [PubMed] [Google Scholar]
- Brange J, Andersen L, Laursen ED, Meyn G, Rasmussen E. Toward understanding insulin fibrillation. J Pharm Sci. 1997;86:517–525. doi: 10.1021/js960297s. [DOI] [PubMed] [Google Scholar]
- Nicol DS, Smith LF. Amino-acid sequence of human insulin. Nature. 1960;187:483–485. doi: 10.1038/187483a0. [DOI] [PubMed] [Google Scholar]
- Ryle AP, Sanger F, Smith LF, Kitai R. The disulphide bonds of insulin. Biochem J. 1955;60:541–556. doi: 10.1042/bj0600541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EN, Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DM, Hubbard RE, Isaacs NW, Reynolds CD, Sakabe K, Sakabe N, Vijayan NM. The structure of 2Zn pig insulin crystals at 1.5 A resolution. Philos Trans R Soc Lond B Biol Sci. 1988;319:369–456. doi: 10.1098/rstb.1988.0058. [DOI] [PubMed] [Google Scholar]
- Bentley G, Dodson E, Dodson G, Hodgkin D, Mercola D. Structure of insulin in 4-zinc insulin. Nature. 1976;261:166–168. doi: 10.1038/261166a0. [DOI] [PubMed] [Google Scholar]
- Derewenda U, Derewenda Z, Dodson EJ, Dodson GG, Reynolds CD, Smith GD, Sparks C, Swenson D. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature. 1989;338:594–596. doi: 10.1038/338594a0. [DOI] [PubMed] [Google Scholar]
- Brange J, Havelund S, Hommel E, Sørensen E, Kühl C. Neutral insulin solutions physically stabilized by addition of Zn2+ Diabet Med. 1986;3:532–536. doi: 10.1111/j.1464-5491.1986.tb00809.x. [DOI] [PubMed] [Google Scholar]
- Huus K, Havelund S, Olsen HB, van de Weert M, Frokjaer S. Thermal dissociation and unfolding of insulin. Biochemistry. 2005;44:11171–11177. doi: 10.1021/bi0507940. [DOI] [PubMed] [Google Scholar]
- Vinther TN, Norrman M, Strauss HM, Huus K, Schlein M, Pedersen TA, Kjeldsen T, Jensen KJ, Hubalek F. Novel covalently linked insulin dimer engineered to investigate the function of insulin dimerization. PLoS One. 2012;7:e30882. doi: 10.1371/journal.pone.0030882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SJ, Cao QP, Steiner DF. Evolution of the insulin superfamily: cloning of a hybrid insulin/insulin-like growth factor cDNA from amphioxus. Proc Natl Acad Sci USA. 1990;87:9319–9323. doi: 10.1073/pnas.87.23.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SG, Choi KD, Jang SH, Shin HC. Role of disulfide bonds in the structure and activity of human insulin. Mol Cells. 2003;16:323–330. [PubMed] [Google Scholar]
- Guo ZY, Feng YM. Effects of cysteine to serine substitutions in the two inter-chain disulfide bonds of insulin. Biol Chem. 2001;382:443–448. doi: 10.1515/BC.2001.054. [DOI] [PubMed] [Google Scholar]
- Katsoyannis PG, Okada Y, Zalut C. Synthesis of a biologically active insulin analog lacking the intrachain cyclic system. Biochemistry. 1973;12:2516–2525. doi: 10.1021/bi00737a023. [DOI] [PubMed] [Google Scholar]
- Vinther TN, Norrman M, Ribel U, Huus K, Schlein M, Steensgaard DB, Pedersen TA, Pettersson I, Ludvigsen S, Kjeldsen T, Jensen KJ, Hubalek F. Insulin analog with additional disulfide bond has increased stability and preserved activity. Protein Sci. 2013;22:296–305. doi: 10.1002/pro.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Pangborn WA, Blessing RH. The structure of T6 human insulin at 1.0 A resolution. Acta Crystallogr D. 2003;59:474–482. doi: 10.1107/s0907444902023685. [DOI] [PubMed] [Google Scholar]
- Biologics Suite. New York Schrödinger 2014–1: BioLuminate, Version 1.4, LLC; 2014.
- Biologics Suite 2014–1: BioLuminate, Version 1.7. New York Schrödinger, LLC; 2014.
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. 2013. New York Schrödinger Schrödinger Release 2014–1: Maestro, Version 9.7 , LLC; 2014.
- Schrödinger Release 2014–1: Maestro, Version 9.7. New York Schrödinger, LLC; 2014.
- Glendorf T, Sørensen AR, Nishimura E, Pettersson I, Kjeldsen T. Importance of the solvent-exposed residues of the insulin B chain alpha-helix for receptor binding. Biochemistry. 2008;47:4743–4751. doi: 10.1021/bi800054z. [DOI] [PubMed] [Google Scholar]
- Kristensen C, Kjeldsen T, Wiberg FC, Schäffer L, Hach M, Havelund S, Bass J, Steiner DF, Andersen AS. Alanine scanning mutagenesis of insulin. J Biol Chem. 1997;272:12978–12983. doi: 10.1074/jbc.272.20.12978. [DOI] [PubMed] [Google Scholar]
- Pullen RA, Lindsay DG, Wood SP, Tickle IJ, Blundell TL, Wollmer A, Krail G, Brandenburg D, Zahn H, Gliemann J, Gammeltoft S. Receptor-binding region of insulin. Nature. 1976;259:369–373. doi: 10.1038/259369a0. [DOI] [PubMed] [Google Scholar]
- Schäffer L. A model for insulin binding to the insulin receptor. Eur J Biochem. 1994;221:1127–1132. doi: 10.1111/j.1432-1033.1994.tb18833.x. [DOI] [PubMed] [Google Scholar]
- Dani VS, Ramakrishnan C, Varadarajan R. MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 2003;16:187–193. doi: 10.1093/proeng/gzg024. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2014–1: Desmond Molecular Dynamics System, Version 3.7. New York: D.E.Shaw Research, 2014. Maestro-Desmond Interoperability Tools, Version 3.7. New York: Schrödinger, 2014.
- Sowdhamini R, Srinivasan N, Shoichet B, Santi DV, Ramakrishnan C, Balaram P. Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein Eng Des Sel. 1989;3:95–103. doi: 10.1093/protein/3.2.95. [DOI] [PubMed] [Google Scholar]
- Bjørk A, Dalhus B, Mantzilas D, Eijsink VG, Sirevåg R. Stabilization of a tetrameric malate dehydrogenase by introduction of a disulfide bridge at the dimer-dimer interface. J Mol Biol. 2003;334:811–821. doi: 10.1016/j.jmb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Clarke J, Henrick K, Fersht AR. Disulfide mutants of barnase. I: Changes in stability and structure assessed by biophysical methods and X-ray crystallography. J Mol Biol. 1995;253:493–504. doi: 10.1006/jmbi.1995.0568. [DOI] [PubMed] [Google Scholar]
- Das M, Kobayashi M, Yamada Y, Sreeramulu S, Ramakrishnan C, Wakatsuki S, Kato R, Varadarajan R. Design of disulfide-linked thioredoxin dimers and multimers through analysis of crystal contacts. J Mol Biol. 2007;372:1278–1292. doi: 10.1016/j.jmb.2007.07.033. [DOI] [PubMed] [Google Scholar]
- Guzzi R, Andolfi L, Cannistraro S, Verbeet MP, Canters GW, Sportelli L. Thermal stability of wild type and disulfide bridge containing mutant of poplar plastocyanin. Biophys Chem. 2004;112:35–43. doi: 10.1016/j.bpc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Le QA, Joo JC, Yoo YJ, Kim YH. Development of thermostable Candida antarctica lipase B through novel in silico design of disulfide bridge. Biotechnol Bioeng. 2012;109:867–876. doi: 10.1002/bit.24371. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Becktel WJ, Levitt M, Matthews BW. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc Natl Acad Sci USA. 1989;86:6562–6566. doi: 10.1073/pnas.86.17.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoliano MW, Ladner RC, Bryan PN, Rollence ML, Wood JF, Poulos TL. Protein engineering of subtilisin BPN: enhanced stabilization through the introduction of two cysteines to form a disulfide bond. Biochemistry. 1987;26:2077–2082. doi: 10.1021/bi00382a002. [DOI] [PubMed] [Google Scholar]
- Robinson CR, Sauer RT. Striking stabilization of Arc repressor by an engineered disulfide bond. Biochemistry. 2000;39:12494–12502. doi: 10.1021/bi001484e. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Hehir K, Stearman RS, Weiss MA, Jeitler-Nilsson A, Suchanek EG, Pabo CO. An engineered intersubunit disulfide enhances the stability and DNA binding of the N-terminal domain of lambda repressor. Biochemistry. 1986;25:5992–5998. doi: 10.1021/bi00368a024. [DOI] [PubMed] [Google Scholar]
- Dombkowski AA. Disulfide by design: a computational method for the rational design of disulfide bonds in proteins. Bioinformatics. 2003;19:1852–1853. doi: 10.1093/bioinformatics/btg231. [DOI] [PubMed] [Google Scholar]
- Salam NK, Adzhigirey M, Sherman W, Pearlman DA. Structure-based approach to the prediction of disulfide bonds in proteins. Protein Eng Des Sel. 2014;27:365–374. doi: 10.1093/protein/gzu017. [DOI] [PubMed] [Google Scholar]
- Craig DB, Dombkowski AA. Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14:346. doi: 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory PJ. Theory of elastic mechanisms in fibrous proteins. J Am Chem Soc. 1956;78:5222–5235. [Google Scholar]
- Doig AJ, Williams DH. Is the hydrophobic effect stabilizing or destabilizing in proteins? The contribution of disulphide bonds to protein stability. J Mol Biol. 1991;217:389–398. doi: 10.1016/0022-2836(91)90551-g. [DOI] [PubMed] [Google Scholar]
- Betz SF. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Sternberg MJ. Analysis and classification of disulphide connectivity in proteins. The entropic effect of cross-linkage. J Mol Biol. 1994;244:448–463. doi: 10.1006/jmbi.1994.1742. [DOI] [PubMed] [Google Scholar]
- Kerp L, Steinhilber S, Kasemir H, Han J, Henrichs HR, Geiger R. Changes in immunospecificity and biologic activity of bovine insulin due to subsequent removal of the amino acids B1, B2, and B3. Diabetes. 1974;23:651–656. doi: 10.2337/diab.23.8.651. [DOI] [PubMed] [Google Scholar]
- Nakagawa SH, Zhao M, Hua QX, Hu SQ, Wan ZL, Jia W, Weiss MA. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry. 2005;44:4984–4999. doi: 10.1021/bi048025o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen T, Brandt J, Andersen AS, Egel-Mitani M, Hach M, Pettersson AF, Vad K. A removable spacer peptide in an alpha-factor-leader/insulin precursor fusion protein improves processing and concomitant yield of the insulin precursor in Saccharomyces cerevisiae. Gene. 1996;170:107–112. doi: 10.1016/0378-1119(95)00822-5. [DOI] [PubMed] [Google Scholar]
- Kjeldsen T, Pettersson AF, Hach M. The role of leaders in intracellular transport and secretion of the insulin precursor in the yeast Saccharomyces cerevisiae. J Biotechnol. 1999;75:195–208. doi: 10.1016/s0168-1656(99)00159-5. [DOI] [PubMed] [Google Scholar]
- Vinther TN, Ribel U, Askov PT, Kjeldsen TB, Jensen KJ, Hubalek F. Identification of anchor points for chemical modification of a small cysteine-rich protein by using a cysteine scan. ChemBioChem. 2011;12:2448–2455. doi: 10.1002/cbic.201100464. [DOI] [PubMed] [Google Scholar]
- Vølund A. Application of the four-parameter logistic model to bioassay: comparison with slope ratio and parallel line models. Biometrics. 1978;34:357–365. [PubMed] [Google Scholar]