

Abstract
V(D)J recombination assembles functional antigen receptor genes during lymphocyte development. Formation of the recombination complex containing the recombination activating proteins, RAG1 and RAG2, is essential for the site-specific DNA cleavage steps in V(D)J recombination. However, little is known concerning how complex formation leads to a catalytically-active complex. Here, we combined limited proteolysis and mass spectrometry methods to identify regions of RAG1 that are sequestered upon association with RAG2. These results show that RAG2 bridges an interdomain boundary in the catalytic region of RAG1. In a second approach, mutation of RAG1 residues within the interdomain boundary were tested for disruption of RAG1:RAG2 complex formation using fluorescence-based pull down assays. The core RAG1 mutants demonstrated varying effects on complex formation with RAG2. Interestingly, two mutants showed opposing results for the ability to interact with core versus full length RAG2, indicating that the non-core region of RAG2 participates in binding to core RAG1. Significantly, all of the RAG1 interdomain mutants demonstrated altered stoichiometries of the RAG complexes, with an increased number of RAG2 per RAG1 subunit compared to the wild type complex. Based on our results, we propose that interaction of RAG2 with RAG1 induces cooperative interactions of multiple binding sites, induced through conformational changes at the RAG1 interdomain boundary, and resulting in formation of the DNA cleavage active site.
Keywords: V(D)J recombination, RAG1, RAG2, limited proteolysis, mass spectrometry, protein-protein interactions, flow cytometry
Introduction
The foundation for the adaptive immune system is the ability of immunoglobulins and T cell receptors to recognize a large array of foreign antigens. The large repertoires of these antigen-binding proteins are produced at the genetic level during lymphocyte development by V(D)J recombination, which assembles the antigen receptor genes from constituent V, J, and sometimes, D gene (coding) segments.1 Defects in V(D)J recombination results in a block in lymphocyte development and fatal immunodeficiency diseases. Conversely, aberrant V(D)J recombination activity may result in genetic instability contributing to lymphomagenesis.1
In the first stage of V(D)J recombination, the DNA is cleaved between each selected coding segment and its flanking recombination signal sequence (RSS) by the lymphoid specific proteins, RAG1 and RAG2, which together constitute the V(D)J recombinase.1 The RSS consists of a conserved heptamer and nonamer separated by either 12 or 23 base pairs, termed the 12-RSS and 23-RSS. Recombination is restricted between two coding segments that are flanked by RSSs of differing spacer lengths. The initial site-specific interaction of the RAG proteins with the RSSs is crucial for the V(D)J recombination process, as aberrant activity may arise from mis-assembly of the RAG proteins with non-RSS DNA.2–5
The RAG proteins are relatively large in size, with 1040 and 527 residues in murine RAG1 and RAG2, respectively. Both proteins consist of a core region, which is the minimal region required for double-stranded DNA cleavage activity.1 The core regions are sufficient for tight association of the RAG proteins, RSS binding, and DNA cleavage activity.1,6 The noncore regions of the RAG proteins may serve to enhance or regulate the catalytic activity.7
Core RAG1 (residues 384–1008) contains both the RSS heptamer and nonamer binding domains, as well as a DDE active site motif consisting of residues D600, D708, and E962.1,6 The RSS nonamer-binding domain (NBD) is located at the N-terminal region of core RAG1 and includes residues 384−460.8 The DDE active site in RAG1 is encompassed by two structural domains.6,9 The central domain (residues 528−760) selectively binds the RSS heptamer,9,10 and contains a putative C2H2 zinc finger, termed ZFB.11 The C-terminal domain (residues 761−980) binds DNA nonspecifically, and is proposed to contact coding segments flanking the RSS heptamer in RAG-RSS complexes.9,12,13
The core region of RAG2 consists of six kelch-like repeats, and is predicted to assume a six-bladed beta propeller fold.14 Mutation of specific residues throughout core RAG2 has been shown to reduce DNA cleavage activity, illustrating its essential role in V(D)J recombination.15–18
An active RAG1-RAG2 complex has been shown to consist of a heterotetramer,19,20 although other stoichiometries of higher oligomers have been detected.21,22 It is likely that the binding of RAG2 induces conformational changes in RAG1 that facilitates DNA cleavage activity, as well as promoting sequence-specific interactions with the RSS over nonsequence specific DNA.23,24 While a peptide that contained the ZFB was shown to be sufficient for binding RAG2,25 regions outside this motif appear to affect complex formation as well.26,27 Altogether, our knowledge of the RAG1-RAG2 interface, and how complex formation promotes DNA cleavage activity, is limited.
To identify regions of the RAG proteins that are critical for mediating complex formation, we combined limited proteolysis and mass spectrometry assays to gain insight into formation of the RAG1-RAG2 complex. We determined that regions of core RAG1 sequestered upon complex formation with RAG2 bordered the interdomain boundary between the central and C-terminal domains (residues 735−775), which includes the ZFB. Mutation of residues at this interdomain boundary of core RAG1 led to defects in the interaction with RAG2, as well as formation of complexes with altered RAG1:RAG2 stoichiometries compared with the wild type RAG complex, as measured by fluorescence-based pull down assays. Another region of core RAG1, containing the active site residue E962, was partially sequestered from proteolysis upon addition of RAG2. From the combination of these results, we propose a model for RAG1-RAG2 complex formation that requires cooperative interactions mediated through the central/C-terminal interdomain boundary of RAG1, which yields formation of the catalytically competent active site.
Results
The surface topology of core RAG1
The surface topology of core RAG1, in the absence and presence of RAG2, was probed using proteomic methods. The core regions of the RAG proteins fused to either MBP (MBP-core RAG1) or GST (GST-core RAG2) were used in these experiments, as the fusion proteins are soluble and readily purified to homogeneity (Supporting Information Figure S1). In addition, DNA cleavage activity and assembly of these fusion proteins have been extensively reported.1,6
To show regions of the protein that are solvent-exposed and likely more conformationally disordered, we performed limited tryptic proteolysis of core RAG1 in conjunction with mass spectrometry. The rationale for this approach is that the limited proteolysis will initially cleave the polypeptide backbone of native proteins at the most accessible regions.28 Additionally, conformationally disordered regions of folded proteins tend to be targeted, as these unstructured regions are more easily accommodated in the protease active sites.28
We first performed an exhaustive digestion of MBP-core RAG1, followed by analysis of the cleavage products by MALDI-TOF mass spectrometry. This showed that ∼94% of the core RAG1 sequence (corresponding to fragments from enzymatic digestion) was assigned to peaks in the 0.5-5 kDa m/z region of the mass spectrum (Supporting Information Figure S2A). Thus, proteolyic cleavage of the core RAG1 was efficient throughout all regions of the protein.
For the limited proteolysis experiments, samples were maintained at 4°C in 0.2 M NaCl, conditions where MBP-core RAG1 is dimeric with little detectable aggregation.29 The MBP-core RAG1 was subjected to proteolysis at a protease:RAG1 ratio of 1:50 (w/w). Trypsin initially cleaved between the MBP and the core RAG1 portions of the fusion protein, as judged by SDS-PAGE (not shown). Significant detection of peaks by MALDI-TOF mass spectrometry (predominantly in the m/z range of <5 kDa) was evident following 1 hr of proteolysis with the number of peaks remaining relatively constant for up to 4 h. Digestion times from 4 to 16 h yielded increased number of peaks, as limiting digestion conditions were exceeded.
A representative spectrum from a limited tryptic digestion that was 3 h in duration is shown in Figure 1(A), and the combined results from all spectra are summarized in Figure 1(C). These data show the production of several trypsin-generated peptides from MBP-core RAG1. Assignment of peaks in the mass spectra to MBP-core RAG1 peptides was based on matching the m/z value of peaks with predicted monoisotopic mass values (for the +1 ion) for peptides in the MBP-core RAG1 protein sequence [labeled a-f in Fig. 1(C)]. Significantly, the majority of the trypsin-generated peptides are clustered within ∼25 residues of either the central or C-terminal domain boundaries of core RAG1.9 Similar results were obtained using either chymotrypsin or endoproteinase-GluC, indicating that sequence specificity of the protease did not skew the results (Supporting Information Figure S3). Given these results, we used 3 h trypsin digestions in the remaining limited proteolysis experiments in this study.
Figure 1.
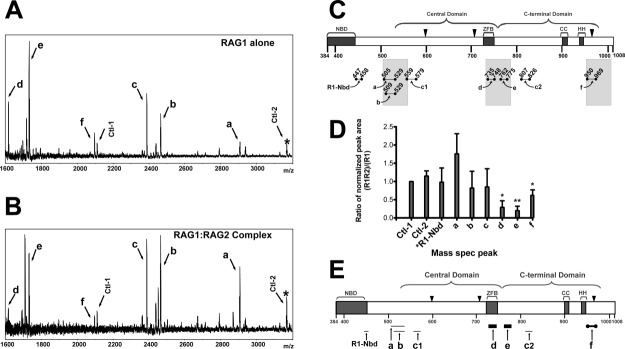
Resolution of RAG2-interacting regions of core RAG1 by MALDI-TOF mass spectrometry of limited proteolysis products. Spectra of MBP-core RAG1 alone (A) and MBP-core RAG1 with GST-core RAG2 (B) measured following treatment of samples with trypsin. Peaks due to MBP peptides are labeled Ctl-1 or Ctl-2, with Ctl-1 serving as an internal standard. (C) Regions of MBP-core RAG1 assigned to spectra peaks in (A) are illustrated. Brackets indicate the locations of both the central and C-terminal domains. NBD and ZFB refer to the nonamer binding domain and zinc finger B, respectively. CC and HH refer to zinc-binding regions in the C-terminal domain.42 The active site residues (D600, D708, and E962) are depicted with black triangles. Proteolytic peptides of core RAG1 generated by trypsin are represented by bars beneath the core RAG1 schematic, and are labeled a, b, c1, c2, d, e, f and R1-Nbd, as described in the text. The shaded bars denote regions within +/- 25 residues of the central and C-terminal domain boundaries. (D) Quantitative analysis of the limited proteolysis experiments. In each MALDI-TOF mass spectrum, the ratio of the peak area for assigned peaks over the peak area for the Ctl-1 peak was determined, and is referred to as normalized peak areas. Plotted on the Y axis is the ratio of the normalized peak areas for the RAG1 only compared to the RAG1:RAG2 samples that were prepared and analyzed in parallel by mass spectrometry (n = 3 experiments). The labels on the X-axis correspond to the peak labels in panels A and B. *R1-Nbd is a peptide in the NBD of RAG1 (at m/z = 1360), as described in the text. *, P < 0.05; **, P < 0.01 determined by student's t test. (E) Posited sites of core RAG1 that contact with core RAG2. The trypsin-generated core RAG1 peptides (labeled a-f) are represented as bars below their respective positions in the core RAG1 schematic. The thickest black bars represent core RAG1 peptides (d and e) that were substantially decreased in abundance upon trypsin digestion in the presence of RAG2. Peptide f (represented as a thick black bar bordered by solid black circles) was also decreased in the presence of RAG2. The relative abundances of peptides R1-Nbd, a, b, c1, and c2 (shown as light gray bars) were not decreased in presence of RAG2.
Assignment of peaks a and d in the limited tryptic digestion of MBP-core RAG1 was clear based on identified m/z values of the peptide fragments. However, additional mass spectrometry experiments were necessary to unambiguously resolve the assignments of the four remaining peptides (b, c, e, and f), since their m/z values corresponded to two separate and nonoverlapping theoretical trypsin-generated MBP-core RAG1 peptides (Table1.). Tandem mass spectrometry using HPLC-MS-MS conclusively showed that peptides b, e, and f accounted for the peptides assigned to their respective peaks. Conversely, separate peptides consisting of residues 559−579 and 807−826 [labeled c1 and c2, Fig. 1(C)] were found to account for the peak with an m/z value of 2387 (Table1.).
Table 1.
Sequence assignment by tandem mass spectrometry of selected peaks generated from limited trypsin digestion of MBP-core RAG1a
| m/z | Peptide Sequencebc | Peptide |
|---|---|---|
| 1730 | 1) 762-SNPYHESVEELRDR-775 | e |
| 2) 90-LYPFTWDAVRYNGK-103 (MBP)d | – | |
| 2094 | 1) 950-DGSIGAWASEGNESGNKLFR-969 | f |
| 2) 622-FSFTVMRITIEHGSQNVK-639 | – | |
| 2387e | 1) 559-YDSALVSALMDMEEDILEGMR-579 | c1 |
| 2) 807-IFQLEIGEVYKHPNASKEER-826 | c2 | |
| 2464 | 1) 509-VLLPGYHPFEWQPPLKNVSSR-529 | b |
| 2) 757-YEVWRSNPYHESVEELRDR-775 | – |
The table lists the m/z value of each analyzed peak (first column), the sequences of two different theoretical trypsin-generated MBP-core RAG1 peptides (labeled 1 and 2) with m/z values within ± 1 D of the value in first column (second column), and the peptide labels b, c1, c2, e, and f consistent with the labels in Figure 1B (third column).
The peptides in boldface indicate the sequences detected by tandem mass spectrometry. The sequences in light gray font were not detected, resulting in unambiguous assignments for three of the peaks (peptides b, e, and f).
The residue numbers are shown for the first and last peptide residue. The numbers for the core RAG1 peptides correspond to that for the full length murine RAG1 protein.
Corresponds to a peptide in the MBP portion of the fusion protein.
The peak with an m/z value of 2387 consisted of a mixture of both of the peptides listed (labeled as c1 and c2).
In addition to peaks from cleavage sites at the central domain and C-terminal domain boundaries, a peak was observed at an m/z of 1360 (not shown) that corresponds to a peptide from tryptic cleavage at residue 446 in the NBD of core RAG1. Residue 446 is highly exposed to solvent according to the crystal structure of the isolated NBD.8 Finally, two fragments from cleavage sites within the MBP domain of the fusion protein were identified [Fig. 1(A)], which also occurred in tryptic digestion of isolated MBP under limiting conditions (not shown). These peptides arise from cleavage at basic residues that are well-exposed in the crystal structure of MBP.30
Whereas peaks a-f were visible at short times following limited tryptic digestion of MBP-core RAG1, very few peaks were detectable upon tryptic digestion of GST-core RAG2 under the same conditions. Following 3 h of tryptic digestion, only peaks from GST fragments were detected (not shown). This is likely due to the relative lack of Lys and Arg residues throughout a large region of core RAG2 resulting in large polypeptides that are not readily detected by MALDI-TOF (Supporting Information Fig. S2B). The resistance of core RAG2 to limited tryptic digestion is also consistent with the predicted structure of core RAG2 being a compact structure consisting of a six bladed propeller structure.14 Although this result did not permit analysis of the surface topology of core RAG2 under these conditions, an advantage was the ability to assess the effect of core RAG2 on the trypsin accessible regions of core RAG1 with little interference from the generation of RAG2 peptides (see below).
Identification of putative core RAG2 contact sites on core RAG1
Next, we examined how complex formation with RAG2 affected proteolytic susceptibility of RAG1. Here, MBP-core RAG1 and GST-core RAG2 were mixed under conditions that show strong interactions by pull down assays, and which show distinct complexes on binding the RSS.9,24 Both MBP-core RAG1 and GST-core RAG2 are predominantly dimeric (Supporting Information Figure S1). A lower ratio of GST-core RAG2 to MBP-core RAG1 was used to favor a 1:1 stoichiometry of complex formation. MBP-core RAG1:GST-core RAG2 mixtures were digested for 3 h under identical conditions as MBP-core RAG1 and analyzed by mass spectrometry [Fig. 1(B)]. The MBP-derived peak at an m/z of 2110 [labeled Ctl-1 in Fig. 1(A,B)] was used as the internal standard. In each spectrum, the ratio of each peak area relative to that of Ctl-1 was determined, and the values compared between samples with GST-core RAG2 absent or present [Fig. 1(D)]. Serving as a separate control, the peak area ratio of a second MBP peak [at m/z = 3168; labeled Ctl-2 in Fig. 1(A,B)] to the Ctl-1 peak is similar regardless of the absence or presence of core RAG2 [Fig. 1(D)].
The combined results for the effect of limited trypsin digestion on core RAG1 in the presence of RAG2 is summarized in Figure 1(E). The addition of GST-core RAG2 significantly decreased peaks d and e, which correspond to peptides 735−748 (d) and 762−775 (e) in the ZFB and near the N-terminal end of the C-terminal domain, respectively. While the 762−775 fragment is adjacent to the ZFB sequence, this region was not included in the ZFB fragment shown to bind core RAG2.25 Production of peptide 950−969 (peak f), which contains the active site residue E962, was also decreased in the presence of core RAG2, although to a lesser extent than the peptides at the interdomain boundary between the central and C-terminal domains.
The production of other trypsin-generated core RAG1 peptides was not significantly influenced by the presence of core RAG2. For example, while the production of peptide a (residues 505−529) trended towards an increase in all trials, this was not a significant change based on the student t test. Furthermore, the production of peptide b (residues 509−529), as well as a peptide to the N-terminal side of peptide a (see next section), was independent of the presence of RAG2. Other regions that were not affected by the presence of RAG2 include peptides 559−579 and 807−826 (c1 and c2, combined in peak c), as well as a peptide at the C-terminal end of the NBD [residues 447−458; R1-Nbd in Fig. 1(E)].
Isotope coding results confirm core RAG2 effects on core RAG1 surface topology
Isotope coding experiments were performed as an alternate approach to test the effects of RAG2 on the surface topology of RAG1 [Fig. 2(A)]. In this approach, limited proteolysis assays for MBP-core RAG1 alone and the MBP-core RAG1:GST-core RAG2 mixture were performed in parallel as described above. Following proteolytic cleavage, the peptides in each sample were labeled at primary amines with the acetylating reagent N-acetoxysuccinimide (h3-NAS) or its d3-deuterated analogue (d3-NAS). The RAG1-only sample was labeled with nondeuterated NAS (to give the h3-Ac form) and the RAG1:RAG2 sample labeled with deuterated NAS (to give the d3-Ac form). Subsequently, the samples were combined and analyzed by MALDI-TOF mass spectrometry. In the combined mixture, the h3-Ac RAG1-only peptides are 3 a.m.u. per labeled-amine less than the corresponding peptide from the d3-Ac RAG1:RAG2 sample. This allowed simultaneous analysis of the RAG1-only and the RAG1:RAG2 samples.
Figure 2.
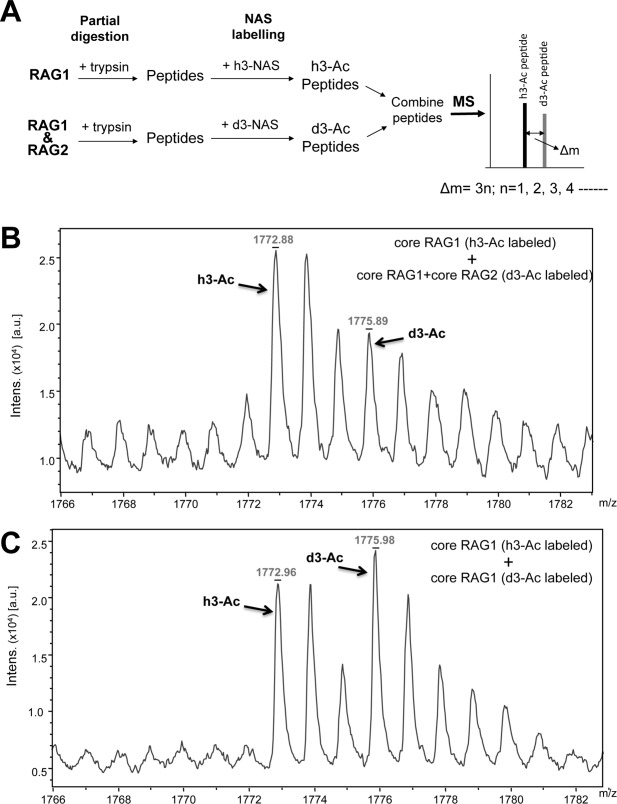
Isotope coding experiment of core RAG1 binding to RAG2. A: MBP-core RAG1 alone or MBP-core RAG1 combined with GST-core RAG2 were labeled with either h3-NAS or d3-NAS following limited tryptic proteolysis. The samples were then combined and measured by MALDI-TOF mass spectrometry. The difference in m/z value of an h3-Ac versus d3-Ac labeled peptide is Δm = 3n, where n is the number of labeled amine groups in the peptide. B: The monoisotopic peaks from MALDI-TOF mass spectrometry for peptide e (core RAG1 residues 762−775) are indicated for the RAG1 only sample labeled with a single acetyl group (h3-Ac) and for the RAG1:RAG2 sample labeled with a single d3-acetyl group (d3-Ac). Assignments of the peaks are described in the text. C: Same as in panel B, except that both the h3- and d3-acetylated forms were RAG1 only samples.
As shown in Figure 2(B), we identified peaks that corresponded to the singly acetylated RAG1 peptide consisting of residues 762−775 (peptide e). The two peaks between the monoisotopic h3-Ac and d3-Ac peaks in Figure 2(B) are the +1 and +2 isotope peaks of the h3-Ac form. Note that the peak area of the monoisotopic d3-Ac peak also has contributions from the +3 isotope peak of the h3-Ac form. The +3 isotope peak is predicted to have a peak area at 18.8% of the monoisotopic h3-Ac peak (http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msisotope). Thus, the peak area of the d3-Ac monoisotopic peak was corrected by subtracting contributions from the +3 isotope peak of the h3-Ac form. The ratio of the corrected d3-Ac peak to the h3-Ac peak was 0.56.
As a control, we also combined separate aliquots of RAG1 only samples that were either in the h3-Ac or the d3-Ac form to control for the efficiency of the labeling reaction [Fig. 2(C)]. The h3-Ac and d3-Ac-labeled RAG1 peptides should give peaks of equal area if the labeling reaction is similarly efficient. The peak area of the d3-Ac peak was corrected as described above. The ratio of the corrected d3-Ac peak to the h3-Ac peak was 0.85.
In addition to peptide e, we detected an acetylated peptide in both samples that originated from the region between the NBD and the central domain (residues 494−504) (Supporting Information Fig. S4). Markedly, the area of the peak corresponding to residues 494−504 did not change upon RAG2 addition (Supporting Information Fig. S4). This is consistent with the lack of effect of RAG2 on the production of peptide b [Fig. 1(D)], indicating that the N-terminal border of the central domain does not form extensive interactions with RAG2.
Altogether, the isotopic coding experiments confirm that peptide e is affected by complex formation with RAG2, and that a major RAG1 site interacting with RAG2 extends into the C-terminal domain. These results suggest a larger interface with RAG2, which bridges a domain boundary between the central and C-terminal domains.
Effect of mutations in the central/C-terminal domain boundary of RAG1 on complex formation with RAG2
To determine if point mutations in the interdomain boundary between the central and C-terminal domains of RAG1 (encompassing peptides d and e) affected RAG1-RAG2 complex formation, we tested binding of the core RAG1 mutants C727,730A, W760A,R761Q, and E767K to RAG2. C727,730A removes two putative zinc-coordinating ligands in the ZFB,11 W760A,R761Q alters residues at the border between the central and C-terminal domains, and E767K alters the charge of a residue in the middle of peptide e. Each of the mutants was defective in cellular recombination assays (Fig. 3).
Figure 3.
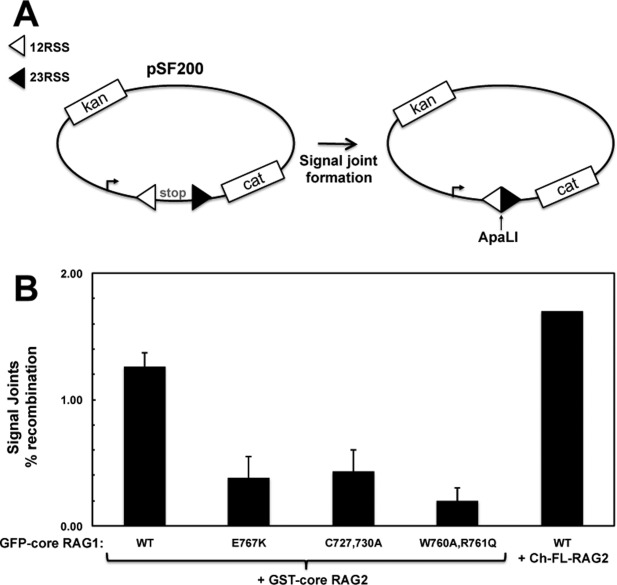
Mutant RAG1 constructs show deficient V(D)J recombination activity. A: Cellular recombination assays were performed using the plasmid substrate pSF200. V(D)J recombination of this substrate removes a transcription terminator allowing transcription of the CAT gene in bacterial cells.43 Precise signal formation introduces a new ApaLI restriction site in the recombined plasmid. B: Recombination activity was determined as described in Materials and Methods. In the first 5 lanes (n = 2 experiments), pSF200 was combined with GFP-core RAG1-expressing and GST-core RAG2-expressing constructs. In the last lane, Ch-FL-RAG2 was used in place of GST-core RAG2 to confirm that the cherry fusion does not impede RAG2 function.
We assessed binding of core RAG1 to both full length and core RAG2 using an RFP-trap fluorescence pull down assay. This approach is based on the binding of mCherry-labeled RAG2 to beads containing antibody specific to RFP, which recognizes mCherry. Binding consisted of incubating the RFP trap beads with lysate from cells transiently expressing RAG2 (either full length or core) fused at its N-terminus to mCherry (Ch-FL-RAG2 or Ch-core RAG2), and core RAG1 with GFP fused at its N-terminus (GFP-core RAG1). The wild type (WT) RAG proteins fused to fluorescent proteins were functional in cellular recombination assays (Fig. 3). GFP-core RAG1 binding to Ch-FL-RAG2 or Ch-core RAG2 was subsequently analyzed by flow cytometry. A major advantage of this approach is rapid quantification of protein-protein interactions through direct measurement of emission from the fluorescent proteins.31
To first test the specificity of the RFP-trap pull down assay, RFP-trap beads were incubated with Ch-FL-RAG2, Ch-core RAG2, or GFP-core RAG1 alone. This showed efficient binding by the Cherry-tagged RAG2 proteins, but no binding by GFP-core RAG1 [Fig. 4(A)]. Thus, binding was specific to expression of Cherry-fused RAG2. Importantly, both the red and green channels showed significant signal when beads were incubated with lysate of cells co-expressing GFP-core RAG1 with either Ch-core RAG2 or Ch-FL-RAG2 [Fig. 4(B,C), left dot plots], with >90% of beads double-labeled in each experiment. The double labeling was due to interaction between the RAG proteins, since co-expression of either GFP with Cherry-tagged RAG2, or WT GFP-core RAG1 with mCherry, did not double-label the beads (Supporting Information Figure S6A).
Figure 4.

Assessment of WT and mutant core RAG1 binding to RAG2 using a fluorescent RFP-trap pull down assay. A: Representative flow cytometry of RFP-trap beads incubated with cell lysates containing either Ch-core-RAG2 (left plot), Ch-FL-RAG2 (middle plot), or WT GFP-core RAG1 (right plot). B: RFP-trap assay results using cell lysates containing Ch-core-RAG2 coexpressed with either WT, C727,730A, or W760A,R761Q GFP-core RAG1 as indicated. Bar graph (at left) plotting the relative amount of complexes containing GFP-core RAG1 (cR1) and Ch-core-RAG2 (cR2) as detected by the RFP-trap assay. The y-axis is the fraction of beads in the upper right quadrant of the flow cytometry dot plots, normalized to sample containing WT GFP-core RAG1. n = 2. C: Same as in panel B, except cell lysates contained Ch-FL-RAG2 coexpressed with the indicated GFP-core RAG1 fusion proteins. Bar graph (at left) is as described above in panel B, but with samples containing Ch-FL-RAG2 (FLR2). n = 3.
Next, we measured the binding of the core RAG1 mutants to RAG2. The GFP-core RAG1 mutants showed similar levels of expression as the WT protein, as detected by fluorescence microscopy (Supporting Information Fig. S5), and by measurement of GFP fluorescence intensities in cell lysates (not shown). In addition, the GFP fusion proteins showed negligible degradation (Supporting Information Fig. S6B). First, the RFP-trap pull down assay was used to test binding between core RAG1 mutants and Ch-core RAG2. Core RAG2 bound effectively to the W760A,R761 and E767K mutant RAG1 proteins [Fig. 4(B) and Supporting Information S6C], but showed severe defects in binding to the C727,730A mutant with less than 5% of beads double-labeled [Fig. 4(B)]. Second, the assays were repeated using Ch-FL-RAG2. In comparison to Ch-core RAG2, pull down assays using Ch-FL-RAG2 as bait yielded contrasting results. Namely, Ch-FL-RAG2 showed effective binding (>90% beads double-labeled) to the C727,730A and E767K mutants [Fig. 4(C) and Supporting Information S6C], and moderate defects in complex formation (∼35% of beads double-labeled) with the W760A,R761Q mutant [Fig. 4(C)]. These results suggest that the non-core region of RAG2 can compensate for the deficiency of core RAG2 in binding to the C727,730A RAG1 mutant. Moreover, FL RAG2 may modulate the binding surface with core RAG1, given the moderate defects in binding to the W760A,R761Q mutant, although in a manner that is not yet clear. These findings are consistent with a previous study, which indicated that the non-core region of RAG2 could enhance complex formation with the core region of RAG1.32 The enhancement in binding may occur through direct interactions of the non-core region of RAG2 with RAG1, or via an indirect effect. It has been previously proposed that an in cis interaction occurs between the non-core and core regions of RAG2,33 which may in turn affect binding interactions with RAG1.
The RFP-trap binding assay used different RAG fusion proteins (mCherry and GFP) than the limited proteolysis experiments (MBP and GST). Nevertheless, in Figure 3 we show that the recombination activity is similar for GFP-core RAG1 when combined with RAG2 tagged with either GST or Cherry. Thus, the N-terminal tag did not affect formation of recombination products in the plasmid substrate, and likely did not affect the outcome of the limited proteolysis experiments. Our results are in line with previous studies, which have shown the RAG proteins maintain catalytic activity regardless of the presence or the identity of an N-terminal fusion tag.1,16,24,34,35
Mutant core RAG1 proteins bind with altered stoichiometry to RAG2
Although mutation of RAG1 residues within the putative RAG2 interaction region showed varying effects in apparent affinity for RAG2, the relative fluorescent signals in the flow cytometry results consistently differed for all of the mutants as compared with the wild type complexes (Fig. 5). We interpret these differences as indicating an altered stoichiometry for RAG1:RAG2 complexes containing the mutant core RAG1 proteins versus the WT core RAG1 protein. The stoichiometric differences occur with complexes containing either core or full length RAG2 (Fig. 5), indicating that the non-core region of RAG2 does not impact the stoichiometry of the RAG1:RAG2 complex. For example, flow cytometry plots for WT GFP-core RAG1 bound to either FL or core RAG2 yielded similar relative intensities of green versus red channels, as demonstrated by the overlay in Figure 5(A). However, double-labeled beads containing RAG2 complexed with mutant core RAG1 proteins produced greater signals in the mCherry channel by RFP-trap than beads containing WT core RAG1 [Fig. 5(B), Supporting Information Fig. S7A]. This is plotted in Figure 5(C), which was generated by measuring the mean Cherry fluorescence signal at specific GFP fluorescence intensities (as illustrated in Supporting Information Fig. S7B). In summary, the mutants show 2-3 times more Ch-RAG2 signal per GFP-core RAG1.
Figure 5.
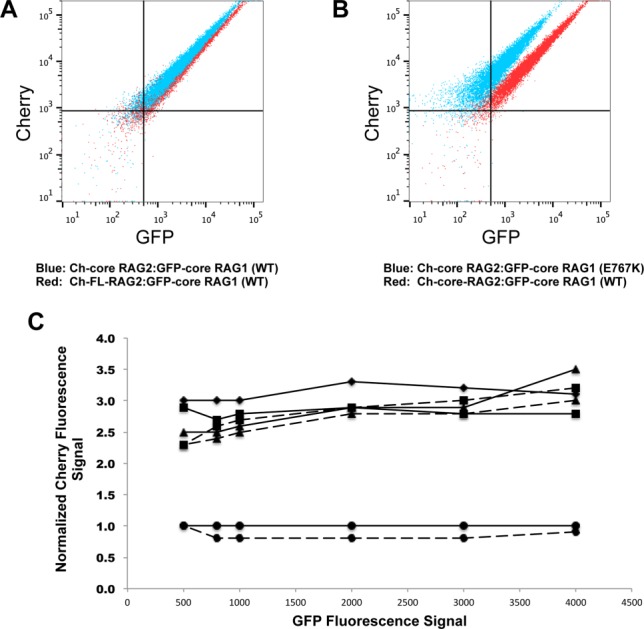
Stoichiometric differences in RAG1 to RAG2 in V(D)J recombinase complexes containing mutant versus WT core RAG1. A: Overlay of the flow cytometry data where RFP-trap beads were incubated with cell lysates coexpressing WT GFP-core RAG1 with either Ch-core-RAG2 (blue dots) or Ch-FL-RAG2 (red dots). B: Overlay of RFP-trap flow cytometry data comparing Ch-core RAG2 co-expressed with either WT (red dots) or E767K (blue dots) GFP-core RAG1. C: Relative stoichiometries of Ch-fused RAG2 to GFP-core RAG1 from representative RFP-trap flow cytometry experiments. Each point is the Cherry fluorescence signal (normalized to the Cherry signal from the coexpressed Ch-FL-RAG2:WT GFP-core RAG1 experiment) at selected GFP fluorescence signals, as illustrated in Figure S7B. Solid and dashed lines are from experiments using Ch-FL RAG2 and Ch-core-RAG2, respectively. Experiments using the different GFP-core RAG1 proteins are represented with the following symbols: WT (circles), C727,730A (diamonds), W760A,R761Q (triangles), and E767K (squares). Results from an RFP-trap experiment using co-expressed Ch-core RAG2 and GFP-core RAG1 (C727,730A) was not shown due to weak complex formation.
As further evidence for the altered stoichiometry of complexes containing mutant RAG1 proteins with RAG2, a reverse pull down assay using GFP-trap beads was performed. The flow cytometry plots revealed an increase in the mCherry signal for the mutant RAG1 complexes in comparison to WT core RAG1 when bound to either FL or core RAG2 (Supporting Information Figure S7C). Although the GFP-trap pull down was less effective than the RFP-trap assay in binding the RAG1:RAG2 complex (with <10% of the beads double-labeled for any combination of proteins used), the results consistently showed that the mutant RAG1 proteins bound two to three times more RAG2 subunits. Altogether the combined results from the RFP-trap and GFP-trap assays are consistent with an increased number of RAG2 subunits bound in the mutant versus the WT RAG1:RAG2 complexes, but inconsistent with other explanations, such as differing amounts of RAG1:RAG2 complexes versus uncomplexed RAG proteins on the respective beads for the mutant versus WT complexes.
Discussion
In this study we employed limited proteolysis combined with isotope coding methods to map the surface topology of core RAG1, and subsequently the regions of core RAG1 that lie in the RAG1-RAG2 interface. The limited proteolysis approach is feasible since it is understood that regions of protein interaction can become inaccessible to digest, such as we have shown here for the RAG1 core as its binds RAG2. Importantly, this approach provides information regarding regions of protein interaction without modification of proteins through labeling or mutagenesis, and with the protein complex in solution at approximately physiological ionic strengths.
We identified regions near or bordering the central/C-terminal domain boundary of RAG1 that are sequestered upon interaction with RAG2. In addition, we identified an additional region in core RAG1, located near the E962 active site residue in the C-terminal domain that either directly contacts RAG2, or is conformationally affected by interaction with RAG2. Previous mutagenesis studies suggested additional regions of RAG1 that are important for interactions with RAG2,18,27 these being D546, E547, and D560. However, mutation of each of these residues did not completely eliminate RSS cleavage activity, suggesting that complex formation remains. RAG1 residue R558 (R561 in human RAG1) has also been implicated in RAG2 interactions, although these results are disputed.27 Our assay could not determine changes in accessibility of these residues since peptide c1, which contained or was adjacent to these residues, was poorly resolved from peptide c2, a separate and unrelated peptide (Table1.).
We tested whether selected point mutations in peptide d (C727,730A), peptide e (E767K), and the border of the two RAG1 domains (W760A,R671Q) impacted RAG1-RAG2 interactions. In comparison to WT RAG1, all three mutants showed significantly decreased V(D)J recombination activity (Fig. 3). A fluorescence-based pull down assay was used to test if the decreased recombination activity observed with each RAG1 mutant was due to defects in complex formation with RAG2. The most deleterious effect in complex formation was with the inability of the GFP-core RAG1 C727,730A mutant to bind Ch-core RAG2, which is consistent with the mass spectrometry results showing that peptide d was sequestered in the protein complex.
Most notably, all three interdomain mutants showed an increased ratio of Cherry to GFP signals in the fluorescent pull down assays as compared with WT RAG1, consistent with an increased number of RAG2 subunits bound to each mutant RAG1 subunit. To account for these results, we propose that RAG2 induces conformational changes in the interdomain boundary of each RAG1 subunit, thus facilitating cooperative interactions of multiple binding sites in RAG1 with a single subunit of RAG2 [Fig. 6(A)]. Mutation of residues within the interdomain boundary eliminates the cooperative interactions resulting in individual RAG1 binding sites complexing with separate RAG2 subunits. Analogously, it has been proposed that multiple binding sites for RAG1 exist on RAG2,16 which would also be a necessary component of our proposed model [Fig. 6(A)].
Figure 6.
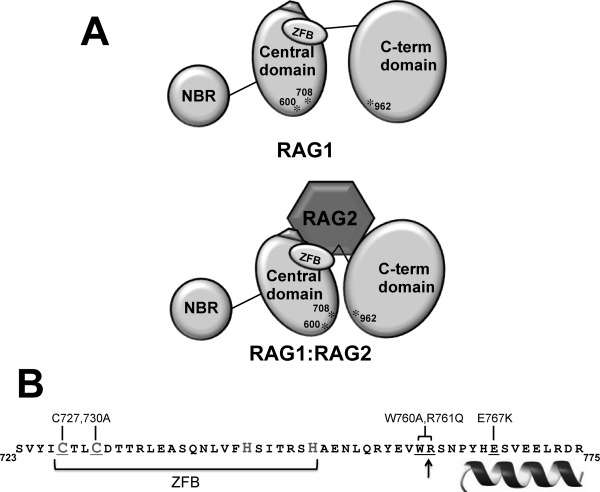
A: Model of the RAG1-RAG2 complex. The upper schematic shows the three domains in core RAG1 (NBD, central, and C-terminal domains). The ZFB is denoted as an oval, and the three active site residues (D600, D708, and E962) labeled and indicated with asterisks. The pentagon adjacent to the ZFB represents a region of charged residues in the central domain that upon mutation were previously shown to disrupt complex formation with RAG2.18,27 For simplicity, only a monomer of core RAG1 is represented. The lower schematic depicts RAG2 bridging the central and C-terminal domains, resulting in a closer juxtaposition of D600 and D708 with E962. B: The sequence of RAG1 that showed decreased susceptibility to proteases in the presence of RAG2. The border between the central and C-terminal domains is marked with an arrow. The sequence of the putative ZFB is bracketed, and likely Cys and His zinc-coordinating residues are denoted in a larger gray font. The helix denotes the region at the C-terminal end of this peptide predicted to be alpha helical. The core RAG1 mutants are indicated above the sequence, and the corresponding residues within the sequence that were mutated are underlined.
Overall, we have determined that the interdomain border between the RAG1 central and C-terminal domains is highly accessible in unbound RAG1, and is a major interaction site with RAG2. Significantly, the structural integrity of the ZFB is essential to form the catalytically active complex. We postulate that association of RAG2 with RAG1 through cooperative interactions forms a complex that is optimized for catalytic activity [Fig. 6(A)]. Cooperative interactions mediated by multiple domains in the formation of functional protein-protein complexes have been increasingly documented in recent years.36,37 Future determination of how the region of RAG1 encompassing this interdomain boundary, including the ZFB and flanking C-terminal residues [Fig. 6(B)], is folded in the absence and presence of RAG2 will shed significant insight into formation of the catalytically competent V(D)J recombinase.
Materials and Methods
Plasmid constructs
Plasmids encoding for MBP fused to core RAG1 (MBP-core RAG1),38 GST fused to core RAG2 (GST-core RAG2),39 and GFP fused to core RAG1 (GFP-core RAG1)40 have been previously reported. GFP-core RAG1 point mutants, C727,730A, W760A,R761Q, and E767K, were generated using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. The entire region of the gene corresponding to the RAG1 coding sequence was examined by DNA sequencing to confirm that only the residue(s) of interest was modified in each mutant. The plasmid encoding for Cherry fluorescent protein fused to full length (FL) RAG2 (Ch-FL-RAG2) was constructed as follows. The gene encoding for murine FL RAG2 was amplified by polymerase chain reaction (pcr) using primers that introduced an EcoRI site at the 5′ end of the product and a BamHI site following the stop codon at the 3′ end of the product. A gene encoding Ch-FL-RAG2 fusion protein was created by inserting the EcoRI and BamHI digested pcr product into the corresponding restriction sites of the pmCherry-C1 vector (Clontech). The sequence of the RAG2 gene was confirmed by DNA sequencing. Plasmids used for expression of GFP and mCherry in control experiments were pWay541 and pmCherry-C1 (Clontech).
Protein purification
MBP-core RAG1 was transformed into Escherichia coli BL21 cells, and expressed and purified as previously described.9 GST-core RAG2, was expressed transiently in 293T cells and purified as previously described,39 with the exception that the detergent NP40 was not included in the final purification step. Concentrations of the purified proteins were determined as previously described.3
Limited proteolysis experiments and Isotope coding experiments
MBP-core RAG1 (1 μg) was incubated with porcine pancreatic trypsin, bovine pancreatic chymotrypsin, and Staphylococcus aureus endoproteinase Glu-C at 4°C for 3 h at a ratio of 1:50 (w/w) protease to MBP-core RAG1. The trypsin and endoproteinase Glu-C digestions were performed in 20 mM Tris, pH 8.0, 200 mM NaCl, 50 μM ZnCl2, and 5 mM β-mercaptoethanol, and the chymotrypsin digestion was in 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, and 10 mM CaCl2. All proteases were mass spectrometry grade reagents (Sigma). The digested products were desalted and purified by a C18 ZipTip (Millipore) and the peptides subsequently eluted in 50% acetonitrile and 0.1% TFA for analysis by MALDI-TOF mass spectrometry.
For limited proteolysis experiments on RAG1:RAG2 samples, purified MBP-core RAG1 (0.8 μM) and GST-core RAG2 (0.4 μM) were incubated 10 min at 4°C, which are at stoichiometries previously used for investigating specific RSS binding activity.24 Subsequently, samples were incubated with trypsin (at 16 nM) as above. Samples were prepared for MALDI-TOF mass spectrometry as described above.
Isotope coding experiments were conducted as follows. Following limited proteolysis as described above, the purified peptides generated from RAG1 alone and RAG1:RAG2 together were subjected to labeling by h-N-Acetoxy-succinimide (h3-NAS) and/or d3-N-Acetoxy-succinimide (d3-NAS) [NAS reagents generously provided by the Oklahoma Center for Medical Glycobiology (OCMG)] as follows. The RAG peptides were combined with a 3-fold molar excess of either h3-NAS or d3-NAS at pH 7.5, and incubated for 3 h at room temperature. Samples were desalted and purified as above. Resulting peptides labeled with h3-Ac and d3-Ac were combined as appropriate prior to analysis by mass spectrometry.
Mass spectrometry
MALDI-TOF mass spectrometry: Peptide samples were spotted on a grid with an equal volume of α-cyano-4-hydroxycinnamic acid. MALDI-TOF mass spectrometry experiments were performed in the OCMG on an Ultraflex II (Bruker Daltonics, Billerica, MA). The instrument was operated in reflectron positive ion mode with an accelerating voltage of 25 kV. Mass spectra were analyzed using flexAnalysis (Bruker Daltonics). The peaks in the spectrum were identified based on matching m/z values with the predicted monoisotopic masses of protease (trypsin, chymotrypsin, or endoproteinase Glu-C) generated peptides from the MBP-core RAG1 sequence with 0-3 missed cleavage sites. Tandem mass spectrometry: Peak m/z values that corresponded to two separate MBP-core RAG1 peptides were assigned by HPLC-MS-MS using a Dionex UltiMate 3000 and ABI MDS Sciex Qstar Elite in the Laboratory for Molecular Biology and Cytometry Research core facility at the University of Oklahoma Health Sciences Center.
Cellular V(D)J recombination assay
The transient cellular V(D)J recombination assay was performed using a slightly modified protocol as that previously described.16 Briefly, 4 μg of the recombination substrate pSF200 (generously provided by S. Fugmann) was combined with 8 μg each of constructs encoding GFP-core RAG1 and either GST-core RAG2 or Ch-FL-RAG2. The plasmids were transfected into HEK 293T cells grown at ∼80% confluency on 10 cm dishes using Fugene 6. Following 48 hrs, plasmid DNA was purified by alkaline lysis methods, treated with DpnI, and subsequently transformed into DH5α cells. Equal aliquots of the transformed cells were plated on LB agar plates containing 25 μg/ml kanamycin only or 25 μg/ml each of kanamycin and chloramphenicol. Colonies were counted following incubation at 37°C for 16 hours. Recombination activity was derived from the ratio of (Kan + Chl)R to KanR colonies, as previously described.16 Signal joint formation of purified plasmids recombined by WT GFP-core RAG1 combined with either GST-core RAG2 or Ch-FL-RAG2 were confirmed by ApaLI restriction digests (not shown).
Flow cytometry protein-interaction assay
Plasmids encoding GFP-core RAG1 (WT or mutant) and Ch-FL-RAG2 were transfected separately or together (2 μg of plasmid DNA total) into 293T cells at 80% confluency in 6-well plates using Fugene X-tremeGENE 9 (Roche). Following 24 h, cells were washed with cold PBS containing 100 mM PMSF, and lysed in buffer containing 20 mM Tris pH 7.6, 200 mM NaCl, 2 mM MgCl2, 0.5 mM DTT, 0.5% NP-40, 5% glycerol, and DNAse I (to 2 U/ml) for 10 min at 4°C. To ensure nuclear lysis, samples were subjected to sonication (15 s pulses, 5 s rests for 1.5 min at 20% power). Lysates were centrifuged to clear debris, and fluorescence intensities of supernatants were measured using the POLARstar Omega plate reader (BMG Labtech). Aliquots of lysates containing equivalent amounts of Ch-FL-RAG2 (based on readout from the fluorescence microplate reader) were incubated overnight at 4°C with 10 μL magnetic beads that were linked by camelidae antibody specific to red fluorescence protein, referred to as RFP-trap magnetic beads (Chromotek). Beads were blocked with 1 mg/mL BSA for 1 h prior to incubation with cell lysates. Following incubation with cell lysates, the beads were washed and resuspended in 300 μL of buffer containing 125 mM Tris, 150 mM NaCl, pH 8.0. In control experiments, beads were also incubated with lysates from 293T cells that were either untransfected or transfected with constructs encoding (1) GFP-core RAG1 only, (2) mCherry and WT GFP-core RAG1, and (3) Ch-FL-RAG2 and GFP.
Flow cytometry measurements were performed using a LSR II Flow Cytometer (BD Biosciences), where GFP and Cherry fluorophores were excited using 488 nm and 561 nm lasers, respectively. Post acquisition analysis was performed using FlowJo software (Tree Star, OR).
Acknowledgments
The authors thank LeAnn Godderz, Bruce Baggenstoss, Christa Feasley (in the Oklahoma Center for Medical Glycobiology core facility), and Stephen Snow (in the OUHSC Laboratory for Molecular Biology and Cytometry Research core facility) for assistance with the mass spectrometry measurements; Augen Pioszak for assistance with the fluorescence microplate reader, and Jacob Bass in the OMRF Flow Cytometry Core Facility for assistance with the flow cytometry measurements.
Glossary
- FL
full length
- GST
glutathione S-transferase
- GFP
green fluorescence protein
- MBP
maltose binding protein
- NAS
N-Acetoxy-succinimide
- NBD
nonamer binding domain
- RAG
recombination activating gene
- RFP
red fluorescence protein
- ZFB
zinc finger B
- RSS
recombination signal sequence
- WT
wild type
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- Rahman NS, Godderz LJ, Stray SJ, Capra JD, Rodgers KK. DNA cleavage of a cryptic recombination signal sequence by RAG1 and RAG2: implications for partial VH gene replacement. J Biol Chem. 2006;281:12370–12380. doi: 10.1074/jbc.M507906200. [DOI] [PubMed] [Google Scholar]
- Nambiar M, Raghavan SC. Mechanism of fragility at BCL2 gene minor breakpoint cluster region during t(14;18) chromosomal translocation. J Biol Chem. 2012;287:8688–8701. doi: 10.1074/jbc.M111.307363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki N, Askary A, Swanson PC, Lieber MR. Mechanistic basis for RAG discrimination between recombination sites and the off-target sites of human lymphomas. Mol Cell Biol. 2012;32:365–375. doi: 10.1128/MCB.06187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De P, Rodgers KK. Putting the pieces together: identification and characterization of structural domains in the V(D)J recombination protein RAG1. Immunol Rev. 2004;200:70–82. doi: 10.1111/j.0105-2896.2004.00154.x. [DOI] [PubMed] [Google Scholar]
- Jones JM, Simkus C. The roles of the RAG1 and RAG2 “non-core” regions in V(D)J recombination and lymphocyte development. Arch Immunol Ther Exp. 2009;57:105–116. doi: 10.1007/s00005-009-0011-3. [DOI] [PubMed] [Google Scholar]
- Yin FF, Bailey S, Innis CA, Ciubotaru M, Kamtekar S, Steitz TA, Schatz DG. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat Struct Mol Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuckle JL, Fauss LJ, Simpson R, Ptaszek LM, Rodgers KK. Identification of two topologically independent domains in RAG1 and their role in macromolecular interactions relevant to V(D)J recombination. J Biol Chem. 2001;276:37093–37101. doi: 10.1074/jbc.M105988200. [DOI] [PubMed] [Google Scholar]
- Peak MM, Arbuckle JL, Rodgers KK. The central domain of core RAG1 preferentially recognizes single-stranded recombination signal sequence heptamer. J Biol Chem. 2003;278:18235–18240. doi: 10.1074/jbc.M302041200. [DOI] [PubMed] [Google Scholar]
- Rodgers KK, Bu Z, Fleming KG, Schatz DG, Engelman DM, Coleman JE. A zinc-binding domain involved in the dimerization of RAG1. J Mol Biol. 1996;260:70–84. doi: 10.1006/jmbi.1996.0382. [DOI] [PubMed] [Google Scholar]
- De P, Peak MM, Rodgers KK. DNA cleavage activity of the V(D)J recombination protein RAG1 is autoregulated. Mol Cell Biol. 2004;24:6850–6860. doi: 10.1128/MCB.24.15.6850-6860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo X, Bailin T, Sadofsky MJ. A C-terminal region of RAG1 contacts the coding DNA during V(D)J recombination. Mol Cell Biol. 2001;21:2038–2047. doi: 10.1128/MCB.21.6.2038-2047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I, Mornon J-P. The V(D)J recombination activating protein RAG2 consists of a six-bladed propeller and a PHD fingerlike domain, as revealed by sequence analysis. Cell Mol Life Sci. 1998;54:880–891. doi: 10.1007/s000180050216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez CA, Ptaszek LM, Villa A, Bozzi F, Sobacchi C, Brooks EG, Notarangelo LD, Spanopoulou E, Pan ZQ, Vezzoni P, Cortes P, Santagata S. Mutations in conserved regions of the predicted RAG2 kelch repeats block initiation of V(D)J recombination and result in primary immunodeficiencies. Mol Cell Biol. 2000;20:5653–5664. doi: 10.1128/mcb.20.15.5653-5664.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugmann SD, Schatz DG. Identification of basic residues in RAG2 critical for DNA binding by the RAG1-RAG2 complex. Mol Cell. 2001;8:899–910. doi: 10.1016/s1097-2765(01)00352-5. [DOI] [PubMed] [Google Scholar]
- Qiu J-X, Kale SB, Yarnal Schultz H, Roth DB. Separation-of-function mutants reveal critical roles for RAG2 in both the cleavage and joining steps of V(D)J recombination. Mol Cell. 2001;7:77–87. doi: 10.1016/s1097-2765(01)00156-3. [DOI] [PubMed] [Google Scholar]
- Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, Gatta LB, Ochs HD, Schwarz K, Notarangelo LD, Vezzoni P, Spanopoulou E. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93:885–896. doi: 10.1016/s0092-8674(00)81448-8. [DOI] [PubMed] [Google Scholar]
- Swanson PC. A RAG-1/RAG-2 tetramer supports 12/23-regulated synapsis, cleavage and transposition of V(D)J recombination signals. Mol Cell Biol. 2002;22:7790–7801. doi: 10.1128/MCB.22.22.7790-7801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy GJ, Ramon-Maiques S, Dimitriadis EK, Kotova S, Biertumpfel C, Heymann JB, Steven AC, Gellert M, Yang W. Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the postcleavage complex. Mol Cell. 2009;35:217–227. doi: 10.1016/j.molcel.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godderz LJ, Rahman NS, Risinger GM, Arbuckle JL, Rodgers KK. Self-association and conformational properties of RAG1: implications for formation of the V(D)J recombinase. Nucleic Acids Res. 2003;31:2014–2023. doi: 10.1093/nar/gkg281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhtenko LS, Gilmore J, Kriatchko AN, Kumar S, Swanson PC, Lyubchenko YL. Molecular mechanism underlying RAG1/RAG2 synaptic complex formation. J Biol Chem. 2009;284:20956–20965. doi: 10.1074/jbc.M109.028977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson PC, Desiderio S. RAG-2 promotes heptamer occupancy by RAG-1 in the assembly of a V(D)J initiation complex. Mol Cell Biol. 1999;19:3674–3683. doi: 10.1128/mcb.19.5.3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Gwyn LM, De P, Rodgers KK. A non-sequence-specific DNA binding mode of RAG1 is inhibited by RAG2. J Mol Biol. 2009;387:744–758. doi: 10.1016/j.jmb.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aidinis V, Dias DC, Gomez CA, Bhattacharyya D, Spanopoulou E, Santagata S. Definition of minimal domains of interaction within the recombination-activating genes 1 and 2 recombinase complex. J Immunol. 2000;164:5826–5832. doi: 10.4049/jimmunol.164.11.5826. [DOI] [PubMed] [Google Scholar]
- McMahan CJ, Sadofsky MJ, Schatz DG. Definition of a large region of RAG1 that is important for coimmunoprecipitation of RAG2. J Immunol. 1997;158:2202–2210. [PubMed] [Google Scholar]
- Ko JE, Kim CW, Kim DR. Amino acid residues in RAG1 responsible for the interaction with RAG2 during the V(D)J recombination process. J Biol Chem. 2004;279:7715–7720. doi: 10.1074/jbc.M311471200. [DOI] [PubMed] [Google Scholar]
- Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Polon. 2004;51:299–321. [PubMed] [Google Scholar]
- De P, Zhao S, Gwyn LM, Godderz LJ, Peak MM, Rodgers KK. Thermal dependency of RAG1 self-association properties. BMC Biochem. 2008;9:5. doi: 10.1186/1471-2091-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurlino JC, Lu GY, Quiocho FA. The 2.3-A resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J Biol Chem. 1991;266:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- Blazer LL, Roman DL, Muxlow MR, Neubig RR. Use of flow cytometric methods to quantify protein-protein interactions. Curr Protoc Cytometry Chapter 13:Unit 13. 2010;11:11–15. doi: 10.1002/0471142956.cy1311s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy GJ, Yang W, Gellert M. Autoinhibition of DNA cleavage mediated by RAG1 and RAG2 is overcome by an epigenetic signal in V(D)J recombination. Proc Natl Acad Sci USA. 2010;107:22487–22492. doi: 10.1073/pnas.1014958107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, Yang W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci USA. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchuk DJ, Mansilla-Soto J, Alarcon C, Singha NC, Langen H, Bianchi ME, Lees-Miller SP, Nussenzweig MC, Cortes P. Ku70/Ku80 and DNA-dependent protein kinase catalytic subunit modulate RAG-mediated cleavage: implications for the enforcement of the 12/23 rule. J Biol Chem. 2004;279:29821–29831. doi: 10.1074/jbc.M403706200. [DOI] [PubMed] [Google Scholar]
- Ciubotaru M, Ptaszek LM, Baker GA, Baker SN, Bright FV, Schatz DG. RAG1-DNA binding in V(D)J recombination: specificity and DNA-induced conformational changes revealed by fluorescence and CD spectroscopy. J Biol Chem. 2003;278:5584–5596. doi: 10.1074/jbc.M209758200. [DOI] [PubMed] [Google Scholar]
- Wang RS, Wang Y, Wu LY, Zhang XS, Chen L. Analysis on multi-domain cooperation for predicting protein-protein interactions. BMC Bioinform. 2007;8:391. doi: 10.1186/1471-2105-8-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moza B, Buonpane RA, Zhu P, Herfst CA, Rahman AK, McCormick JK, Kranz DM, Sundberg EJ. Long-range cooperative binding effects in a T cell receptor variable domain. Proc Natl Acad Sci USA. 2006;103:9867–9872. doi: 10.1073/pnas.0600220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers KK, Villey IJ, Ptaszek L, Corbett E, Schatz DG, Coleman JE. A dimer of the lymphoid protein RAG1 recognizes the recombination signal sequence and the complex stably incorporates the high mobility group protein HMG2. Nucleic Acids Res. 1999;27:2938–2946. doi: 10.1093/nar/27.14.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanopoulou E, Zaitseva F, Wang F-H, Santagata S, Baltimore D, Panayotou G. The homeodomain region of Rag-1 reveals the parallel mechanisms of bacterial and V(D)J recombination. Cell. 1996;87:263–276. doi: 10.1016/s0092-8674(00)81344-6. [DOI] [PubMed] [Google Scholar]
- Arbuckle JL, Rahman NS, Zhao S, Rodgers W, Rodgers KK. Elucidating the domain architecture and functions of non-core RAG1: the capacity of a non-core zinc-binding domain to function in nuclear import and nucleic acid binding. BMC Biochem. 2011;12:23. doi: 10.1186/1471-2091-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W, Rodgers W, Hughes T. Making genes green: creating green fluorescent protein (GFP) fusions with blunt-end PCR products. Biotechniques. 1998;25:94–96, 98. doi: 10.2144/98251st05. [DOI] [PubMed] [Google Scholar]
- Gwyn LM, Peak MM, De P, Rahman NS, Rodgers KK. A zinc site in the C-terminal domain of RAG1 is essential for DNA cleavage activity. J Mol Biol. 2009;390:863–878. doi: 10.1016/j.jmb.2009.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse JE, Lieber MR, Gellert M, Mizuuchi K. Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J joining signals. Cell. 1987;49:775–783. doi: 10.1016/0092-8674(87)90615-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information