

Abstract
A potential new photosensitizer based on a dissymmetric porphyrin derivative bearing a thiol group was synthesized. 5-[4-(11-Mercaptoundecyloxy)-phenyl-10,15,20-triphenylporphyrin (PR-SH) was used to functionalize gold nanoparticles in order to obtain a potential drug delivery system. Water-soluble multifunctional gold nanoparticles GNP-PR/PEG were prepared using the Brust–Schiffrin methodology, by immobilization of both a thiolated polyethylene glycol (PEG) and the porphyrin thiol compound (PR-SH). The nanoparticles were fully characterized by transmission electron microscopy and 1H nuclear magnetic resonance spectroscopy, UV/Vis absorption spectroscopy, and X-ray photoelectron spectroscopy. Furthermore, the ability of GNP-PR/PEGs to induce singlet oxygen production was analyzed to demonstrate the activity of the photosensitizer. Cytotoxicity experiments showed the nanoparticles are nontoxic. Finally, cellular uptake experiments demonstrated that the functionalized gold nanoparticles are internalized. Therefore, this colloid can be considered to be a novel nanosystem that could potentially be suitable as an intracellular drug delivery system of photosensitizers for photodynamic therapy.
Keywords: drug delivery, gold nanoparticles, photodynamic therapy, photosensitizers, porphyrins
Introduction
Porphyrins and their derivatives are components present in several complexes in nature such as haemoglobin, chlorophyll, cobalamine, and bacterioclorophylls.1 They are also one of the most popular aromatic macrocycles in supramolecular chemistry because of their multiple functions.2, 3 Recently, the synthesis of new porphyrin derivatives has been increasingly investigated owing to their great potential in such diverse areas as photochemistry, molecular recognition, sensors, and molecular rotors, as well as in photodynamic cancer therapy.4–8 Also, the development of a synthetic porphyrin methodology for nonsymmetric systems is one of great interest because the existing procedures to synthesize them are low yielding.9–11 Especially, the synthesis of dissymmetrical porphyrins presents an extra difficulty because a mixture of porphyrins is generally obtained, a fact that usually implies the need for complex purification processes.12–14
Porphyrins are photosensitizers (PS) that can promote the formation of singlet oxygen or free radicals that are highly toxic locally at the cellular level.15–17 The target tissue is typically irradiated with red light. PS can cause significant cellular damage, destroy tumor blood vessels, and induce immunity against the damaged tissues.18 The accumulation of the PS in the target region is one of the main difficulties associated with photodynamic therapy (PDT) that makes use of the photosensitizing capability of the porphyrins. More specifically, one of the drawbacks of PDT is that the porphyrin structures that are used generally are quite hydrophobic, which makes them difficult to formulate. However, the most serious disadvantage of PDT is the nonspecific distribution of the PS in the body, which leads to adverse effects upon a patients exposure to sunlight. On the other hand, gold nanoparticles (GNPs) have raised huge interest in the biomedical field because of their biocompatibility, their ready and versatile synthesis, as well as their ability to act as drug delivery vehicles whereby suitable functionalization can lead to site-specific accumulation.19, 20
Therefore, photosensitizer-coated nanoparticles (NPs) are a promising platform currently investigated to improve specificity in PDT, while at the same time allowing the hydrophobicity of the photosensitizer to be circumvented.21 Despite the interest in such systems, only a few examples combine NPs and photosensitizers, specifically phtalocyanines and porphyrins,22–27 as well as incorporate an antibody against a receptor present in breast cancer cells.28 In particular, monopodal porphyrins containing a thiol group are amenable to be linked covalently to gold surfaces29–31 as well as be coated on GNPs.32–34 Depending on the solubility of the photosensitizer, either the Brust–Schiffrin35 or the Turkevich36 methods can be used for GNPs preparation. Systems based on porphyrin-functionalized GNPs for PDT have been reported where porphyrin-coated GNPs were prepared, and their PDT efficacy was demonstrated in vitro and in vivo.37–39 In these published works, protoporphyrin, hematoporphyrin, and brucine–porphyrin derivatives were used to functionalize GNPs, indicating that these porphyrin derivatives are more efficient for PDT when they are immobilized on the GNPs, in comparison with the free ligands. The Turkevich method was used in these examples, because the porphyrin derivatives used are water-soluble.
As an alternative to the method that generated these earlier particles, the Brust–Schiffrin method has been employed in this work due to the possibility to immobilize non-water-soluble ligands onto the GNPs, because a biphasic solution is used. Thus, the capability to incorporate a lipophilic PS onto the GNP surface was proven by preparing a double coating with a porphyrin derivative and a polyethylene glycol (PEG) that increases the water solubility of the nanosystem.
In this context, this study describes first the synthesis of a new dissymmetrical porphyrin composed of four phenyl groups at the meso positions, with one of them incorporating an alkyl chain with a thiol end group. Thus, 5-[4-(11-mercaptoundecyloxy)-phenyl-10,15,20-triphenylporphyrin (PR-SH) was prepared, and its capability to coat and stabilize water-soluble GNPs have been investigated. The porphyrin was chosen because of its similarity to second generation photosensitizers, previously studied in solution for PDT,40–42 and the thiol group allows its incorporation onto GNPs. Therefore, water-soluble GNPs (GNP-PR/PEG) have been synthesized, coadsorbing the thiol-modified porphyrin and a PEG-modified thiol chain to the surface of the metal. GNPs were prepared by the Brust–Schiffrin method providing a potential water-soluble drug delivery system in order to localize the activity of the photosensitizer. The same methodology was used to synthesize GNP-PEG, which were used for the control experiments. The GNPs were characterized using a variety of techniques such as proton nuclear magnetic resonance spectroscopy (1H NMR), UV/Vis absorption spectroscopy, transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS). Furthermore, the production of singlet oxygen after irradiation was measured, and the cytotoxicity of the GNPs was also analyzed in human mammary epithelial cell cultures using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) proliferation assay kit. Internalization and GNP localization inside cells were assessed by inductively coupled plasma mass spectrometry (ICP-MS) and TEM, respectively.
Results and Discussion
Synthesis and characterization of the photosensitizer PR-SH
The formation of the dissymmetrical porphyrin PR-OH, incorporating three phenyl groups and one hydroxyphenyl group at the meso positions, was achieved by reaction of pyrrole with a mixture of benzaldehyde and 4-hydroxybenzaldehyde in propionic acid (Scheme 1), that gives a mixture of all possible meso-substituted porphyrins.
Scheme 1.
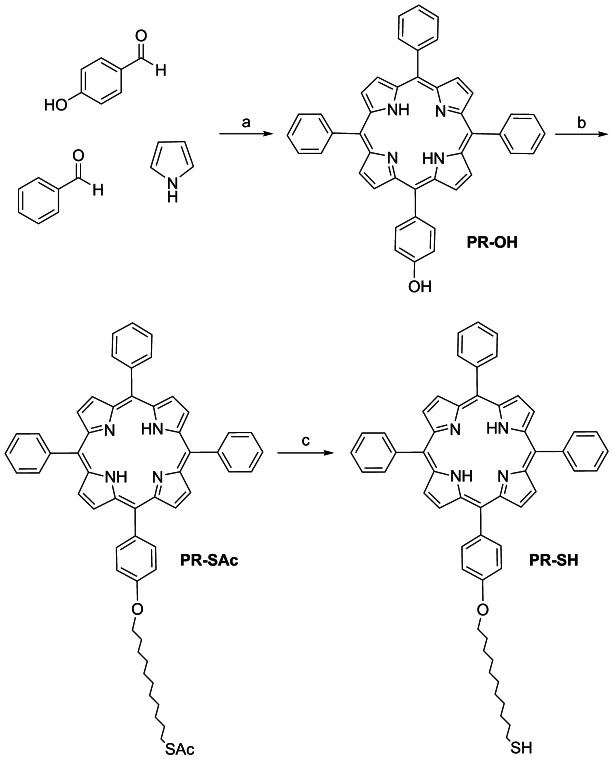
Stepwise synthesis of PR-SH. Reagents and conditions: a) propionic acid, reflux, 2 h, 5 %; b) 11-bromoundecyl thioacetate, K2CO3, CH3CN, 82 °C, 24 h, 67 %; c) KOH, MeOH/H2O, rt, 3 h, 80 %.
Two factors facilitated the isolation of PR-OH: first, acetonitrile washing allowed the elimination of all the impurities and pyrrole secondary products, and secondly, two rounds of silica gel column chromatography using dichloromethane and consequently chloroform, gave good separation of the desired porphyrin PR-OH (5.4 %).43
PR-OH was isolated as a purple solid, and this material was further purified by crystallization from acetonitrile, giving crystals suitable for single-crystal X-ray characterization to know the order and packing of the structure (see Supporting Information).
The thiolated chain was incorporated by the reaction of PR-OH with 11-bromoundecyl thioacetate under basic conditions yielding PR-SAc (67 %). Subsequently, thioacetate PR-SAc was deprotected using potassium hydroxide in a mixture of methanol and water, giving the desired compound PR-SH as a purple oil (80 %) (Scheme 1). The thiol chain incorporated into the porphyrin acts as a linker between the photosensitizer and the GNP.
The free porphyrins were identified and characterized using 1H NMR spectroscopy. Deprotection of porphyrin PR-SAc to obtain PR-SH was monitored in deuterated chloroform. The peak corresponding to the methyl group of the protected thiol at 2.32 ppm was not present in the spectrum of the deprotected porphyrin (PR-SH), confirming the efficiency of the deprotection protocol. Furthermore, a shift of the peak corresponding to the S−CH2 (from 2.88 to 2.48 ppm) was observed, indicating the presence of the SH group in PR-SH (Figure S1 in the Supporting Information).
Preparation of gold nanoparticles (GNP-PR/PEG)
The preparation of water-soluble GNPs was performed using the biphasic method described by Brust and Schiffrin because of the low hydrophilicity of the porphyrin PR-SH (Scheme 2). Tetraoctyl ammonium bromide (TOAB) was used as a phase-transfer agent in order to allow the interaction between the PR-SH, soluble in toluene, and the gold dissolved in water. Thiol-terminated PEG was also incorporated to the aqueous phase to increase the polarity of the final colloidal system. GNP-PR/PEG, water-soluble NPs functionalized with the PR-SH and PEG, were isolated from the aqueous phase and were purified by sequential washing and centrifugation. In order to provide a model colloid for the biological control experiments, GNP-PEG colloids were synthesized using the same methodology described above, but without the incorporation of the photosensitizer.
Scheme 2.
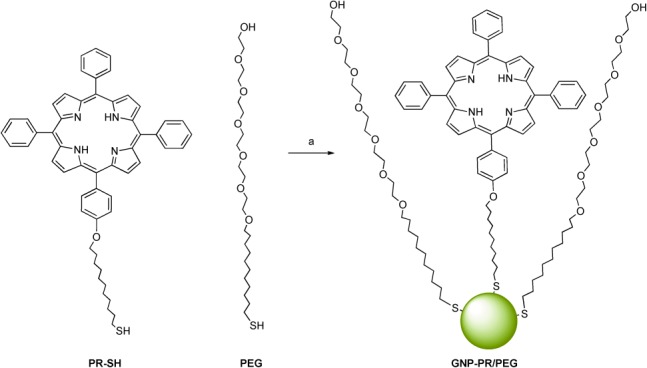
Synthesis of GNP-PR/PEG. Reagents and conditions: a) HAuCl4⋅3H2O, NaBH4, TOAB, toluene/H2O, rt, 16 h.
UV/Vis absorption spectroscopy
GNP formation and the presence of the porphyrin on the GNP were confirmed by UV/Vis absorption spectroscopy. The GNPs prepared here have a characteristic peak near 520 nm originating from their surface plasmon absorption.35 Moreover, free-base porphyrins also present five characteristic absorption bands: the Soret band (near 420 nm) and four Q bands between 500 and 700 nm.2 Figure 1 shows the UV/Vis spectra of the free-base porphyrin PR-SH and GNP-PR/PEG. PR-SH shows the typical porphyrin bands: the Soret band at 421 nm and the Q bands at 515, 551, 593, and 646 nm. In the case of the GNPs, the Soret band still appears at 421 nm, and although the spectra are quite broad, weak absorption bands corresponding to the four Q bands of the porphyrin can be identified. The typical surface plasmon resonance (SPR) peak of the GNP at 520 nm coincides with the lowest wavelength absorption peak of the Q bands of the porphyrin. UV/Vis absorption spectroscopy results also demonstrate that the core of the porphyrin is still metal-free once attached to the GNPs. During the synthesis of the GNPs, gold atoms might be incorporated into the pyrrole ring because it can complex the gold(III) ion.44 When the porphyrin complexes with gold, only one Q band appears; although in the UV/Vis spectra for GNP-PR/PEG the four Q bands were identified, demonstrating a metal-free porphyrin pyrrole ring. Fluorescence of the GNP-PR/PEG was also examined, indicating a low-intensity emission.
Figure 1.
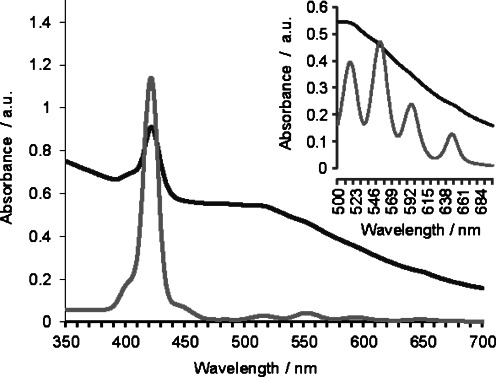
UV/Vis absorption spectra of solutions of PR-SH (0.25 mm) (light grey) and GNP-PR/PEG (0.4 mg mL−1) (dark grey). The spectra of PR-SH and GNP-PR/PEG were recorded in CH2Cl2 and H2O, respectively. For PR-SH (0.5 mm), magnification of the Q band region of the spectrum is also shown.
Quantification of the amount of porphyrin per GNP was attempted using the absorption spectra and taking into account the diameter of the colloid determined by TEM (see section below). First, a calibration line of PR-SH was obtained using a range of concentrations between 25 μm and 0.5 μm (Figure S3 in the Supporting Information) in order to calculate its extinction coefficient (ɛ418=38 940). The wavelength selected was that corresponding to the Soret band (418 nm) because it was the most intense peak confirming the porphyrin that appeared in the GNP spectrum.45 The UV/Vis absorption spectrum of GNP-PR/PEG shows quite broad absorption bands, and in order to normalize the absorbance value of the Soret band, the background signal of the GNP core at 442 nm was subtracted from the Soret band peak (Figure S4 in the Supporting Information).
Accordingly, we calculated that the molarity of the porphyrin present on the surface of the GNPs corresponds to 7.30 μm for GNP-PR/PEG. In order to find the porphyrin to NP ratio, the concentration of GNP was calculated using the diameter obtained by TEM (3–4 nm) and its absorbance value at 450 nm (0.56) obtaining a mean value of about 0.22 μm.46 Using a volume of 2 mL and Avogadros number, we get a ratio of about 33 porphyrin adducts per GNP for GNP-PR/PEG.
X-ray photoelectron spectroscopy analysis
To confirm the formation of the GNPs and their functionalization with PR-SH, XPS experiments were performed. Initially, in order to detect the gold atoms, a sputtering of the sample of GNP-PR/PEG was necessary to identify the peaks clearly.47 The sputtering is normally used when the NP is completely coated, and the photoelectrons need to penetrate to detect the gold atoms. Figure S5 in the Supporting Information shows the binding energies of the XPS spectrum of GNP-PR/PEG, and the most intense peaks correspond to elements C and O, indicating the presence of the PEG chain on the GNPs. Furthermore, typical peaks for the presence of Au appear at 88.5 and 85 eV corresponding to the double peaks of Au 4f5/2 and Au 4f7/2. These are typical values for Au0. Also, very-low-intensity peaks appear in the S and the N1s regions, showing the presence of pyrrolic nitrogen atoms (N−H) that confirm the presence of the porphyrin core.48
Transmission electron microscopy analysis
A morphologic and size study of the GNPs described here was done using TEM. A drop of the GNP solutions was placed on a carbon-coated copper grid for the analysis. Figure 2 a shows a representative TEM image of the porphyrin-bearing GNP-PR/PEG. Spherical NPs with a narrow distribution, in terms of size polydispersity and polymorphism, were observed. The corresponding histogram shows diameters in the range of 3.5 to 4 nm (Figure 2 b). The water-soluble NPs containing only PEG (GNP-PEG) give a practically identical average core size between 3.5 and 4 nm (Figure S6 in the Supporting Information). In both cases, the particles are well separated and in very few cases show short distances between particles, a good indication that they are well dispersed in water and can be used as essentially single-particle delivery systems.
Figure 2.
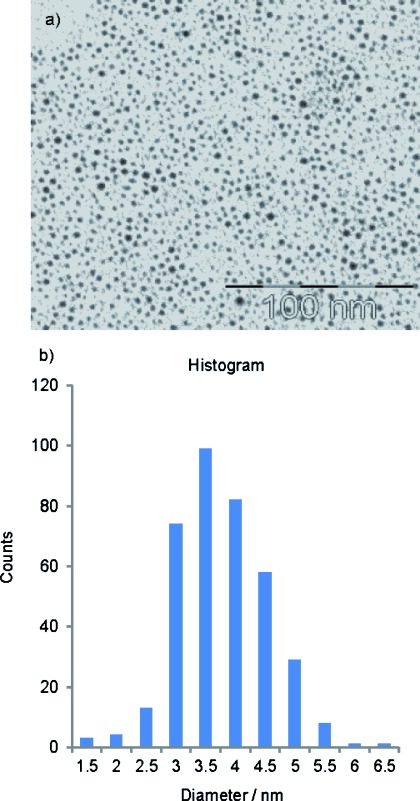
Transmission electron microscopy (TEM) images of a) GNP-PR/PEG and b) their corresponding size-distribution histogram.
Measure of singlet oxygen production
In order to demonstrate the promising use of the GNP-PR/PEG in PDT, its capacity to induce the production of reactive oxygen species after laser irradiation was tested. The singlet oxygen production was examined by measuring the fluorescence decay of 9,10-anthracenedipropionic acid (ADPA), because it is photobleached in the presence of singlet oxygen.49–51
Thus, ADPA was added to a water solution of GNP-PR/PEG (0.25 mg mL−1) and the sample was irradiated at 647 nm for 30 min. The sample was irradiated at 647 nm because it corresponds into the porphyrin Q band nearest to the red region, which is a wavelength region commonly used in PDT.16 Figure 3 shows the fluorescence spectra of ADPA, taken in 5 min intervals over 30 min, as well as the corresponding fluorescence intensity decay in the typical 407 nm peak of the ADPA. After 10 min of irradiation, a significant decay in fluorescence intensity started, demonstrating the capacity of porphyrin-modified GNP to induce singlet oxygen production, making it a promising candidate for PDT.
Figure 3.
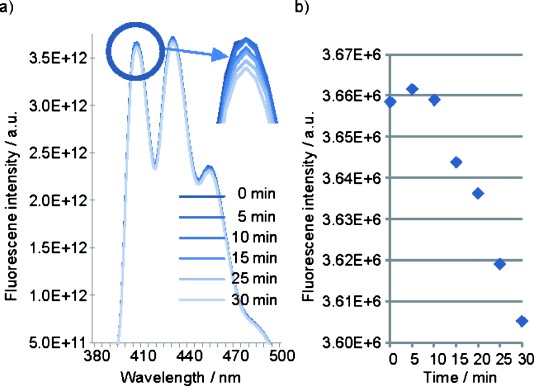
a) Fluorescence emission spectra of ADPA after irradiation of GNP-PR/PEG at 647 nm for 30 min, and b) decay of ADPA fluorescence intensity at 407 nm.
Cytotoxicity of GNP-PEG and GNP-PR/PEG
To assess the suitability of GNP-PEG and GNP-PR/PEG for PDT, it is necessary to evaluate their potential cytotoxicity in living cells. For this reason, MCF10A cells were chosen. The MTT assay has been widely used52, 53 and recommended for the evaluation of the cytotoxicity of NPs.54
After 24 and 72 h of culture, there was a clear and significant dose-dependent cytotoxic effect for both types of NPs, starting at 5 μg mL−1 (Figure 4). GNP-PEG was significantly more cytotoxic than the GNP-PR/PEG after incubation at both 24 and 72 h. After 24 h of incubation, the IC50 was found at 15 μg mL−1 for GNP-PEG whereas for GNP-PR/PEG, it was at 200 μg mL−1. At 72 h, the IC50 was found at 15 μg mL−1 for GNP-PEG, and at 20 μg mL−1 for GNP-PR/PEG. According to literature,54–56 the cytotoxicity of GNPs depends on their size and surface properties (charge, ligand density, hydrophobicity) and on their concentration. As each published study has been done with different GNP sizes, surface properties, concentrations, and different cell lines, it is extremely difficult to compare the results obtained in the present article with the published ones in a meaningful way, although some trends can be found.
Figure 4.
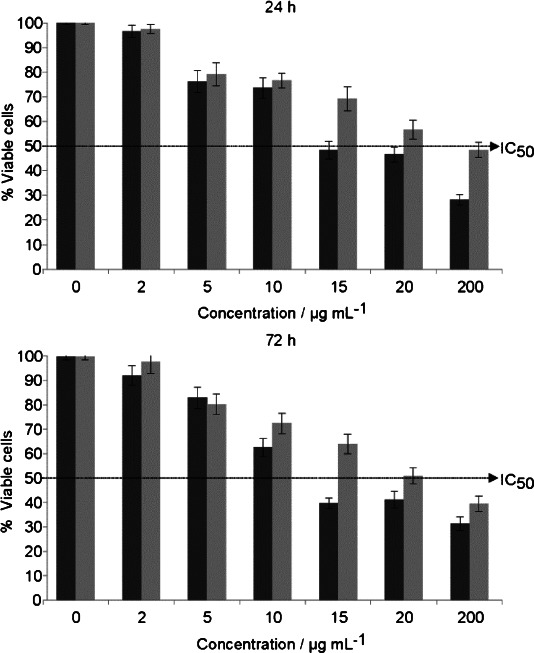
Toxicity of GNP-PEG (dark grey) and GNP-PR/PEG (light grey). Cells were incubated in the presence of different concentrations of GNPs for 24 h (top) or 72 h (bottom). The discontinuous line corresponds to 50 % of viable cells. Data represent the mean ± SEM (n=3).
Regarding the size of the GNPs, on the one hand, citrate-stabilized GNPs of 5 nm have been shown to be cytotoxic at a concentration higher than 50 μm (10 μg mL−1), when incubated for 72 h with Balb/3T3 cells in an assay for colony forming efficiency. However, using the same concentration of GNPs of 15 nm did not show cytotoxicity even at the highest concentration tested—300 μm (58.8 μg mL−1).57 On the other hand, it has been described that cell viability of MCF-7 and PC-3 cells was not decreased when incubated with citrate-capped GNPs of different sizes (5, 6, 10, 17 and 45 nm) and only slightly decreased when incubated with similar sizes (3, 8 and 30 nm). Thus, in that work GNP size was not related with viability at least in a size-dependent manner.58 In the present study, the IC50 for GNP-PR/PEG is 200 μg mL−1 at 24 h and 20 μg mL−1 at 72 h in contrast with the GNP-PEG which is more cytotoxic (IC50=15 μg mL−1 at 24 and 72 h). In this case, the size of the particles are similar, although GNP-PEG is slightly smaller than GNP-PR/PEG; consequently, in the present study the differences in cytotoxicity are not related to GNP size.
Taking into account the surface properties, it must be pointed out that GNP-PEG has been used in several studies. It has been described in literature that 30, 50, and 90 nm plain GNPs stimulated cell proliferation, whereas PEGylated GNP spheres of the same size did not interfere with proliferation of PC-3 cells at a concentration of 1.5 nm for 88 h.59 Other works, using GNP@MPA-PEG of 3.5 nm diameter at different concentrations (0.08–10 μm) found that cytotoxicity was dose-dependent, obtaining a 70 % viability when incubated with HeLa cells for 72 h using the highest concentration.60 On the other hand, no cytotoxicity was observed when CT26 cells were incubated with 4.7 nm GNP-PEG for 48 h at high concentrations (500–3000 μm).61 Finally, GNPs functionalized with different PS have also been tested for cell viability. GNP-porphyrin–brucine (14.7 nm) was not cytotoxic at 1 or 2.5 μm using PE/CA-PJ34 cells,32 whereas GNP-hematoporphyrin (15 and 45 nm) incubated with MT-4 cells was cytotoxic in a dose-dependent manner (different percentages of hematoporphyrin and Au were tested).33 Our results are in agreement with a dose-dependent cytotoxicity of GNPs. However, a lower cytotoxicity was found for GNP-PR/PEG when compared with GNP-PEG, that could be explained, at least in part, by the presence of porphyrin which reduces the number of PEG molecules and probably their cytotoxicity, because it is known that photosensitizers are not toxic by themselves.22 Taking all these results, we can confirm that GNP-PR/PEG does not affect cell viability at low concentrations of NPs (2–10 μg mL−1), and thus it is a good candidate for PDT.
Uptake of GNP-PEG and GNP-PR/PEG
The uptake of GNP-PEG and GNP-PR/PEG was analyzed by means of TEM and ICP-MS. Whereas TEM determines the localization of NPs inside cells, ICP-MS enables quantification of NPs internalized by the cells. The total amount of Au present in the cells (pellet) and in the culture medium (together with phosphate-buffered saline washes) was quantified using ICP-MS. When cells were incubated for 24 h with 200 μg mL−1 of GNP-PEG, 1.97 % of the total amount of Au was detected in the pellet, indicating that some GNP-PEG was internalized by cells. In the same conditions, the cells were able to internalize 0.72 % of the total amount of the gold present in 200 μg mL−1 of GNP-PR/PEG. Thus, the cells internalized more Au from GNP-PEG than from GNP-PR/PEG. This could explain, in part, the higher cytotoxicity of GNP-PEG in relation to GNP-PR/PEG. However, the IC50 of GNP-PEG was 15 μg mL−1, whereas the IC50 of GNP-PR/PEG was more than 10 times this value (200 μg mL−1).
Concerning cell uptake of GNP-PEG, it has been described that PEG prevents nonspecific interactions of serum proteins with NPs and, in turn, their recruitment and internalization,46 but at least two studies have reported the internalization of 3–5 nm GNP-PEG.60, 61 In both cases, as in the present work, internalization was analyzed by ICP-MS and TEM, demonstrating that these GNPs can be internalized by different types of cells: HeLa,60 CT26,61 and MCF10A (present work). The amount of GNPs internalized varies within studies, probably due to type of GNP-PEG used and its concentration.
Analysis of the ultrathin sections of MCF10A cells incubated with 100 μg mL−1 GNPs for 24 h has shown that both kinds of NPs, GNP-PEG and GNP-PR/PEG, have been taken up by the cells, and that, once inside, they are not aggregated. This is relevant because it has been described that the cytotoxicity of GNPs biofunctionalized with polyvinyl pyrrolidone (GNP-PVP) on HeLa cells was caused by NP aggregation rather than particle size.62 Small size (2 nm) NPs were not cytotoxic and did not aggregate inside HeLa cells in contrast with 25 nm GNP-PVP, which caused 50 % cell death after 1 day in culture and presented large aggregates in TEM sections. NP clustering is an added problem when internalization uptake is compared since it is not known if aggregates are formed prior to or during interaction with the membrane.55
On the other hand, it has been suggested that it is necessary to produce enough free energy to bind NPs to the surface of the cell membranes; NPs smaller than 14 nm need to aggregate with other NPs in order to be able to wrap the membrane, while NPs larger than 50 nm can reach the membrane and be endocytosed individually by cells.63 That would explain why GNP-PEG and GNP-PR/PEG, which are smaller than 50 nm, are internalized less efficiently. Owing to the small size and nonaggregation of the NPs of the present study, it was difficult to observe these particles inside the cells, but both types, GNP-PEG and GNP-PR/PEG, were found either in the cytoplasm, in vesicles, in the Golgi, and also in the nucleus (Figure 5). Some authors have found large aggregates of 5 nm and 15 nm GNPs inside vesicles, but not in other subcellular organelles.57 Others have found that 3.5 nm GNP@MPA-PEG can reach the nuclear envelope forming large aggregates, clusters of few NPs, and even some individual ones.60 It has also been described that plain spherical GNPs of 30 nm are internalized mainly in clusters, and, once inside the cells, NPs can be seen as single particles or as clusters inside vesicles, free in the cytosol, or even in the nucleus.59 Finally, individual particles and aggregations of GNP-PVP of 2, 10, and 25 nm in the cytoplasm, mitochondria, and vesicles have been also described.62 Overall, we can conclude that the surface properties of the GNP-PEG and GNP-PR/PEG prevent their clustering, hampering their uptake by MCF10A cells. However, we have demonstrated that they can be internalized, and that some particles of GNP-PR/PEG were individually localized in the cytoplasm.
Figure 5.
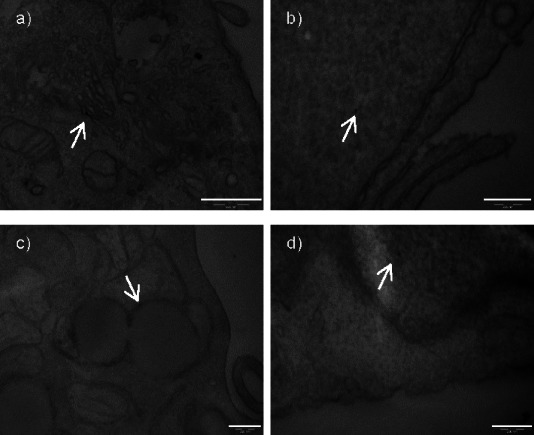
Transmission electron microscopy (TEM) images of MCF10A cells incubated with GNP-PEG (a,b) or GNP-PR/PEG (c,d) at 24 h. Intracellular distribution: Golgi (a), cytoplasm (b), vesicles (c,d). Bar=200 nm.
Conclusions
A new thiolated porphyrin (PR-SH) has been synthesized in only a few steps. The capability of the porphyrin to be attached to gold nanoparticles (GNPs) as well as to stabilize them has been demonstrated. The corresponding GNP-PR/PEG gold colloid was prepared using the Brust–Schiffrin method incorporating the photosensitizer and a polyethylene glycol (PEG) chain in order to make the nanoparticles water soluble. Transmission electron microscopy images of GNP-PR/PEG have been obtained showing well-dispersed spherical nanoparticles with a size near 3–4 nm. The coverage of the functionalized tetrapyrrole on the GNPs is approximately 30 porphyrins per nanoparticle. The capability of the porphyrin-modified GNP to induce singlet oxygen production was tested by observing 9,10-anthracenedipropionic acid (ADPA) photobleaching upon irradiation.
Cytotoxicity experiments were carried out with the GNP-PR/PEG with GNP-PEG as a control. IC50 for GNP-PR/PEG and GNP-PEG after 24 h were 200 μg mL−1 and 15 μg mL−1, respectively, demonstrating that they are nontoxic. Finally, uptake experiments using MCF10A cells demonstrated the internalization of the modified GNPs. Therefore, GNP-PR/PEG is novel nanosystem that could be potentially suitable as a drug delivery system for photodynamic therapy.
Experimental Section
Solvents and reagents: propionic acid, EtOAc, hexane, distilled H2O, CH2Cl2, CHCl3, CH3CN, MeOH, toluene, and absolute EtOH were analytical grade. Commercial compounds: 9,10-anthracenedipropionic acid (ADPA), pyrrole, p-hydroxybenzaldehyde, benzaldehyde, S-(11-bromoundecyl) thioacetate, HAuCl4⋅3 H2O, tetraoctyl ammonium bromide (TOAB), and NaBH4 were purchased from Sigma– Aldrich. NaHCO3, K2CO3, and KOH were purchased from Panreac. α-Thio-ω-hydroxy polyethylene glycol (487 Da) (PEG) was purchased from Prochimia.
Instrumentation: 1H NMR and 13C NMR spectra were recorded using a 400 MHz spectrometer (Varian, Palo Alto, USA). Chemical shifts (δ) are expressed in ppm relative to the solvent peak (CDCl3). Thin-layer chromatography (TLC) was performed on F254-silica-gel-coated plates from Merck. Column chromatography was carried out on silica gel 60 (Merck 9385, 230–400 mesh).UV/Vis absorption spectra were obtained using a UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) analysis was performed using a Voyager-DE-RP (Appplied Biosystems, Framingham, USA) mass spectrometer, and high-resolution mass spectra (HRMS) were obtained with electrospray ionization (ESI) on a liquid chromatograph/mass-selective detector time-of-flight LC/MSD-TOF mass spectrometer (Agilent Technologies, Santa Clara, USA) at the Centres Cientifics i Tècnològics de la Universitat de Barcelona (CCiT-UB). MS analysis was operated in the delayed-extraction mode using 2,5-dihydroxybenzoic acid as a matrix. TEM experiments were performed at the CCiT-UB. The samples were observed with a Tecnai SPIRIT microscope (FEI Co., Hillsboro, USA) at 120 kV. The images were captured by a Megaview III camera and digitalized with the iTEM program. The size of the gold core of the NPs was measured with the analysis software (Olympus). XPS experiments were performed in a PHI 5500 Multitechnique System (Physical Electronics, Chanhassen, USA) with a monochromatic X-ray source (aluminum K-alpha line of 1486.6 eV energy and 350 W), placed perpendicular to the analyzer axis and calibrated using the 3 d5/2 line of Ag with a full width at half maximum (FWHM) of 0.8 eV. The analyzed area was a circle of 0.8 mm diameter. The selected resolution for the spectra was 187.5 eV of pass energy and 0.8 eV/step for the general spectra and 23.5 eV of pass energy and 0.1 eV/step for the spectra of the different elements. All measurements were made in an ultra-high vacuum (UHV) chamber with a pressure between 5×10−9 and 2×10−8 torr. Fluorescence spectroscopy measurements were performed using a Jobin–Yvon SPEX Nanolog-TM spectrofluorometer (Horiba, Kyoto, Japan).
5-[4-Hydroxyphenyl]-10,15,20-triphenylporphyrin (PR-OH): Dry pyrrole (4.57 mL, 65.6 mmol) was added to propionic acid (100 mL, 140 °C) at reflux. Subsequently, p-hydroxybenzaldehyde (2 g, 16.4 mmol) and benzaldehyde (5 mL, 49.2 mmol) were added to the solution at reflux, and the reaction mixture was stirred for 2 h. Then, propionic acid was evaporated in vacuo, and the crude mixture was redissolved in EtOAc and washed with H2O (3×100 mL) and with a solution of 5 % NaHCO3 (3×100 mL). Then the organic phase was dried with MgSO4, filtered, and the solvent was evaporated in vacuo. CH3CN (70 mL) was added and a purple solid was obtained by filtration. The mixture of porphyrins was separated by silica gel column chromatography using CH2Cl2 as the eluent, followed by column chromatography in CHCl3 giving PR-OH as a purple solid (560 mg, 5.4 %);43 mp>300 °C; 1H NMR (300 MHz, CDCl3): δ=8.87 (d, J=4.8 Hz, 2 H, pyr′.), 8.83 (s, 6 H, pyr.), 8.21 (dd, J=7.3, 1.9 Hz, 6 H, H2,6), 8.05 (d, J=8.1 Hz, 2 H, H2′,6′), 7.78–7.70 (m, 9 H, H3,4,5), 7.15 (d, J=8.0 Hz, 2 H, H4′,6′), −2.69–−2.83 ppm (m, 2 H, NH); UV/Vis (CH2Cl2): λmax=412.5, 515, 550.0, 590, 647.5 nm; MS-MALDI-TOF: m/z=631.2 [M+H]+.
5-[4-(11-Undecyloxythioacetate)-phenyl]-10,15,20- triphenylporphyrin (PR-SAc): Dry CH3CN (60 mL) was heated to 82 °C in an Ar atmosphere, and K2CO3 (55.2 mg, 0.8 mmol) was added. The solution was left to stir, and after 30 min, PR-OH (165 mg, 0.262 mmol) and 11-bromoundecyl thioacetate (243 μL, 0.8 mmol) were added. After 24 h, the reaction was cooled to rt, filtered, and evaporated in vacuo. The crude mixture was dissolved in CH2Cl2 and washed with H2O (3×100 mL). Then, the organic phase was dried with MgSO4 and after filtration the solvent was evaporated in vacuo. The product was isolated by silica gel column chromatography using hexane/EtOAc (7:3) as eluent, obtaining PR-SAc as a purple oil (151 mg, 67 %); 1H NMR (400 MHz, CDCl3): δ=8.89 (d, J=4.8 Hz, 2 H, pyr′.), 8.84 (d, J=2.4 Hz, 6 H, pyr.), 8.23–8.20 (d, J=7.2, 6 H, H2,6), 8.13–8.09 (d, J=8.5 Hz, 2 H, H2′,6′), 7.79–7.74 (m, 9 H, H3,4,5), 7.29–7.26 (d, J=8.4 Hz, 2 H, H4′,6′), 4.25 (t, J=6.5 Hz, 2 H, O-CH2), 2.88 (t, J=7.3 Hz, 2 H, CH2-S), 2.32 (s, 3 H, SCOCH3), 2.03–1.93 (m, 2 H, CH2CH2O-), 1.60 (m, 4 H), 1.40–1.32 (m, 12 H, -CH2-), −2.76 ppm (s, 2 H, NH); UV/Vis (CH2Cl2): λmax=412.5, 517, 556.5, 597.5, 649.5 nm; MS-MALDI-TOF: m/z=859.4 [M+H]+.
5-[4-(11-Mercaptoundecyloxy)-phenyl-10,15,20-triphenylporphyrin (PR-SH): Porphyrin PR-SAc (151 mg, 0.175 mmol) was dissolved in a mixed solution of CHCl3 (50 mL) and MeOH (15 mL). KOH (1 g) was dissolved in H2O/MeOH solution (5 mL, 50 % MeOH), and the basic solution was added to the porphyrin solution and was left to stir at rt. The degree of hydrolysis was monitored by TLC. After the completion of the hydrolysis (2 h), H2O was added to the solution, and the reaction was stirred for 1 h. The organic phase was then washed with H2O until the pH of the aq phase became neutral. The final product was obtained by evaporating the solvent from the organic phase, giving PR-SH as a purple oil (114 mg, 80 %); 1H NMR (400 MHz, CDCl3): δ=8.90 (d, J=4.8 Hz, 2 H, pyr′.), 8.84 (s, 6 H, pyr.), 8.22 (d, J=7.2, 6 H, H2,6), 8.12 (d, J=8.5 Hz, 2 H, H2′,6′), 7.76 (m, 9 H, H3,4,5), 7.29 (d, J=8.4 Hz, 2 H, H3′,5′), 4.25 (t, J=6.3 Hz, 2 H, O-CH2), 2.56 (t, J=7.3 Hz, 2 H, CH2-SH), 2.05–1.92 (m, 2 H, CH2CH2O-), 1.68–1.35 (m, 16 H), −2.75 ppm (s, 2 H, NH); 13C NMR (101 MHz, CDCl3): δ=136.58, 133.51, 129.58, 126.41, 125.62, 125.43, 124.12, 117.56, 67.28, 48.42, 36.08, 33.26, 31.73, 30.91, 29.60, 29.01, 28.68, 28.64, 28.56, 28.47, 28.34, 26.95, 26.06, 23.44, 21.67, 18.70, 18.64, 16.62, 13.09 ppm; UV/Vis (CH2Cl2): λmax=412.5, 514.5, 551, 592.5, 645.5 nm; MS-MALDI-TOF: m/z=817.4 [M+H]+; HRMS-ESI m/z [M+H]+ calcd for C55H52N4OS: 817.3856, found: 817.3935.
Water-soluble porphyrin/PEG-functionalized GNPs (GNP-PR/PEG): Water-soluble GNPs were synthesized using the Brust–Schiffrin method.35 HAuCl4⋅3 H2O (63 mg, 0.16 mmol) was dissolved in H2O (5 mL) and added to a stirred solution of tetraoctyl ammonium bromide (202 mg, 0.37 mmol) in toluene (10 mL). Next, PR-SH (20 mg, 0.021 mmol) was dissolved in toluene (1.5 mL) and added to the stirred solution, and subsequently thiol-PEG (20 mg, 0.042 mmol) was added to the biphasic solution. An excess of aqueous reducing agent NaBH4 (45 mg, 1.26 mmol) in H2O (5 mL) was also added to the mixture dropwise. The reaction was allowed to occur while stirring at rt overnight. The aq phase of the resulting dark red solution was separated using an extraction funnel and was subjected to H2O removal in a rotary evaporator, followed by multiple washings with EtOH (3×5 mL) using centrifugation. Finally, a red solid (137 mg) was obtained which was dissolved in H2O (5 mL) giving GNP-PR/PEG that was characterized by UV-Vis, TEM, XPS, and 1H NMR. Synthesis of the GNP-PEG was performed as was described before for the GNP-PR/PEG but the porphyrin was not added.
Singlet oxygen experiments: Singlet oxygen formation was monitored using fluorescence spectroscopy determining the fluorescent decay of ADPA upon irradiation. ADPA (100 μL of a 1.2 mm solution in MeOH) was added to GNP-PR/PEG (3 mL of 0.25 mg mL−1 in H2O). The mixture was added to a quartz cuvette with a length of 1 cm, and it was irradiated at 647 nm using the laser source of the fluorimeter (5 slits) for a duration of 30 min. Every 5 min, fluorescence emission spectra of the mixture was recorded between 365 and 600 nm (λex=355 nm).
Cell culture: The MCF10A nontumorigenic epithelial cell line was purchased from the American Type Culture Collection (ATCC) and maintained in Dulbeccos modified eagle medium F12 (DMEM/F12) (Gibco) supplemented with 5 % horse serum (Gibco), 20 ng mL−1 epidermal growth factor (Gibco), 0.5 mg mL−1 hydrocortisone (Sigma), 100 ng mL−1 cholera toxin (Sigma), and 10 μg mL−1 insulin (Gibco) at 37 °C and 5 % CO2. Cells were split 1:3 every 72 h.
Cytotoxicity of GNP-PEG and GNP-PR/PEG: Cytotoxicities of GNP-PEG and GNP-PR/PEG were determined using the Vybrant MTT Cell Proliferation Assay Kit (Molecular Probes), following the manufacturers instructions. MCF10A cells were seeded in 96-well dishes at a density of 5000 cells/well and grown for 72 h when confluence was reached. Then, cells were incubated with GNP-PEG or GNP-PR/PEG at six different concentrations: 2, 5, 10, 15, 20, or 200 μg mL−1. After 24 or 72 h, the medium was replaced with fresh culture medium (100 μL) and a stock solution of MTT (10 μL of 12 mm) and incubated for 2 h. Afterwards, the medium was removed leaving only 25 μL, and then DMSO (50 μL) was added. Cells were incubated for 10 min at 37 °C and 5 % CO2 to solubilize the formazan crystals. The measurement of absorbance was done at 570 nm using a Victor 3 Multilabel counter (PerkinElmer, Waltham, USA). For each treatment, viability was calculated as the absorbance of treated cells divided by the absorbance of untreated control cells. IC50 values (concentration of NPs that resulted in 50 % of cell viability) were obtained from three independent experiments.
ICP-MS analysis: Cells were seeded in 24-well dishes at a density of 3×104 cells/well. After 48 h, GNP-PR/PEG or GNP-PEG was added to the culture medium at a concentration of 200 μg mL−1 and incubated for 24 h. After co-incubation, the cell culture medium containing GNPs was collected, and the attached cells were washed with phosphate-buffered saline (PBS) twice to remove the GNPs that adhered to the plasma membrane. Then, cells were harvested by trypsinization and collected in a separate tube. Cells were collected by centrifugation, and the cell pellet was digested in aqua regia (HNO3:HCl 1:3) at 150 °C for 20 min. After that, the amount of gold in the cells and culture medium was determined on an Agilent 7500ce ICP-MS (Agilent, Santa Clara, USA).
TEM analysis: To analyze the intracellular location of GNPs, 1×106 MCF10A cells were seeded in a 9 cm2 plate and grown for 48 h. Afterwards, GNP-PEG or GNP-PR/PEG (100 μg mL−1) was added to the culture. After 24 h in the presence of the GNPs, cells were fixed with 2.5 % glutaraldehyde in phosphate buffer (PB) for 1 h at 4 °C, washed with PB, and postfixed with 1 % OsO4 in PB containing 0.8 % K3[Fe(CN)6] at 4 °C. Then, samples were dehydrated in acetone, infiltrated with Epon resin (Electron Microscopy Sciences) for 48 h, embedded in the same resin, and polymerized at 60 °C for 48 h. Ultrathin sections were obtained using a Leica Ultracut UC6 ultramicrotome (Leica Microsystems, Wetzlar, Germany) and mounted on Formvar-coated copper grids. They were gently stained with 2 % uranyl acetate in H2O for 10 min. Finally, sections were observed in a Jeol-J1010 electron microscope (Jeol Ltd, Tokyo, Japan) equipped with a SIS Megaview III CCD camera and with a Jeol-JEM 2011 electron microscope (Jeol Ltd, Tokyo, Japan) (operated at 200 kV) equipped with energy dispersive X-ray (EDX) compositional analysis.
Statistical analysis: Data were analyzed using IBM SPSS Statistics (Ver. 19.0; Oregon, USA). First, all data were tested for normality and homoscedasticity by using the Kolmogorov–Smirnov and Levene tests. In the case of data at 24 h, a previous arcsin √x transformation on viability percentages was applied to accomplish the parametric assumptions. Then, one-way analysis of variance (ANOVA) was run followed by a post-hoc Scheffé test for pairwise comparisons. For data at 72 h, no linear transformation remedied the heteroscedasticity, and in this case the Kruskal–Wallis and Mann–Whitney tests were applied as an alternative to parametric statistics. In all the cases, the level of significance was set at p <0.05, and data are shown as means ± standard error of the mean (SEM).
Acknowledgments
The authors acknowledge financial support from the Ministerio de Ciencia e Innovación (MICINN), Spain (project TEC2011–29140-C03–02 and 03) and the Generalitat de Catalunya, Spain (2009 SGR 158, 2009 SGR 00282). T.P. thanks the Generalitat de Catalunya for the AGAUR FI-DGR 2011 predoctoral grant. The authors wish to thank the staff at the Servei de Microscòpia of the Universitat Autònoma de Barcelona and the staff at the Unitat de Microscòpia Electrònica i Reconeixement Molecular in Situ. Special thanks goes to Carmen López at the Centres Cientifics i Tècnològics de la Universitat de Barcelona (CCiT-UB) and Dr. Laura Rodríguez from the Universitat de Barcelona.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1.Lemberg R. In: Fortschritte Der Chemie Org. Naturstoffe/Prog. Chem. Org. Nat. Prod./Progrés Dans La Chim. Des Subst. Org. Nat. Zechmeister L, editor. Vienna: Springer; 1954. pp. 299–349. [Google Scholar]
- 2.Kadish KM, Smith KM, Guilard R. Handbook of Porphyrin Science: Supramolecular Chemistry. Singapure: World Scientific Publishing Company; 2010. [Google Scholar]
- 3.Beletskaya I, Tyurin VS, Tsivadze AY, Guilard R, Stern C. Chem. Rev. 2009;109:1659–1713. doi: 10.1021/cr800247a. [DOI] [PubMed] [Google Scholar]
- 4.Costa SMB, Andrade SM, Togashi DM, Paulo PMR, Laia CAT, Isabel Viseu M, Gonçalves da Silva AM. J. Porphyrins Phthalocyanines. 2009;13:509–517. [Google Scholar]
- 5.Drain CM, Russell KC, Lehn J-M. Chem. Commun. 1996:337–338. [Google Scholar]
- 6.Purrello R, Gurrieri S, Lauceri R. Coord. Chem. Rev. 1999;190–192:683–706. [Google Scholar]
- 7.Puigmartí-Luis J, Saletra WJ, González A, Amabilino DB, Pérez-García L. Chem. Commun. 2014;50:82–84. doi: 10.1039/c3cc44794d. [DOI] [PubMed] [Google Scholar]
- 8.Lang K, Mosinger J, Wagnerová DM. Coord. Chem. Rev. 2004;248:321–350. [Google Scholar]
- 9.Geier GR, Ciringh Y, Li F, Haynes DM, Lindsey JS. Org. Lett. 2000;2:1745–1748. doi: 10.1021/ol005917h. [DOI] [PubMed] [Google Scholar]
- 10.Li F, Kexin Y, Tyhonas JS, MacCrum KA, Lindsey JS. Tetrahedron. 1997;53:12339–12360. [Google Scholar]
- 11.Moonsin P, Sirithip K, Jungsuttiwong S, Keawin T, Sudyoadsuk T, Promarak V. Tetrahedron Lett. 2011;52:4795–4798. [Google Scholar]
- 12.Xiaoquan L, Zaixin G, Jingwan K, Zhihua W, Jinzhang G. Synth. Commun. 2000;30:2213–2221. [Google Scholar]
- 13.Peng C-L, Lai P-S, Chang C-C, Lou P-J, Shieh M-J. Dye. Pigment. 2010;84:140–147. [Google Scholar]
- 14.Iavicoli P, Xu H, Feldborg LN, Linares M, Paradinas M, Stafström S, Ocal C, Nieto-Ortega B, Casado J, López Navarrete JT, Lazzaroni R, De Feyter S. J. Am. Chem. Soc. 2010;132:9350–9362. doi: 10.1021/ja101533j. [DOI] [PubMed] [Google Scholar]
- 15.Bonnett R, Martínez G. Tetrahedron. 2001;57:9513–9547. [Google Scholar]
- 16.Juarranz Á, Jaén P, Sanz-Rodríguez F, Cuevas J, González S. Clin. Transl. Oncol. 2008;10:148–154. doi: 10.1007/s12094-008-0172-2. [DOI] [PubMed] [Google Scholar]
- 17.Swavey S, Tran M. Porphyrin and Phthalocyanine Photosensitizers as PDT Agents: A New Modality for the Treatment of Melanoma. Manhattan, NY: InTech; 2013. [Google Scholar]
- 18.Macdonald IJ, Dougherty TJ. J. Porphyrins Phthalocyanines. 2001;5:105–129. [Google Scholar]
- 19.Boisselier E, Astruc D. Chem. Soc. Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh P, Han G, De M, Kim CK, Rotello VM. Adv. Drug Delivery Rev. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Y, Samia AC, Meyers JD, Panagopoulos I, Fei B, Burda C. J. Am. Chem. Soc. 2008;130:10643–10647. doi: 10.1021/ja801631c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee DK, Fong LS, Zhang Y. Adv. Drug Delivery Rev. 2008;60:1627–1637. doi: 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Bekalé L, Barazzouk S, Hotchandani S. J. Mater. Chem. 2012;22:2943–2951. [Google Scholar]
- 24.Bechet D, Couleaud P, Frochot C, Viriot M-L, Guillemin F, Barberi-Heyob M. Trends Biotechnol. 2008;26:612–621. doi: 10.1016/j.tibtech.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Konan YN, Berton M, Gurny R, Allémann E. Eur. J. Pharm. Sci. 2003;18:241–249. doi: 10.1016/s0928-0987(03)00017-4. [DOI] [PubMed] [Google Scholar]
- 26.Obaid G, Chambrier I, Cook MJ, Russell DA. Angew. Chem. Int. Ed. 2012;51:6158–6162. doi: 10.1002/anie.201201468. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012;124 [Google Scholar]
- 27.Camerin M, Magaraggia M, Soncin M, Jori G, Moreno M, Chambrier I, Cook MJ, Russell DA. Eur. J. Cancer. 2010;46:1910–1918. doi: 10.1016/j.ejca.2010.02.037. [DOI] [PubMed] [Google Scholar]
- 28.Stuchinskaya T, Moreno M, Cook MJ, Edwards DR, Russell DA. Photochem. Photobiol. Sci. 2011;10:822–831. doi: 10.1039/c1pp05014a. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Li M, Li H, Yang Y, Kashimura Y, Wang C, Torimitsu K, Lu X, Hu W. J. Phys. Chem. C. 2010;114:12320–12324. [Google Scholar]
- 30.Zuo G, Lu X, Xue Z, Lv B, Wang Y, Kang J. Synth. React. Inorganic, Met. Nano-Metal Chem. 2006;36:589–594. [Google Scholar]
- 31.Chan Y-H, Schuckman AE, Pérez LM, Vinodu M, Drain CM, Batteas JD. J. Phys. Chem. C. 2008;112:6110–6118. [Google Scholar]
- 32.Ohyama J, Hitomi Y, Higuchi Y, Shinagawa M, Mukai H, Kodera M, Teramura K, Shishido T, Tanaka T. Chem. Commun. 2008:6300–6302. doi: 10.1039/b815100h. [DOI] [PubMed] [Google Scholar]
- 33.Canitez F, Yavuz M, Ozturk R. J. Nanopart. Res. 2011;13:7219–7228. [Google Scholar]
- 34.Hasobe T, Imahori H, Kamat PV, Ahn TK, Kim SK, Kim D, Fujimoto A, Hirakawa T, Fukuzumi S. J. Am. Chem. Soc. 2005;127:1216–1228. doi: 10.1021/ja047768u. [DOI] [PubMed] [Google Scholar]
- 35.Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. J. Chem. Soc. Chem. Commun. 1994:801–802. [Google Scholar]
- 36.Kimling J, Maier M, Okenve B, Kotaidis V, Ballot H, Plech A. J. Phys. Chem. B. 2006;110:15700–15707. doi: 10.1021/jp061667w. [DOI] [PubMed] [Google Scholar]
- 37.Záruba K, Králová J, Řezanka P, Poučková P, Veverková L, Král V. Org. Biomol. Chem. 2010;8:3202–3206. doi: 10.1039/c002823a. [DOI] [PubMed] [Google Scholar]
- 38.Gamaleia NF, Shishko ED, Dolinsky GA, Shcherbakov AB, Usatenko AV, Kholin VV. Exp. Oncol. 2010;32:44–47. [PubMed] [Google Scholar]
- 39.Hayden SC, Austin LA, Near RD, Ozturk R, El-Sayed MA. J. Photochem. Photobiol. A. 2013;269:34–41. [Google Scholar]
- 40.Allison RR, Downie GH, Cuenca R, Hu X-H, Childs CJH, Sibata CH. Photodiagnosis Photodyn. Ther. 2004;1:27–42. doi: 10.1016/S1572-1000(04)00007-9. [DOI] [PubMed] [Google Scholar]
- 41.Vicente MGH, Osterloh J. J. Porphyrins Phthalocyanines. 2002;6:305–324. [Google Scholar]
- 42.Banfi S, Caruso E, Buccafurni L, Murano R, Monti E, Gariboldi M, Papa E, Gramatica P. J. Med. Chem. 2006;49:3293–3304. doi: 10.1021/jm050997m. [DOI] [PubMed] [Google Scholar]
- 43.Brulé E, de Miguel YR, Hii KK. Tetrahedron. 2004;60:5913–5918. [Google Scholar]
- 44.Antipas A, Dolphin D, Gouterman M, Johnson EC. J. Am. Chem. Soc. 1978;100:7705–7709. [Google Scholar]
- 45.Imahori H, Norieda H, Nishimura Y, Yamazaki I, Higuchi K, Kato N, Motohiro T, Yamada H, Tamaki K, Arimura M, Sakata Y. J. Phys. Chem. B. 2000;104:1253–1260. [Google Scholar]
- 46.Haiss W, Thanh NTK, Aveyard J, Fernig DG. Anal. Chem. 2007;79:4215–4221. doi: 10.1021/ac0702084. [DOI] [PubMed] [Google Scholar]
- 47.Baer DR, Gaspar DJ, Nachimuthu P, Techane SD, Castner DG. Anal. Bioanal. Chem. 2010;396:983–1002. doi: 10.1007/s00216-009-3360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katsonis N, Vicario J, Kudernac T, Visser J, Pollard MM, Feringa BL. J. Am. Chem. Soc. 2006;128:15537–15541. doi: 10.1021/ja065823o. [DOI] [PubMed] [Google Scholar]
- 49.Lindig BA, Rodgers MAJ, Schaap AP. J. Am. Chem. Soc. 1980;102:5590–5593. [Google Scholar]
- 50.Knop K, Mingotaud A-F, El-Akra N, Violleau F, Souchard J-P. Photochem. Photobiol. Sci. 2009;8:396–404. doi: 10.1039/b811248g. [DOI] [PubMed] [Google Scholar]
- 51.Guo Y, Kumar M, Zhang P. Chem. Mater. 2007;19:6071–6072. doi: 10.1021/cm7028454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fraga S, Faria H, Soares ME, Duarte JA, Soares L, Pereira E, Costa-Pereira C, Teixeira JP, de Lourdes Bastos M, Carmo H. J. Appl. Toxicol. 2013;33:1111–1119. doi: 10.1002/jat.2865. [DOI] [PubMed] [Google Scholar]
- 53.Martinez Paino IM, Marangoni VS, de Cássia Silva de Oliveira R, Antunes LMG, Zucolotto V. Toxicol. Lett. 2012;215:119–125. doi: 10.1016/j.toxlet.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 54.Pan Y, Bartneck M, Jahnen-Dechent W. In: Methods Enzymol. Nejat D, editor. Waltham: Academic Press; 2012. pp. 225–242. [DOI] [PubMed] [Google Scholar]
- 55.Lévy R, Shaheen U, Cesbron Y, Sée V. Nano Rev. 2010;1:4889. doi: 10.3402/nano.v1i0.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khlebtsov N, Dykman L. Chem. Soc. Rev. 2011;40:1647–1671. doi: 10.1039/c0cs00018c. [DOI] [PubMed] [Google Scholar]
- 57.Coradeghini R, Gioria S, García CP, Nativo P, Franchini F, Gilliland D, Ponti J, Rossi F. Toxicol. Lett. 2013;217:205–216. doi: 10.1016/j.toxlet.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 58.Vijayakumar S, Ganesan S. Toxicol. Environ. Chem. 2013;95:277–287. [Google Scholar]
- 59.Arnida, Malugin A, Ghandehari H. J. Appl. Toxicol. 2010;30:212–217. doi: 10.1002/jat.1486. [DOI] [PubMed] [Google Scholar]
- 60.Gu Y-J, Cheng J, Lin C-C, Lam YW, Cheng SH, Wong W-T. Toxicol. Appl. Pharmacol. 2009;237:196–204. doi: 10.1016/j.taap.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 61.Liu C-J, Wang C-H, Chien C-C, Yang T-Y, Chen S-T, Leng W-H, Lee C-F, Lee K-H, Hwu Y, Lee Y-C, Cheng C-L, Yang C-S, Chen YJ, Je JH, Margaritondo G. Nanotechnology. 2008;19:295104. doi: 10.1088/0957-4484/19/29/295104. [DOI] [PubMed] [Google Scholar]
- 62.Cui W, Li J, Zhang Y, Rong H, Lu W, Jiang L. Nanomedicine. 2012;8:46–53. doi: 10.1016/j.nano.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 63.Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary