

Abstract
Actinonin is a pseudotripeptide that displays a high affinity towards metalloproteases including peptide deformylases (PDFs) and M1 family aminopeptidases. PDF and M1 family aminopeptidases belong to thermolysin-metzincin superfamily. One of the major differences in terms of substrate binding pockets between these families is presence (in M1 aminopeptidases) or absence (in PDFs) of an S1 substrate pocket. The binding mode of actinonin to PDFs has been established previously; however, it is not clear how the actinonin, without a P1 residue, would bind to the M1 aminopeptidases. Here we describe the crystal structure of Escherichia coli aminopeptidase N (ePepN), a model protein of the M1 family aminopeptidases in complex with actinonin. For comparison we have also determined the structure of ePepN in complex with a well-known tetrapeptide inhibitor, amastatin. From the comparison of the actinonin and amastatin ePepN complexes, it is clear that the P1 residue is not critical as long as strong metal chelating head groups, like hydroxamic acid or α-hydroxy ketone, are present. Results from this study will be useful for the design of selective and efficient hydroxamate inhibitors against M1 family aminopeptidases.
Keywords: actinonin, amastatin, M1 aminopeptidase, peptide deformylase, matrix metalloproteinase
Introduction
Actinonin is a natural antibiotic produced by the Gram-positive actinobacterium Streptomyces roseopallidus. The compound is a pseudotripeptide hydroxamate inhibitor derived from L-prolinol, a known inhibitor of the peptide deformylase (PDF) family of proteins (Fig. 1).1 In bacteria, PDF proteins are emerging as attractive antibacterial targets.1 A derivative of actinonin, GSK1322322, has recently completed Phase-II clinical trials as a potential new antimicrobial agent.2 In humans, actinonin was also shown to target the mitochondrial PDF, offering a potential route to new cancer therapeutics.3,4 At least 30 PDF enzymes from various species have been crystallized in complex with actinonin and reveal that the hydroxamate group acts to chelate the metal binding group found in PDF proteins.5,6 PDF proteins, unlike classical proteases, do not possess a typical S1 substrate-binding pocket.7 Crystal structures of PDF in complex with actinonin show that the P1' n-pentyl side chain binds in the S1' pocket.8
Figure 1.

Schematic representation of chemical composition of actinonin and amastatin. The side chains are labeled as per the sub-site occupancy in the enzyme pocket.
The PDF proteins belong to the thermolysin-metzincin superfamily and are “clan MA and MB” metalloproteases.9,10 Both MA and MB proteases contain a HEXXH motif in which two histidine residues coordinate the zinc and the conserved glutamate is required for catalytic function.7 The HEXXH motif is common in peptidases and can be found in the M9A neuron-specific aminopeptidases,11 M12 meprin alpha and beta subunits,12 M17 family of leucyl aminopeptidases (EC 3.4.11.1), and the M1 class aminopeptidases.13 The M1 aminopeptidases that are sensitive to the actinonin include CD13/Aminopeptidase N (APN/PepN)14 and puromycin sensitive aminopeptidase (PSA).15 Actinonin significantly blocks CD13 activity, resulting in the migration of eosinophils across HUVEC monolayers16 and PSA is known to have an important role in the neuromodulation within the central nervous system and in reproductive biology.15
M1 class aminopeptidases are multi-domain enzymes with a thermolysin like catalytic domain with the buried and a well-defined S1 pocket that plays an important role in enzyme specificity and inhibitor affinity.17–19 M1 aminopeptidases generally prefer peptide substrates of four to six amino acids in length that contain a hydrophobic or basic amino acid at the P1 position.17,19–22 Actinonin effectively lacks a P1 residue and therefore its mechanism of action upon the M1 aminopeptidases is difficult to predict. Here we describe the first crystal structure of actinonin in complex with aminopeptidase N from Escherichia coli (ePepN) and compare its' binding mode with a classical and well-characterized peptidomimetic, amastatin. We further investigated the inhibitory properties of both actinonin and amastatin against four members of M1 family enzymes from different species. Structure-activity relationships (SAR) from these studies have established a structural and biochemical basis for activity and specificity of actinonin towards the M1 aminopeptidase family.
Results and Discussion
Actinonin, a pseudotripeptide, interacts with the PDF-active site zinc via its hydroxamate head group.7 Inhibition of microbial PDFs can kill gram-positive bacteria and therefore there is much interest in the pharmacological properties of actinonin and its derivatives.23–25 In addition to PDF enzymes, actinonin also inhibits the M1 family aminopeptidases and some Matrix Metalloproteinases (MMPs) in the nanomolar range.26 In contrast to the PDFs, M1 aminopeptidases have well-defined S1 pocket that is buried in the enzyme structure.27 To date, the lack of the crystal structure of actinonin in the complex with any of the M1 family aminopeptidases has limited our understanding of the molecular basis for inhibition. The M1 family aminopeptidases are important enzymes that can be found in all living cells.28,29 If actinonin and its derivatives are to be considered as potential new antibacterial drug candidates, attention should be given to the possibility of off-target effects through inhibition of other important enzymes, specifically other metallopeptidases. In our current study, we have provided the structural and biochemical basis for the inhibition of M1 family aminopeptidases by actinonin.
ePepN in complex with actinonin
X-ray diffraction data were collected on the ePepN-actinonin complex at 1.9 Å resolution. The overall structure was similar to that of the wild type enzyme (0.26 Å r.m.s.d. over 870 Cα-atoms). There were no gross structural changes observed when compared with the unbound wild-type enzyme (PDB ID: 2HPO). The position of the Cα-atoms within the active site were also unchanged. Several side chains of active site residues (Met260, Tyr275, Arg293, Glu382, Arg783, and Arg825) showed rearrangement to accommodate the inhibitor.
The actinonin binds to the enzyme through its hydroxamate head group, interacting with the active site zinc in a bidentate mode (Fig. 2). Strong electron density was observed for the entire actinonin molecule, which adopts a relatively linear conformation [Fig. 2(a)]. The hydroxamate group orients perpendicular to the direction of the rest of the peptide chain. In addition to binding to the zinc ion in the active site, the hydroxamate also makes a series of hydrogen bonds with proximal side chains. Glu298 forms strong hydrogen bonds with hydroxyl and the amine groups [Fig. 2(b,c)]. On the other side, the Tyr381 interacts with the carbonyl portion of the head group. The hydroxamate group has been proposed to represent the reaction intermediate during the peptide hydrolysis by metalloenzymes.30 Glu298 and Tyr381 have also been proposed to play critical roles in the catalytic mechanism of ePepN enzyme, where Glu298 activates the nucleophilic water to attack the scissile peptide bond and Tyr381 stabilizes the oxyanion tetrahedral transition state.27,31
Figure 2.

Crystal structure of actinonin in complex with ePepN at 1.9 Å. a) 2Fo-Fc = 1.1 σ electron density map representation around the zinc bound actinonin. Molecule binds in the extended conformation making two strong interactions with a zinc ion. A cover radius of 2.0 Å was used during the map generation. b) Stereo representation of the actinonin bound in the enzyme pocket with surrounding residues. Note that hydroxamate head group makes a series of hydrogen bonds in the active site. c). A two-dimensional schematic representation of all residues surrounding the actinonin in LigPlot diagram.45 Apart from hydrogen bonds, several hydrophobic interactions are noticed between the ligand and the protein. An interactive view is available in the electronic version of the article
The active site of the M1 family aminopeptidases is described by a well-defined cylindrical S1 pocket that leads into a larger cavity, which accommodates the rest of the binding site responsible for substrate recognition.27 Actinonin lacks a P1 side chain. The head group hydroxamate is the only region that occupies the S1 pocket while the rest of the molecule binds in the large cavity of active site. The n-pentyl side chain in the P1' position sits in a deep and relatively hydrophobic pocket in an extended conformation [Fig. 2(b)]. The carbonyl in the peptide bond that links the P1' and P2' (Val) residues forms a hydrogen bond with the amide nitrogen of Gly261 [Fig. 2(c)]. The prolinol ring at the P3' position in actinonin makes hydrophobic contacts with Met260, Gly261 and Tyr275. Due to these hydrophobic interactions, the prolinol is well ordered. The Met260 side chain is usually disordered in the absence of any substrate or inhibitor in the S1 pocket.27 Here, it is noticed that the side chain of Met260 swings out into the interface of P2' (Val) and P3' (prolinol) side chains of the actinonin providing the much-needed hydrophobic surface for the inhibitor (Fig. 2). Apart from Met260, several other generally disordered residues (specifically Arg783 and Arg825) attain an ordered conformation upon binding of actinonin.
ePepN in complex with amastatin
Amastatin and bestatin are peptidomimetic inhibitors that inhibit M1 aminopeptidases.32 Since actinonin lacks a P1 residue, we determined the crystal structure of the ePepN-amastatin complex at 2.3 Å resolution to compare active site interactions between the two inhibitors (Fig. 3). The hydroxyl group (O2) and the carbonyl group (O3) interact with the zinc ion increasing its coordination to five.22,27 Like in the bestatin structure (PDB ID: 2HPT), the α-amino group positions itself into a negatively charged depression formed by Glu121, Glu264, and Glu320 forming hydrogen bonds with all three carboxylates (Fig. 3). The P1 leucine side chain is placed in the S1 pocket with Met260 forming a hydrophobic cushion at the top. Other residues that contribute to the hydrophobic nature of the S1 pocket are Met263, and the Cβ and Cγ atoms of Glu121. Note that Met263 is part of the G261AMEN substrate specificity motif common to all M1 class aminopeptidases.33 Similar to that found in the bestatin structure, the O3 of amastatin, apart from interacting with the zinc ion, also forms a hydrogen bond (2.7 Å) with the hydroxyl group of Tyr38127 [Fig. 3(b,c)]. Good density was observed for the P1' (Val) and P2' (Val) side chains occupying their respective enzyme pockets. The peptide carbonyl between P1 and P1' residues makes a hydrogen bond (2.7 Å) with Gly261 amide nitrogen. The P3' residue is an aspartic acid surrounded by Lys274, Tyr275 and Arg825. Both the carboxylates make decent hydrogen bonds with the enzyme pocket [Fig. 3(c)].
Figure 3.

Crystal structure of amastatin in complex with ePepN at 2.3 Å resolution. a). 2Fo-Fc Electron density map surrounding the amastatin and the zinc in the active site at 1.1 σ. Similar to actinonin, amastatin forms two coordinate bonds with the active site zinc ion. A cover radius of 2.0 Å was used during the map generation. b). Extended conformation of the amastatin is represented in the stereo diagram. Amastatin makes several hydrogen bonds with residues that surround it. c). LigPlot diagram of amastatin (green sticks) in the active site of ePepN. Note that the orientation of the amastatin in this panel is rotated by 180° compared with that in (b). Large network of hydrogen bonds were noticed between the ligand and the protein. An interactive view is available in the electronic version of the article
Comparison between actinonin and amastatin bound ePepN structures
Both actinonin and amastatin molecules possess metal chelating groups that enable them to bind strongly to the M1 family aminopeptidases with inhibition in the nanomolar range (Table1). Both molecules adopt a linear and extended conformation in the ePepN complex (Fig. 4). The r.m.s. deviation of 0.25 Å for 866 Cα-atoms were observed between the two structures.34 The P1' side chain of the actinonin is linear and longer compared with the valine in the amastatin. In both inhibitor complexes the P2' residue is a valine and their side chains are placed about 2 Å apart. Neither side chain position makes a good interaction with protein atoms. This suggests an ill-defined S2' pocket. The terminal P3' residues of each compound are different (prolinol in the actinonin vs aspartic acid in the amastatin); however, occupy the same sub-site making several but non-identical contacts with the protein [Figs. 2(c) and 3(c)]. Overall, amastatin makes four hydrophilic interactions in the form of hydrogen bonds than actinonin molecule. Due to these strong interactions, amastatin binds with higher affinity to ePepN compared with actinonin and would account for the increased potency of this compound (Table1).
Table 1.
Enzyme Inhibition (IC50 Value in µM)
| Enzyme | Class | Actinonin | Amastatin |
|---|---|---|---|
| ePepN | M1 aminopeptidase | 0.19 ± 0.04 | 0.047 ± 0.008 |
| PmAPN | M1 aminopeptidase | 0.29 ± 0.06 | 0.53 ± 0.02 |
| hPSA | M1 aminopeptidase | 0.09 ± 0.01 | 0.28 ± 0.01 |
| PfAPN | M1 aminopeptidase | 2.46 ± 0.39 | 0.81 ± 0.12 |
| hMMP-7 | Matrix metalloproteinase | 2.10 ± 0.002 | >100 |
ePePN—E. coli aminopeptidase N, PmAPN—porcine microsomal aminopeptidase N, hPSA—human puromycin sensitive aminopeptidase N, PfAPN—Plasmodium falciparum aminopeptidase N, hMMP-7—human matrix metalloprotease-7.
Aminopeptidase controls included Mycobacterium tuberculosis methionine aminopeptidase 1c and hMetAP1b- human methionine aminopeptidase 1b. Nonmetallo enzyme controls were performed using human dihydrofolate reductase. No inhibitory activity was noted for any control up to 100 µM of compound.
Figure 4.

Superposition of actinonin (brown sticks) and amastatin (green sticks) bound ePepN structures near the active site are depicted (0.25 Å rmsd between two structures). Except for the P2' residue (valine in both structures), most parts of ligand align well. The presence or absence of P1 residue side chain does not seem to affect the mode of binding of ligands in this case.
Comparison of the actinonin bound E. coli PDF and ePepN
PDFs and M1 class aminopeptidases both have the metzincin motif that holds the zinc in the active site. A structural alignment was performed, using the catalytic domain of ePepN (residues 200–444) and the entire structure of ePDF, centering on actinonin and the two conserved zinc-binding histidines. This alignment produced an r.m.s.d. of 0.4 Å (Fig. 5). The helix (H1) that carries the HEXXH motif aligns well in both proteins. The third metal-binding residue to coordinate with the zinc is a glutamate in ePepN and a cysteine in ePDF (ref for ePDF). In both structures, the active site zinc ion adopts penta-coordination with two positions contributed by the actinonin. The position of the P1' and P2' residues align well in both structures; however, the P3' residue assumes a different conformation [Fig. 5(b)]. It is generally observed that in PDFs, substrate specificity is limited only to the S1' pocket and there are no defined S2' and S3' subsites.7
Figure 5.
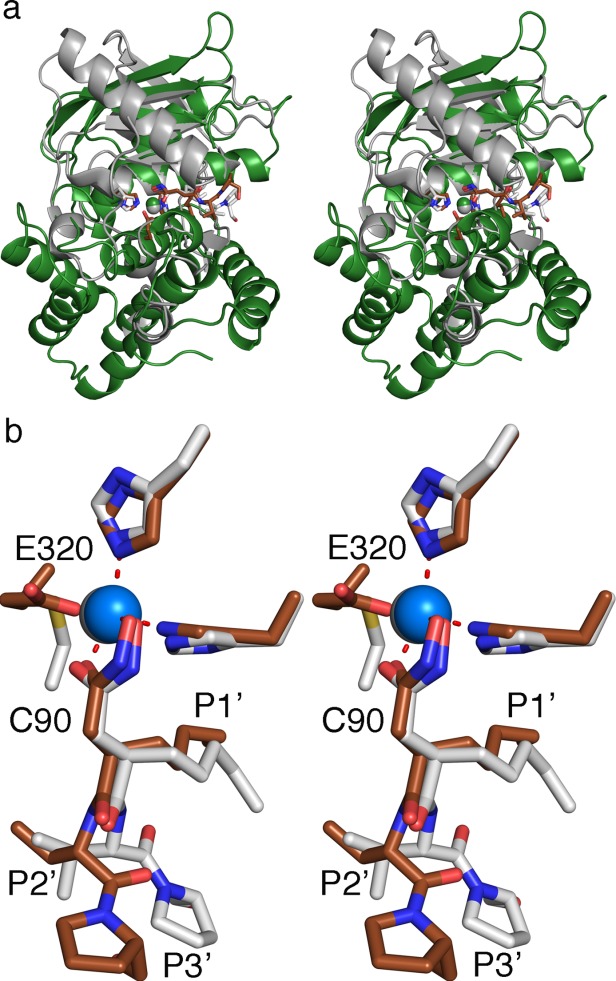
Structural alignment of catalytic domain of ePepN (200–444 residues) and ePDF in complex with actinonin. a). Cartoon representation of ePepN (green) and ePDF (white). The actinonin is shown in the sticks (brown in the ePepN complex while it is white in ePDF structure). Note that the core of the protein structure aligns well. b). Stick representation of actinonin from both the structure in the active site. Molecular trajectory deviates from P2' residue and becomes maximal at P3'.
Biochemical studies of actinonin and amastatin against other M1 aminopeptidases
To understand the behavior of actinonin and amastatin against various metalloenzymes, we have tested their activity against four different M1 enzymes (Table1). The inhibitory properties of either amastatin or actinonin were largely nonselective against the M1 enzymes with all enzymes showing activity in the nanomolar range. Amastatin was a more potent inhibitor than actinonin of both ePepN and PfAPN; however, actinonin show higher affinity and selectivity against hPSA than either the bacterial or parasitic proteins (Table1). The activity of ePepN and PmAPN were comparable to each other for both inhibitors.
We also tested another metzincin superfamily member, hMMP-7, and found that it was also inhibited by the actinonin but surprisingly, not by amastatin (Table1). The potency of the compounds for hMMP-7 is not comparable to the M1 aminopeptidases due to the increased level of hMMP-7 required for the assay; however, the results show that actinonin is a good inhibitor of this protein. For further assay controls we also tested other aminopeptidases, namely a human and Mycobacterium methionine aminopeptidase, and also human dihydrofolate reductase enzyme as a nonmetallo enzyme control. No inhibition was noted for either compound against any of these enzymes (IC50 > 100 µM). Previously it was demonstrated that both actinonin and amastatin inhibited the MetAP from Mycobacterium smegmatis.35 However, we did not observe such behavior under our current assay conditions.
From these results, it does not therefore seem to matter if the P1 residue is present or not for the overall efficiency in the inhibition of M1 class aminopeptidases. From our biochemical data it is clear that the metal binding groups dictate affinity of the inhibitors. Finer differences due to the sub-site specificities of different side chains on the inhibitors appear to contribute little to potency. hMMP-7 was inhibited by actinonin but not by amastatin. As per the substrate specificity defined previously for the hMMP-7, glycine is the preferred residue at the P1 position while the leucine is least preferred.35 We hypothesize that the selective preference of actinonin binding to hMMP-7 can be explained by the actinonin hydroxamate binding in the S1 pocket acting as a glycine mimic (in terms of the size) thereby showing high affinity to hMMP-7. However, the leucine in the P1 position of amastatin will face resistance from the S1 pocket.
Material and Methods
Materials
Actinonin, amastatin, porcine microsomal aminopeptidase N, and substrates for enzyme assays including l-leucine-p-nitroanilide (Leu-pNA), l-methionine-p-nitroanilide (Met-pNA), dihydrofolate, NADPH, p-aminophenylmercuric acetate (APMA), (7-methoxycoumarin-4-yl)acetyl-l-Pro-l-Leu-Gly-l-Leu-[N3-(2,4-dinitrophenyl)-l−2,3-diaminopropionyl]-l-Ala-l-Arg-NH2 [MOCAc-PLGL(Dpa)AR], His select HF Nickel affinity resin and other chemicals used in this study were obtained from Sigma Aldrich (St. Louis).
Expression and purification of proteins
Protein expression and purification methods are described below. All the enzyme reactions were performed in triplicate on the microplate multimode reader (Tecan, Austria).
Cloning, expression, and purification of human PSA
The human PSA (hPSA) gene was cloned into the pET-41 EK/LIC vector with the N-terminal GST tag, as described earlier.21 Enzyme activity assays were carried out in 10 mM Tris-HCl buffer at pH 7.5 with 150 mM NaCl, 75 nM PSA enzyme, 100 µM substrate Leu-pNA in a 100 μL reaction at 37°C. The absorbance was read at 405 nm.
Expression, purification, and enzyme activity of PfAPN
Plasmodium falciparum aminopeptidase N (PfAPN) gene was cloned from genomic DNA of P. falciparum (kind gift from Puran Singh Sijwali, CSIR-Center for Cellular and Molecular Biology, Hyderabad, India) into pET-28a (Novagen, Germany) vector using the restriction enzymes EcoRI and HindIII (New England Biolabs, Ipswich, MA). The protein was expressed in BL21 (DE3) cells (Novagen) using ampicillin selection. The expression and purification protocols were as described for ePepN.36
Enzyme assays were performed in 10 mM Tris-HCl, pH 7.5 buffer in the presence of 150 mM NaCl, 20 nM of enzyme, and 100 µM substrate (Leu-pNA) in a 100 µL reaction at 37°C. The absorbance was monitored at 405 nm.
Cloning, expression, purification, and enzyme activity of hDHFR
Protein expression and purification methods were as described previously.37 Enzyme activity was carried out in a reaction volume of 100 μL containing 20 mM Tris-HCl buffer, pH 7.5, 20 nM hDHFR, 11.25 μM dihydrofolate, 21.5 μM NADPH at 22°C. The conversion of NADPH to NADP+ was monitored at 340 nm.
Cloning, expression, refolding, and enzyme activity of human matrix metalloproteinase 7 (hMMP-7)
A truncated form of the hMMP-7 that lacks the N-terminal 17 amino acids (and consequently the signal peptide) was cloned from A549 cells (adenocarcinomic human alveolar basal epithelial cells) cDNA into the pET15b (Novagen, Darmstadt, Germany) vector using NdeI and BamHI (New England Biolabs, Ipswich, MA). The 28 kDa protein was overexpressed in BL21 (DE3) E. coli cells at 37°C until the O.D600 = 1.0 before addition of 1 mM IPTG. Protein induction proceeded for a further 4 h. The harvested cell pellet was resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 250 mM NaCl and 1% Triton X-100, 10% glycerol, 5 mg lysozyme, 5 mg DNase I, 5 mM MgCl2, and 1 mM PMSF), sonicated and centrifuged (34,541g for 30 min). Supernatant was discarded. The pellet was further washed with wash buffer (2 mM Urea, 20 mM Tris-HCl, pH 8.0, 1% Triton X-100, 15 mM EDTA), sonicated and the inclusion bodies (IB) were separated by centrifugation. IB (500 mg) was solubilized in 1.5 mL of solubilization buffer (0.5M Tris-HCl, pH 7.5, 6M guanidine-HCl) and the solution was diluted with 150 mL of refolding buffer (50 mM Tris-HCl, pH 7.5 0.1 mM ZnCl2, 0.2M NaCl, 1% Brij). The refolded protein solution was centrifuged at 34,541g to remove precipitated protein. The soluble protein was dialyzed into 50 mM Tris-HCl, pH 7.5, 0.2M NaCl, 10 mM CaCl2 buffer at 4°C. Aliquots of the resulting pure protein were stored at −80°C.
Enzyme activity was carried out with 5 μM enzyme after activating with 16 μM APMA (p-aminophenylmercuric acetate) and 1 μM fluorogenic peptide substrate MOCAc-PLGL(Dpa)AR in a 100 μL buffer (50 mM Tris-HCl, pH 7.5, 0.2M NaCl, 10 mM CaCl2) at 37°C and the fluorescence was monitored at excitation and emission maxima of 328 nm, 393 nm respectively.
Porcine microsomal aminopeptidase N (PmAPN)
PmAPN was purchased from Sigma Aldrich (Cat. No. L9776). The 100 µL reaction buffer containing 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 20 nM of PmAPN and incubated at 37°C for 30 min. The reaction was initiated by adding 100 µM of Leu-pNA, while monitoring increase in absorption at 405 nm.
Human methionine aminopeptidase 1b
HsMetAP1b was purified as described earlier.38 The assay was performed in 100 µL solution composed of 25 mM Hepes, pH 8.0, 100 mM KCl, 6 µM of enzyme, 3 molar equivalents of cobalt chloride and 200 µM substrate (Met-pNA) at 30°C. The absorbance was monitored at 405 nm.
Mycobacterium tuberculosis methionine aminopeptidase 1c
Activity of MtMetAP1c was performed in a reaction volume of 100 µL of 25 mM Hepes, pH 7.5, 150 mM KCl, 4 µM enzyme, 150 µM cobalt chloride, and 100 µM of the inhibitor (actinonin or amastatin). The reaction was incubated for 30 min and the reaction was monitored continuously at 405 nm, after adding 200 µM Met-pNA.
Determination of IC50 values
Concentrations ranging from 1 nM to 100 μM of actinonin and amastatin were incubated with each of the enzymes for 30 min at 37°C in a 96-well plate. After addition of the substrate, immediate increase (hPSA, PmAPN, ePepN, PfAPN, MtMetAP1c, hMetAP1b) or decrease (hDHFR) in absorption was measured as described above. Enzymatic assays for hMMP-7 were measured as reported earlier.39 All inhibition assays were performed in triplicates and initial velocities were measured. Percentage inhibition and log values of inhibitor concentrations were plotted using Sigma Plot to determine IC50 values (Table1).
Crystallization and data collection
Crystals were obtained by mixing 3 μL of the enzyme (10 mg/mL) with 3 μL of well solution (2.0M sodium malonate at pH 7.5) in a hanging drop and equilibrating it against the well solution for three to five days at 25°C. A single crystal (in 5 µL) was soaked with 0.5 µL of 1 mM actinonin dissolved in DMSO, incubated for 1 h at 25°C. Then the crystal was cryopreserved under the stream of liquid nitrogen for data collection on Rigaku MicroMax-007HF X-ray generator using R-axis IV++ image plate. Powder of the amastatin was added directly to the drop with crystals in the cryoprotectant solution (22% glycerol, 2.0M sodium malonate, pH 7.5) to obtain a final concentration of 2 mM and incubated overnight at 25°C. A single crystal was cryopreserved in liquid nitrogen and data collected using synchrotron radiation at the 8.2.2 beam line at the Advanced Light Source (ALS, Lawrence Berkeley National Laboratory, Berkeley, CA) using their Quantum 315 ADSC Area Detector.
Structure determination and refinement
X-ray data were processed and scaled using HKL-3000.40 Both the structures were isomorphous with the apo structure in space group P3121 [PDB ID: 2HPO]. Fo-Fo maps for each ligand bound structure using the apo enzyme structure factors from 2HPO clearly demonstrated the presence of inhibitors [Supporting Information Fig. 1(a,b)].41 Structure refinement was performed using Refmac 5 in CCP4 program suite, and COOT was used to visualize the model.42,43 Stereochemical restraints for both the ligands were available within the Refmac 5 ligand dictionary. X-ray data and refinement statistics are summarized for each structure in the Table2. Pymol was used for preparing final figures.44
Table 2.
X-Ray Data Collection and Refinement Statistics
| Cell parameters | ePepN-actinonin | ePepN-amastatin |
|---|---|---|
| Space group | P3121 | P3121 |
| a, b (Å) | 120.45 | 119.39 |
| c (Å) | 170.75 | 170.13 |
| Data collection | ||
| Wavelength (Å) | 1.54 | 1.00 |
| Resolution range (Å) (highest resolution shell) | 16.10–1.90 (1.97–1.90) | 49.72–2.31 (2.39–2.30) |
| Collected reflections total (Unique) | 1,09,675 (10,884) | 62,174 (6,165) |
| Completeness (%) (highest resolution shell) | 97.17 (97.34) | 99.73 (99.60) |
| Mean (I)/(σ (I)) (highest resolution shell) | 16.66 (3.83) | 12.09 (5.32) |
| Rsym (%) (highest resolution shell) | 5.1 (29.3) | 7.5 (22.6) |
| Refinement statistics | ||
| R (R-free) (%) | 0.16 (0.19) | 0.15 (0.18) |
| r.m.s deviations | ||
| r.m.s bond lengths (Å) | 0.02 | 0.02 |
| r.m.s angles (°) | 2.08 | 1.58 |
| Ramachandran statistics | ||
| Favored (%) | 99 | 98 |
| Allowed (%) | 1 | 2 |
| Average B (Å2) | ||
| Wilson B-factor | 20.21 | 24.64 |
| Average B-factor | 21.00 | 26.00 |
| Macromolecules | 20.20 | 24.90 |
| Solvent | 35.70 | 26.50 |
| PDB code | 4Q4E | 4Q4I |
Conclusions
The crystal structure of actinonin in complex with ePepN provides for the first time the molecular basis for the inhibition of M1 family aminopeptidases. While the hydroxamate head group plays a critical role in defining the high affinity, the P1' residues also seem to contribute to the selectivity of the inhibitory compound. Since this study suggests that the mode of binding of actinonin in the PDFs and ePepN active sites is similar, care should be taken in designing actinonin-based inhibitors specific for either class of enzyme.
Acknowledgments
The authors acknowledge 8.2.2 beam line and the beam line scientist Dr. Corie Ralston at the Advanced Light Source (ALS, Berkeley) for facilitating the X-ray data collection ePepN-amastatin complex. The authors thank Dr. Sheena McGowan from Monash University, Australia for helping them with the critical review of the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, Margolis PS, Wu C, Ni ZJ, Trias J, White RJ, Yuan Z. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry. 2000;39:1256–1262. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- Butler MS, Blaskovich MA, Cooper MA. Antibiotics in the clinical pipeline in 2013. J Antibiot. 2013;66:571–591. doi: 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- Lee MD, Antczak C, Li Y, Sirotnak FM, Bornmann WG, Scheinberg DA. A new human peptide deformylase inhibitable by actinonin. Biochem Biophys Res Commun. 2003;312:309–315. doi: 10.1016/j.bbrc.2003.10.123. [DOI] [PubMed] [Google Scholar]
- Xu Y, Lai LT, Gabrilove JL, Scheinberg DA. Antitumor activity of actinonin in vitro and in vivo. Clin Cancer Res. 1998;4:171–176. [PubMed] [Google Scholar]
- Bernstein FC, Koetzle TF, Williams GJ, Meyer EF., Jr Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- Ragusa S, Mouchet P, Lazennec C, Dive V, Meinnel T. Substrate recognition and selectivity of peptide deformylase. Similarities and differences with metzincins and thermolysin. J Mol Biol. 1999;289:1445–1457. doi: 10.1006/jmbi.1999.2832. [DOI] [PubMed] [Google Scholar]
- Yen NT, Bogdanovic X, Palm GJ, Kuhl O, Hinrichs W. Structure of the Ni(II) complex of Escherichia coli peptide deformylase and suggestions on deformylase activities depending on different metal(II) centres. J Biol Inorg Chem. 2010;15:195–201. doi: 10.1007/s00775-009-0583-8. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ. Evolutionary families of metallopeptidases. Methods Enzymol. 1995;248:183–228. doi: 10.1016/0076-6879(95)48015-3. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, O'Brien E, Barrett AJ. MEROPS: the protease database. Nucleic Acids Res. 2002;30:343–346. doi: 10.1093/nar/30.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KS, Saito M, Hui M. A novel neuron-specific aminopeptidase in rat brain synaptosomes. Its identification, purification, and characterization. J Biol Chem. 1998;273:31053–31060. doi: 10.1074/jbc.273.47.31053. [DOI] [PubMed] [Google Scholar]
- Kruse MN, Becker C, Lottaz D, Kohler D, Yiallouros I, Krell HW, Sterchi EE, Stocker W. Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J. 2004;378:383–389. doi: 10.1042/BJ20031163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morty RE, Morehead J. Cloning and characterization of a leucyl aminopeptidase from three pathogenic Leishmania species. J Biol Chem. 2002;277:26057–26065. doi: 10.1074/jbc.M202779200. [DOI] [PubMed] [Google Scholar]
- Umezawa H, Aoyagi T, Tanaka T, Suda H, Okuyama A, Naganawa H, Hamada M, Takeuchi T. Production of actinonin, an inhibitor of aminopeptidase M, by actinomycetes. J Antibiot. 1985;38:1629–1630. doi: 10.7164/antibiotics.38.1629. [DOI] [PubMed] [Google Scholar]
- Brooks DR, Hooper NM, Isaac RE. The Caenorhabditis elegans orthologue of mammalian puromycin-sensitive aminopeptidase has roles in embryogenesis and reproduction. J Biol Chem. 2003;278:42795–42801. doi: 10.1074/jbc.M306216200. [DOI] [PubMed] [Google Scholar]
- Braun RK, Foerster M, Workalemahu G, Haefner D, Kroegel C, Walker C. Differential regulation of aminopeptidase N (CD13) by transendothelial migration and cytokines on human eosinophils. Exp Lung Res. 2003;29:59–77. doi: 10.1080/01902140303766. [DOI] [PubMed] [Google Scholar]
- Addlagatta A, Gay L, Matthews BW. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry. 2008;47:5303–5311. doi: 10.1021/bi7022333. [DOI] [PubMed] [Google Scholar]
- Dalal S, Ragheb DR, Schubot FD, Klemba M. A naturally variable residue in the S1 subsite of M1 family aminopeptidases modulates catalytic properties and promotes functional specialization. J Biol Chem. 2013;288:26004–26012. doi: 10.1074/jbc.M113.465625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag M, Bogyo M, Ellman JA, Salvesen GS. Aminopeptidase fingerprints, an integrated approach for identification of good substrates and optimal inhibitors. J Biol Chem. 2010;285:3310–3318. doi: 10.1074/jbc.M109.060418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niven GW, Holder SA, Stroman P. A study of the substrate specificity of aminopeptidase N from Lactococcus lactis subsp. cremoris Wg2. Appl Microbiol Biotechnol. 1995;44:100–105. doi: 10.1007/BF00164487. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Horowitz PM, Karsten SL, Jackson GR, Geschwind DH, Fu Y, Berry RW, Binder LI. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry. 2006;45:15111–15119. doi: 10.1021/bi061830d. [DOI] [PubMed] [Google Scholar]
- Wong AH, Zhou D, Rini JM. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J Biol Chem. 2012;287:36804–36813. doi: 10.1074/jbc.M112.398842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran CT, Carta F, Scozzafava A. Metalloenzyme inhibitors for the treatment of Gram-negative bacterial infections: a patent review (2009–2012) Expert Opin Therap Pat. 2013;23:777–788. doi: 10.1517/13543776.2013.777042. [DOI] [PubMed] [Google Scholar]
- O'Dwyer K, Hackel M, Hightower S, Hoban D, Bouchillon S, Qin D, Aubart K, Zalacain M, Butler D. Comparative analysis of the antibacterial activity of a novel peptide deformylase inhibitor, GSK1322322. Antimicrob Agents Chemo. 2013;57:2333–2342. doi: 10.1128/AAC.02566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boularot A, Giglione C, Petit S, Duroc Y, Alves de Sousa R, Larue V, Cresteil T, Dardel F, Artaud I, Meinnel T. Discovery and refinement of a new structural class of potent peptide deformylase inhibitors. J Med Chem. 2007;50:10–20. doi: 10.1021/jm060910c. [DOI] [PubMed] [Google Scholar]
- Sina A, Lord-Dufour S, Annabi B. Cell-based evidence for aminopeptidase N/CD13 inhibitor actinonin targeting of MT1-MMP-mediated proMMP-2 activation. Cancer Lett. 2009;279:171–176. doi: 10.1016/j.canlet.2009.01.032. [DOI] [PubMed] [Google Scholar]
- Addlagatta A, Gay L, Matthews BW. Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site. Proc Natl Acad Sci USA. 2006;103:13339–13344. doi: 10.1073/pnas.0606167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina-Osorio P. The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med. 2008;14:361–371. doi: 10.1016/j.molmed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MA, Matthews BW. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediate in catalysis. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- Ito K, Nakajima Y, Onohara Y, Takeo M, Nakashima K, Matsubara F, Ito T, Yoshimoto T. Crystal structure of aminopeptidase N (proteobacteria alanyl aminopeptidase) from Escherichia coli and conformational change of methionine 260 involved in substrate recognition. J Biol Chem. 2006;281:33664–33676. doi: 10.1074/jbc.M605203200. [DOI] [PubMed] [Google Scholar]
- Rich DH, Moon BJ, Harbeson S. Inhibition of aminopeptidases by amastatin and bestatin derivatives. Effect of inhibitor structure on slow-binding processes. J Med Chem. 1984;27:417–422. doi: 10.1021/jm00370a001. [DOI] [PubMed] [Google Scholar]
- Vazeux G, Iturrioz X, Corvol P, Llorens-Cortes C. A glutamate residue contributes to the exopeptidase specificity in aminopeptidase A. Biochem J. 1998;334:407–413. doi: 10.1042/bj3340407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Heinz A, Jung MC, Duca L, Sippl W, Taddese S, Ihling C, Rusciani A, Jahreis G, Weiss AS, Neubert RH, Schmelzer CE. Degradation of tropoelastin by matrix metalloproteinases–cleavage site specificities and release of matrikines. FEBS J. 2010;277:1939–1956. doi: 10.1111/j.1742-4658.2010.07616.x. [DOI] [PubMed] [Google Scholar]
- Gumpena R, Kishor C, Ganji RJ, Jain N, Addlagatta A. Glu121-Lys319 salt bridge between catalytic and N-terminal domains is pivotal for the activity and stability of Escherichia coli aminopeptidase N. Protein Sci. 2012;21:727–736. doi: 10.1002/pro.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapolu S, Alla M, Ganji RJ, Saddanapu V, Kishor C, Bommena VR, Addlagatta A. Synthesis, cytotoxicity and hDHFR inhibition studies of 2H-pyrido[1,2-a]pyrimidin-2-ones. Med Chem Commun. 2013;4:817–821. [Google Scholar]
- Kishor C, Arya T, Reddi R, Chen X, Saddanapu V, Marapaka AK, Gumpena R, Ma D, Liu JO, Addlagatta A. Identification, biochemical and structural evaluation of species-specific inhibitors against type I methionine aminopeptidases. J Med Chem. 2013;56:5295–5305. doi: 10.1021/jm400395p. [DOI] [PubMed] [Google Scholar]
- Oneda H, Inouye K. Refolding and recovery of recombinant human matrix metalloproteinase 7 (matrilysin) from inclusion bodies expressed by Escherichia coli. J Biochem. 1999;126:905–911. doi: 10.1093/oxfordjournals.jbchem.a022533. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific LLC; 2002. http://www.pymol.org. [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inform Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information