

Abstract
Transition metals offer many possibilities in developing potent chemotherapeutic agents. They are endowed with a variety of oxidation states, allowing for the selection of their coordination numbers and geometries via the choice of proper ligands, leading to the tuning of their final biological properties. We report here on the synthesis, physico-chemical characterization, and solution behavior of two gold(III) pyrrolidinedithiocarbamates (PDT), namely [AuIIIBr2(PDT)] and [AuIIICl2(PDT)]. We found that the bromide derivative was more effective than the chloride one in inducing cell death for several cancer cell lines. [AuIIIBr2(PDT)] elicited oxidative stress with effects on the permeability transition pore, a mitochondrial channel whose opening leads to cell death. More efficient antineoplastic strategies are required for the widespread burden that is cancer. In line with this, our results indicate that [AuIIIBr2(PDT)] is a promising antineoplastic agent that targets cellular components with crucial functions for the survival of tumor cells.
Keywords: antitumor, gold(III), mitochondrial permeability transition pore, pyrrolidinedithiocarbamates (PDT), reactive oxygen species (ROS)
Introduction
Metal-based compounds have been largely used and studied as chemotherapeutic drugs since the discovery of the cytotoxic properties of cisplatin (cis-[Pt(NH3)2Cl2]) in the late 1960s;1–3 after its approval by the US Food and Drug Administration in 1978, cisplatin has become a milestone in antineoplastic treatment.3, 4 The therapeutic relevance of platinum-based compounds has led researchers to elucidate their mechanisms of action, with the aim to improve their efficacy on neoplasms and to hamper adverse reactions in healthy tissues.
It is known that the metal center in metal-based drugs is the pivotal moiety that acts as the anticancer “weapon”, whereas the organic part of the molecule (ligand) plays other key roles, such as stabilizing the metal oxidation state, reducing systemic toxicity, and tuning water solubility. Under physiological-like conditions, metal centers, being positively charged, are favored to bind to negatively charged biomolecules, such as enzymes and nucleotides, or undergo redox processes.5–8 On the other hand, the mechanism of biological action of metal compounds is, in several cases, not fully elucidated. As an example, in anticancer metallodrugs, DNA is not always the primary target as it appears for cisplatin.6–9 In addition, the type of ligands (O-, N-, S-donors, monodentate or chelating ones, etc.), the nature, and the oxidation state of the metal center determine the hallmarks of the final compound such as its coordination geometry and stereochemistry and, ultimately, its biological behavior.10–14 These observations, together with the poor toxicological profile of cisplatin, whose clinical use causes adverse effects and the occurrence of resistance,15, 16 have prompted the research into alternative platinum-based compounds, in order to find drugs with a better chemotherapeutic index in terms of toxicity–activity ratio.15, 17, 18
In the last years, we have broken into this research scene with our nonplatinum-based coordination compounds. These molecules are metal–dithiocarbamato (dtc) complexes potentially able to combine the cytotoxic activity of the related metal center (e.g. AuI,III, RuII,III, ZnII, CuII) with lack of toxicity owing to the inherent chemoprotective action of the dtc ligand.19, 20 Overall, our findings provide evidence for the importance of stabilizing the heavy metal center in order to prevent its nonspecific reactivity that would give rise to systemic toxicity.21 Up to now, the most promising results have been obtained with gold(III) derivatives of the type [AuIIIX2(dtc)] (X=Cl, Br; dtc=various dithiocarbamato ligands). These compounds closely reproduce the structural features of cisplatin, and most of them are endowed with good anticancer activity, together with a very low or negligible systemic toxicity.22–26
In parallel to the synthesis of gold chemotherapeutics, we have also been synthetizing and studying coordination compounds of ruthenium and copper with pyrrolidinedithiocarbamate (PDT) as a ligand. In this context, we found that the presence of PDT strongly affects the final biological activity. In fact, we recorded an encouraging cytotoxic activity in the nano and micromolar range against human non-small-cell lung cancer, two estrogen-receptor-α-positive breast cancer cell lines, the triple negative MDA-MB-231 breast cancer cell line, androgen-receptor-independent prostate tumor, ovarian and cervical squamous carcinoma cell lines, and the corresponding cisplatin-resistant subclones.27, 28 Therefore, in the present work we have combined the chelating ligand PDT with gold metal in a 1:1 metal-to-ligand stoichiometry with the remaining coordination sites occupied by two cis-halogen ions which may undergo hydrolysis in the physiological milieu.
In particular, we report on the synthesis of two new gold(III) pyrrolidinedithiocarbamates with the general formula [AuIIIX2(PDT)] as shown in Figure 1, wherein X is either Br (complex 1) or Cl (complex 2). Their physico-chemical characterization was followed by the evaluation of their in vitro cytotoxicity on HeLa (cervical cancer), HCT116 (colon neoplasia), SAOS (osteosarcoma), and MEF-NF1−/− (immortalized primary embryonic fibroblast) cancer cell lines. The solution behavior of both complexes was also studied under physiological-like conditions by UV/Vis spectrophotometry. Then, the cell-death-inducing mechanisms were explored by investigating whether the compounds affect redox equilibrium or influence the mitochondrial permeability transition pore (PTP) activity.
Figure 1.
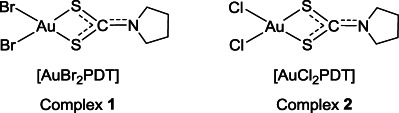
Chemical structures of the synthesized gold(III) complexes.
Results and Discussion
Synthesis and characterization of the gold(III) complexes
Two gold(III)-pyrrolidinedithiocarbamato derivatives were prepared in very good yields via direct reaction of the salt ligand ammonium PDT with the metal precursor potassium tetrabromoaurate(III) or potassium tetrachloroaurate(III) under equimolar conditions at room temperature. The purity of all final compounds was higher than 95 % (by thermogravimetric and elemental analysis). Both the starting ammonium PDT and the corresponding gold(III)-dithiocarbamato derivatives 1 and 2 (Figure 1) have been characterized by means of several techniques in order to confirm the expected stoichiometry as well as to investigate their structural features.
Elemental and thermogravimetric analyses highlighted a very good correlation between calculated and found values. In particular, thermal degradation primarily takes place via two steps, leading to the formation of metallic gold, in agreement with literature data29, 30 (Table 1). The presence of an endothermic differential scanning calorimetry (DSC) peak at about 1064 °C (gold Tmelt) along with no weight loss further confirms the decomposition to elemental gold. The first step is likely due to the formation of gold(I) thiocianate upon reductive elimination as reported in literature for similar gold(III) dithiocarbamato compounds.31, 32
Table 1.
TG and DSC data for the gold(III)-dithiocarbamato compounds 1 and 2.
| Cmpd | Weight loss to Au(0) [%] | Peak temperature (process)[a] [°C] | |
|---|---|---|---|
| Calculated | Found | ||
| 1 | −60.91 | −60.95 | 261 (endo); 497 (exo); 1064 (endo) |
| 2 | −52.21 | −51.91 | 242 (endo); 495 (exo); 1064 (endo) |
[a] Endo=endothermic, exo=exothermic.
The elucidation of the coordination mode and the strength of the bonds between the metal and the ligand donor atoms is of paramount importance in the characterization of metal derivatives designed for medicinal applications. In this regard, medium and far FT-IR spectroscopy are among the best diagnostic tools. Table 2 highlights that the band associated with the N−CSS stretching shifts toward higher energies when passing from the free ligand to the corresponding gold derivatives due to an increased carbon–nitrogen double bond character following the coordination.33–35 The detection of a single band at about 1000 cm−1, attributable to the νa (S−C−S) for both gold(III) complexes, points out that the −NCSS moiety chelates the metal center in a symmetrical bidentate mode in agreement with our data on other complexes.21, 24, 36 A band, detectable in the range 420–630 cm−1 and commonly ascribed to the νs (S−C−S) vibration, is observed for both the complexes and the free dithiocarbamato ligand (Table 2).29, 37–40 It is interesting to underline that this absorption is associated also with some M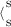 〉C ring deformations, as previously shown by detailed FT-IR isotopic studies on various dithiocarbamato complexes.41 The presence of two new bands in the range 370–420 cm−1, absent in the IR spectrum of the free ligand and ascribable to gold(III)–sulfur vibrations (νs and νa), further accounts for a chelated coordination geometry.38 Notably, on the basis of the energies recorded for the νa/s (S−Au−S), compound 2 is characterized by a slightly stronger metal–ligand bond, which indirectly determines a higher stretching frequency for the N−CSS bond (Table 2).42 Two halides complete the coordination sphere in a cis-geometry as shown by the appearance of two new bands (νa/s (X−Au−X)) at 240/218 and 362/315 cm−1 for the complex 1 and 2, respectively.43, 44 The other main frequencies, ascribable to the organic alkylic backbone, seem to be negligibly affected by metal coordination, except for the disappearance of the broad band at 3102 cm−1 (νa/s (NH4+)) (Table 2).
〉C ring deformations, as previously shown by detailed FT-IR isotopic studies on various dithiocarbamato complexes.41 The presence of two new bands in the range 370–420 cm−1, absent in the IR spectrum of the free ligand and ascribable to gold(III)–sulfur vibrations (νs and νa), further accounts for a chelated coordination geometry.38 Notably, on the basis of the energies recorded for the νa/s (S−Au−S), compound 2 is characterized by a slightly stronger metal–ligand bond, which indirectly determines a higher stretching frequency for the N−CSS bond (Table 2).42 Two halides complete the coordination sphere in a cis-geometry as shown by the appearance of two new bands (νa/s (X−Au−X)) at 240/218 and 362/315 cm−1 for the complex 1 and 2, respectively.43, 44 The other main frequencies, ascribable to the organic alkylic backbone, seem to be negligibly affected by metal coordination, except for the disappearance of the broad band at 3102 cm−1 (νa/s (NH4+)) (Table 2).
Table 2.
Selected FT-IR frequencies of the ligand PDT and the corresponding gold(III)-dithiocarbamato compounds 1 and 2.
| Cmpd | Vibrational mode [cm−1] | ||||
|---|---|---|---|---|---|
| νa/s NH4+ | ν N−CSS | νa/s SCS | νa/s SAuS | νa/s XAuX | |
| NH4(PDT) | 3102[a] | 1412 | 988/565 | — | — |
| 1 | — | 1583 | 943/537 | 411/371 | 240/218[b] |
| 2 | — | 1586 | 942/538 | 413/375 | 362/315[c] |
[a] NH4+; [b] X=Br; [c] X=Cl.
Concerning the NMR characterization, one and two-dimensional spectra (1H,13C HMBC, Table 3) showed an upfield shift (about 20 ppm with respect to the free ligand) for the signal of the dithiocarbamato carbon atom for both complexes (at about 190 ppm), along with the disappearance of the NH4+ proton peak (at 7.33 ppm), confirming the formation of the gold(III) derivatives. In comparison with the spectrum recorded for ammonium PDT, all 13C resonances are shifted at higher fields. Conversely, upon metal coordination the 1H multiplets of all the methylene groups are shifted at lower fields, thus pointing out opposite shield effects on the two investigated NMR-active nuclei. By means of NMR kinetics, we checked the stability over time of both complexes in D6-dimethylsulfoxide (DMSO) as predissolution of our hydrophobic compounds in this organic solvent is necessary to carry out the biological tests in cell culture medium.
Table 3.
13C NMR spectral data of the ligand PDT and compounds 1 & 2.
| Cmpd | δ [ppm] | |
|---|---|---|
| R−N | −CSS | |
| NH4(PDT) | 26.06 (CH2CH2N) 53.00 (CH2CH2N) | 208.81 |
| 1 | 23.35 (CH2CH2N) 50.87 (CH2CH2N) | 188.32 |
| 2 | 23.41 (CH2CH2N) 51.10 (CH2CH2N) | 188.58 |
UV/Vis spectrophotometric studies
When studying metallodrugs, it is necessary to test their stability under physiological conditions.5 Therefore, detailed spectrophotometric studies were carried out for both complexes dissolved in DMSO (reference solvent) and saline solution. In order to better investigate the possible reactivity in both media, several electronic spectra were recorded up to 24 hours. It is worth pointing out that our compounds show a good antiproliferative activity already after a few hours of treatment.22 Accordingly, in this work we highlight only the chemical and biological behavior after a three-hour treatment. The kinetic behavior of the coordination compounds 1 and 2 was first monitored in DMSO (Figure 2) at 37 °C and a concentration of 100 μm (main features collected in Table 4).
Figure 2.
UV/Vis spectra recorded for compounds 1 (a) and 2 (b) in DMSO at 37 °C.
Table 4.
Main absorption bands (evaluated at time zero) in the UV/Vis electronic spectra acquired in DMSO and saline solution at 37 °C.
| Compound | Solvent | λmax [nm] | |
|---|---|---|---|
| Band 1 | Band 2 | ||
| 1 | DMSO | 277.3 | 319.0 |
| Saline solution | 269.3 | 311.6 | |
| 2 | DMSO | 268.6 | 318.3 |
| Saline solution | 269.2 | 311.1 | |
Band 1 and band 2 have been ascribed in literature to intraligand π*←π transitions mainly located in the −NCS and −CSS moieties, respectively.29, 38 Moreover, an intraligand π*←n transition, where n is the in-plane nonbonding sulfur orbital, should be recorded at around 340 nm45 but is not observable owing to the overlap with the more intense adjacent bands. A weak band at about 380 nm is visible only for the coordination compound 1 (labeled as band 3 in Figure 2) and is attributable to an intramolecular L←M charge transfer involving the M nd orbitals and the dithiocarbamato π* system.46, 47 However, owing to the quite high oxidizing power of AuIII, this transition has been ascribed also to an electron transfer of the type u←g from a 4p orbital of the bromide ligands to the lowest unfilled 5d orbital of the metal center.48 Its low intensity can be explained by a poor overlap between the 5d metal orbital and the involved 4p orbital (π symmetry).48
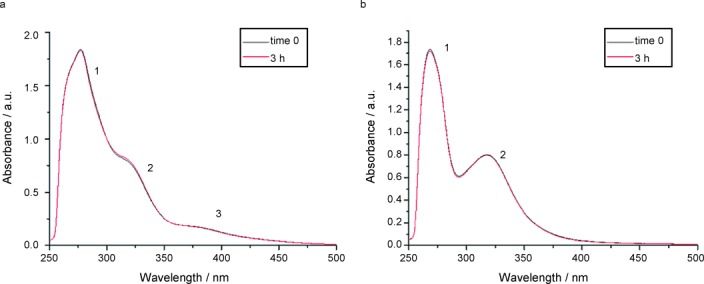
For both complexes, no major alteration of the bands related to the metal–ligand chromophore was observed over time. This behavior is likely due to the large stabilization effects brought about by the dithiocarbamato ligand. Such stability was further confirmed in the same solvent by 1H NMR over three hours. On the basis of these results, DMSO was used to dissolve 1 and 2 prior to performing all the biological tests (<0.5 % v/v) and was taken as reference medium in the discussion of the spectral features detected in saline solution. In this aqueous environment, the evolution with time was likewise followed by UV/Vis spectrophotometry for three hours at 37 °C (Figure 3), with both compounds being previously dissolved in DMSO (final concentration 100 μm, at 0.8 % and 1.3 % v/v content of DMSO for compound 1 and 2, respectively).
Figure 3.
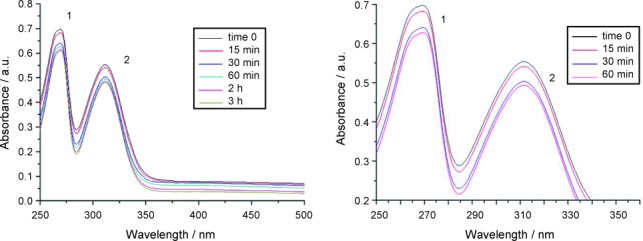
UV/Vis spectra recorded for compound 1 in saline solution at 37 °C over 3 h (left) and the first 60 min (right).
Table 4 reports only the absorption data recorded at the time zero. Similar to other our complexes,14, 49 contrary to DMSO medium, some decreases of the band intensities were quickly observed for both complexes upon dissolution in saline, reaching a steady condition after three hours. Overall, the maximum position of the bands is essentially unchanged over time. This points out that the gold center keeps the +3 oxidation state thanks to the stabilizing effects played by the chelating dithiocarbamato ligand.50 The drop in spectral intensity can be ascribed to the progressive hydrolysis of gold(III)-bromide (compound 1) and gold(III)-chloride (compound 2) bonds, leading to the water-soluble aquo complexes and, ultimately, to the precipitation of the hydroxo derivatives (verified by elemental and far FT-IR analyses). In addition, compound 1 undergoes more rapid hydrolysis compared to the complex 2 with an absorbance decrease over one hour of about 11 % and 1 %, respectively, (Figure 3). This behavior highlights a higher intrinsic reactivity of the bromide derivative with respect to the chloride one under physiological-like conditions. In fact, bromide ranks lower than chloride within the experimental spectrochemical series of ligands,51 thus pointing out a greater capacity for the latter to cause d-orbital splitting. The larger the crystal field stabilization energy, the stronger the metal–ligand bond, in agreement with our previously described FT-IR data on the Au−X bond strength.
Biological assays
Compounds 1 and 2 induce dose-dependent tumor cell death
Treatment with both compounds 1 and 2 resulted in a rapid (three-hour) dose-dependent decrease of cell viability in the following human cancer cell lines: the highly aggressive SAOS-2 osteosarcoma cells, the HeLa cervical adenocarcinoma cells, the HCT116 colorectal carcinoma cells, and the mouse embryonic fibroblasts, obtained from mice where the bona fide tumor suppressor gene NF1 was genetically ablated (MEF-NF1−/−) (Table 5).
Table 5.
Growth inhibition of various cell lines after 3 h treatment with the gold(III) compounds 1 and 2.
| IC50[a] [μM] | ||||
|---|---|---|---|---|
| Cmpd | SAOS-2 | HeLa | HCT116 | MEF NF1−/− |
| 1 | 12.2±1.6 | 17.7±1.6 | 15.8±2.1 | 3.6±0.9 |
| 2 | 26.4±3.1 | 30.8±4.2 | 43.6±5.4 | 7.8±1.1 |
| Cisplatin | NE[b] | NE[b] | NE[b] | NE[b] |
[a] Data represent the mean ± S.D. for n=7. Cells tested were the following: human tumor cell lines SAOS-2 (human osteosarcoma), HeLa (cervical cancer), and HCT116 (colon neoplasia) and the immortalized primary mouse embryonic fibroblast (MEF) model MEF-NF1−/− (isolated from knockout strains for the oncosuppressor gene neurofibromine); [b] NE stands for not evaluable at 3 h treatment.
Our data also show that compound 1 is the most promising anticancer molecule, as it induced a markedly higher cytotoxicity than compound 2 in all tested cell types (Figure 4, a–d), suggesting that the presence of bromides instead of chlorides in the gold coordination sphere profoundly affects the biological efficacy of the potential drug. Cisplatin was tested as a reference compound under the same experimental conditions recording no significant cytotoxicity after three hours of treatment.
Figure 4.
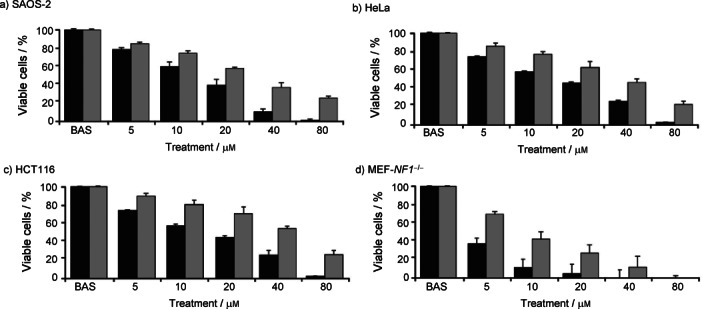
Compounds 1 (black bars) and 2 (grey bars) are cytotoxic to some cancer cell lines. Cell viability was measured on SAOS-2 (a), HeLa (b), and HCT116 (c) human cancer cell lines and on MEF-NF1−/−cells (d). Shown are dose-response experiments after a 3 h treatment with compounds 1 and 2. BAS stands for the control. Values represent the mean ± S.D. of at least n=7.
Compound 1 increases intracellular reactive oxygen species (ROS) and sensitizes the permeability transition pore (PTP) to opening
In the light of the antiproliferation screening, we then conducted the subsequent biological studies on the more active compound 1.
Recently, we have shown52 that the gold(III)-dithiocarbamato complex AuL12 causes a rise of mitochondrial superoxide levels that kills tumor cells by inducing ROS-dependent opening of the PTP. The PTP is a large channel located in the inner mitochondrial membrane, whose opening causes mitochondrial depolarization and swelling, leading to rupture of the outer mitochondrial membrane, release of Ca2+ and of pro-apoptotic proteins, and then cell death.53, 54 Therefore, we evaluated whether the cytotoxicity elicited by compound 1 is mediated by a similar oxidative stress, which would prompt cell death by selectively triggering PTP opening in neoplastic cells. We performed these experiments on MEF-NF1−/−, obtained from knockout mice for the onco-suppressor gene NF1. Such a gene encodes neurofibromin (Nf1), which negatively regulates Ras,55 a master protooncogene whose hyperactivation is the most common oncogenic event, which prompts cell proliferation, apoptosis resistance, and metabolic rearrangements.56 In addition to Ras hyperactivation, the ablation of the NF1 gene also results in the genetic disease neurofibromatosis, characterized by the occurrence of diverse tumor types.57 Based on these considerations, MEF-NF1−/− cells constitute a good model to check the antiblastic activity of our compounds.
The gold(III) compound 1 induced a rise in ROS levels (Figure 5 a). Kinetic experiments showed that the ROS increase was extremely rapid, reaching a maximum after 60 minutes (Figure 5 a). To ascertain whether compound 1 is capable of eliciting PTP opening, we performed a Ca2+ retention capacity (CRC) analysis, which evaluates PTP modulation in a semiquantitative way through the assessment of the amount of Ca2+ taken up by mitochondria before pore opening.58 As shown in Figure 5 b, compound 1 triggered a dose-dependent CRC decrease on isolated liver mitochondria, which constitutes the standard model when setting CRC experiments, because hepatocytes are endowed with a high number of mitochondria. Inhibition of Ca2+ release from mitochondria by a control treatment of cyclosporine A (CSA), a well-known PTP inhibitor, confirmed that CRC experiments were assessing PTP modulation, and that the observed calcium release is due to PTP opening. These data suggest that compound 1 seems to promote cell death by inducing PTP opening, so we also carried out CRC experiments on the MEF-NF1−/− cells.58 We found that a three-hour treatment with the gold(III) complex 1 caused a dose-dependent CRC shortening, that is PTP opening, on this cellular model as well (Figure 5 c). Furthermore, we performed parallel experiments in the presence of the ROS-scavenging compound Trolox, finding no changes in the effect of compound 1 on PTP opening. These experiments suggest that the cell death commitment induced by compound 1 is not only associated with its verified ability to induce ROS, but it is likely due to other ROS-independent pathways, that ultimately lead to PTP opening.
Figure 5.
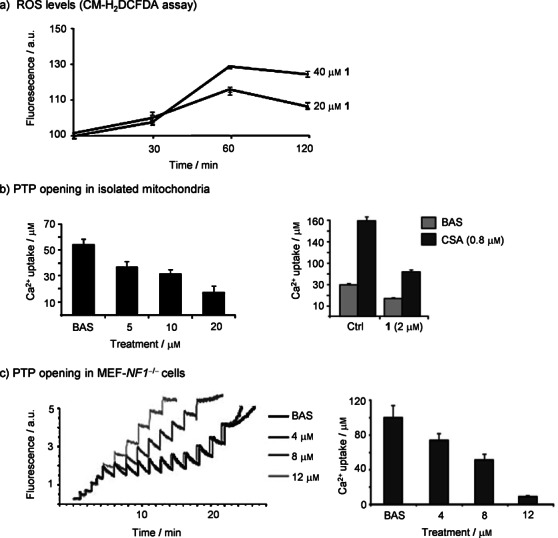
Compound 1 is able to induce reactive oxygen species (ROS) and sensitize the permeability transition pore (PTP). a) Dose-response and ROS kinetics measured on mitochondria of MEF-NF1−/− treated up to 120 min with 20 μm and 40 μm of 1; values represent the mean ± S.D. of at least n=5. b) Left: changes on pore permeability transition induced by compound 1 on purified mitochondria from mouse liver and measured by calcium retention capacity (CRC). Right: same experiments carried out in the presence of the inhibitor cyclosporine A (CSA), added 1 h before treatment. Values represent the mean ± S.D. of at least n=4. c) Left: example of a single experiment of PTP sensibility measured by a CRC assay on whole MEF-NF1−/−cells treated with different doses of 1. Mitochondrial calcium uptake is plotted in terms of Calcium Green-5N fluorescence. Right: average histogram of the total Ca2+ uptake before PTP opening (error bars represent the S.D. for n=3).
Conclusions
In the last four decades, the unquestionable therapeutic success of the anticancer drug cisplatin has prompted the design, synthesis, and biological development of several metal-based potential chemotherapeutics. On the whole, our gold(III) complexes of the type [AuX2(dtc)] (X=Cl, Br; dtc=various dithiocarbamates) stand out among others for their capability to strongly inhibit tumor growth both in vitro and in vivo by mechanisms of action differing from those recognized for the clinically-established platinum drugs.59
In this work, our novel compounds 1 and 2 showed a marked cytotoxicity towards different tumor cell types in a short treatment time (Table 5).
In order to exploit the chemotherapeutic potential of this class of gold(III) derivatives, it is mandatory to dissect their mode of action on target cells. One possibility is that tumor cell death is caused by oxidative stress. It must be underlined that malignant cells are extremely sensitive to oxidative stress as they are characterized by complex metabolic rearrangements, with an enhancement of glucose utilization and a concomitant decrease of mitochondrial respiration (the Warburg effect60–62). As respiratory chain complexes are the major producers of ROS inside cells,63 downmodulation of their activity enhances ROS generation, forcing cancer cells to set a novel homeostatic redox equilibrium by simultaneously inducing antioxidant defenses.64 Thus, neoplasms might be sensitized to oxidative stress with respect to nontransformed tissues, as any increase in ROS levels could unbalance their redox equilibrium by overpowering their residual anti-oxidant capabilities.
In this research, we observed that the great activity of compound 1 on NF1−/− MEFs is paralleled by an increase in ROS levels. This oxidative assault is able to induce the PTP, a mitochondrial channel whose opening commits cells to death.54 However, the presence of the scavenger Trolox does not affect the ability of compound 1 to trigger the pore transition in MEF-NF1−/− cells, thus suggesting the oxidative stress can only partially account for the efficacy of our potential drug. The chemical behavior of both complexes was also studied under physiological-like conditions by UV/Vis spectrophotometry in order to gain insights into the stability of the gold(III) coordination sphere and to connect it with the degree of cytotoxicity of the compounds. In this regard, the organic/inorganic nature of the ligands can play a major role in modulating anticancer properties of the metal ion. On the whole, the analysis of the collected chemical data and their comparison with the biological results brings out a structure–activity relationship. In particular, we found that lability of the halogen–gold bond perfectly matches the biological effects of the complexes. In fact, it turned out that the faster the hydrolysis process of the Au−Br/Cl bond, the greater the chemotherapeutic effect observed, thus accounting for the stronger cytotoxicity of complex 1 in comparison with complex 2. Such a phenomenon is in agreement with the findings reported by Casini et al. for other gold(III) anticancer agents.5 In other words, our compounds undergo an “activation” process (i.e. release of one or both halide ligands from the tetracoordinated gold(III) center), required to react with the possible cellular targets and, hence, to trigger the antitumor effects. On the other hand, based on the redox properties (oxidizing) of the metal ion, a reductive step for the AuIII center cannot be ruled out a priori when studying the mechanism of action of this class of potential pharmaceuticals.
Experimental Section
Chemicals
KAuBr4⋅2H2O and KAuCl4⋅2H2O were purchased from Alfa Aesar, [D6]DMSO from Aldrich, ammonium pyrrolidinedithiocarbamate from Fluka, and P4O10 from AnalaR Normapur–Prolabo. Dulbeccos modified Eagle medium (DMEM) was purchased from Euroclone. All other reagents and solvents (from Aldrich or Fluka) were of high purity and used without further purification. SAOS-2 osteosarcoma cells, HeLa cervical adenocarcinoma cells, and HCT116 colorectal carcinoma cells were purchased from the American Type Culture Collection (ATCC). MEF-NF1−/− cells were kindly provided by Prof. Reuven Stein (George S. Wise Faculty of Life Sciences, Tel Aviv University, Israel).
Instrumentation and general procedures
Elemental analysis was performed using a Carlo Erba 1108 CHNS-O microanalyser (CE Instruments Ltd., Wigan, UK).
FT-IR spectra were recorded at 298 K under N2 atmosphere in Nujol between two polyethylene tablets on a Nicolet Nexus 870 FT-IR spectrophotometer (Nicolet Instrument Technologies, now Thermo Scientific, Waltham, USA) in the range 50–600 cm−1 (200 scansions, 4 cm−1 resolution), and in solid KBr on a Nicolet Nexus 5SXC FT-IR spectrophotometer (32 scansions, 2 cm−1 resolution) in the range 400–4000 cm−1. Data processing was carried out using OMNIC version 5.1 (Thermo Scientific, Waltham, USA).
All 1 D 1H NMR and 2 D spectra were acquired in [D6]DMSO at 298 K on a Avance DRX300 spectrophotometer (Bruker, Billerica, USA) equipped with a broadband inverse (BBI) [1H, X] probehead. Typical acquisition parameters for 1 D 1H NMR spectra (1H: 300.13 MHz): 64–128 transients, spectral width 12 kHz, using 32 k data points and a delay time of 3.0 s. Spectra were processed by exponential weighting with a resolution of 0.5 Hz and a line-broadening threshold of 0.1 Hz. Typical acquisition parameters for 2 D [1H,13C] HMBC NMR spectra (1H: 300.13/13C: 75.48 MHz): 512 transients of 16 scans/block, spectral width 7.5/18.8 kHz, 1 k/1 k data points and a delay time of 2.0 s. Sequences were optimized for 1J(13C,1H)=145 Hz/nJ(13C,1H)=5 Hz with no 1H decoupling. Spectra were processed by using sine-square weighting with a resolution of 1.0/3.0 Hz and a line-broadening threshold of 0.3/1.0 Hz. Data processing was carried out by means of MestReNova version 6.2 (Mestrelab Research, Escondido, USA). Chemical shifts were referenced to the characteristic signal of the solvent by considering external tetramethylsilane (TMS).
Thermogravimetric (TG) and differential scanning calorimetry (DSC) curves were recorded on a thermobalance equipped with a DSC 2929 calorimeter (TA Instruments, New Castle, USA). Measurements were carried out in the range 25–1300 °C in alumina crucibles under air (flux rate 30 cm3 min−1) at a heating rate of 5 °C min−1, using alumina as reference.
Electronic spectra were acquired at 37 °C in the range 250–500 nm using a Cary 100 UV/Vis double beam spectrophotometer (Agilent, Santa Clara, USA) (1 cm optical path, quartz cuvettes), using fresh solutions of the samples in DMSO. With respect to the studies in 0.9 % (w/v) NaCl solution, compounds 1 and 2 were dissolved in DMSO beforehand and subsequently diluted with saline solution (∼1 % v/v DMSO) to yield a final concentration of 100 μm. For the kinetic studies, the initial time (time zero) was set upon the complete dissolution of the complex.
Synthesis of the gold(III) complexes
To an aqueous solution of KAuX4⋅2H2O (X=Br, Cl; 0.6 mmol), the ligand NH4(PDT) (0.6 mmol) was added dropwise while stirring at rt, giving rise to the immediate precipitation of a solid. After 10 min, the crude product was isolated by centrifugation (5,000 rpm, 15 min)and washed with H2O and diethyl ether. Both complexes were then dried in vacuo in the presence of P4O10, with the final yield in the range of 85–90 %.
Dibromido[N-dithiocarboxy-κS,κS′-pyrrolidine]gold-(III) (AuIIIBr2(PDT), 1) Orange powder (89.6 %): soluble in DMSO, acetone, and dimethylformamide (DMF), slightly soluble in CH3CN and CH2Cl2, insoluble in H2O, EtOH, MeOH, isopropanol, CCl4, and diethyl ether; FT-IR (KBr): ṽmax=2971–2849 (ν, CH2), 1583 (ν, N−CSS), 1447 (ν, CH2), 943 cm−1 (νa, S−C−S); FT-IR (Nujol): ṽmax=537 (νs, S−C−S), 411 (νa, S−Au−S), 371 (νs, S−Au−S), 240 (νa, Br−Au−Br), 218 cm−1 (νs, Br−Au−Br); 1H NMR (300.13 MHz, [D6]DMSO, 298 K): δ=2.06 (m, 4 H, CH2CH2N); 3.81 ppm (m, 4 H, CH2N); 13C NMR (75.48 MHz, [D6]DMSO, 298 K): δ=23.35 (CH2CH2N), 50.87 (CH2N), 188.32 ppm (CSS); Anal. calcd for C5H8AuBr2NS2 (MW=503.81): C 11.94, H 1.60, N 2.78, S 12.75, found: C 12.05, H 1.60, N 2.74, S 12.80; TG (air): calcd weight loss to Au(0) −60.91 %, found −59.95 %.
Dichlorido[N-dithiocarboxy-kS,κS′-pyrrolidine]gold-(III) (AuIIICl2(PDT), 2) Yellow ochre powder (85.3 %): soluble in DMSO, acetone, and DMF, slightly soluble in CH3CN and CH2Cl2, insoluble in H2O, EtOH, MeOH, isopropanol, CCl4, and diethyl ether; FT-IR (KBr): ṽmax=2980–2866 (ν, CH2), 1586 (ν, N−CSS), 1439 (ν, CH2), 942 cm−1 (νa, S−C−S); FT-IR (Nujol): ṽmax=538 (νs, S−C−S), 413 (νa, S−Au−S), 375 (νs, S−Au−S), 362 (νa, Cl−Au−Cl), 315 cm−1 (νs, Cl−Au−Cl); 1H NMR (300.13 MHz, [D6]DMSO, 298 K): δ=2.06 (m, 4 H, CH2CH2N), 3.82 ppm (m, 4 H, CH2N); 13C NMR (75.48 MHz, [D6]DMSO, 298 K): δ=23.41 (CH2CH2N), 51.10 (CH2N), 188.58 ppm (CSS); Anal. calcd for C5H8AuCl2NS2 (MW=412.13): C 14.50, H 1.95, N 3.38, S 15.49, found: C 14.56, H 1.92, N 3.34, S 15.48; TG (air): calcd weight loss to Au(0) −52.21 %, found −51.91 %.
Cell viability assay
Cell viability was measured using the Resazurin-based In Vitro Toxicology Assay Kit (Sigma-Aldrich, St. Louis, USA). Cells (40,000/well) were plated into 96-well microplates the day before the experiment. After the 3 h treatments with compounds 1 or 2, the Resazurin-based reagent was added. Then, cells were maintained at 37 °C for an additional 2 h, and the absorbance at 595 nm was detected using the iMark microplate reader (BioRad, Hercules, USA). All treatments were carried out in DMEM supplemented with l-Glutamine (Euroclone), penicillin (100 U mL−1, Euroclone), and streptomycin (100 μg mL−1, Euroclone) distributed into each well (200 μL) of a 96-well cell culture plate. Stock solutions of all complexes were made in DMSO and then diluted with complete medium in such a way that the final amount of solvent in each well did not exceed 0.4 % (v/v). Cytotoxicity data were expressed as IC50 values (Table 5), that is the concentration of the complex inducing a 50 % decrease in cell number compared with control cultures.
ROS measurements
ROS measurements were performed using the probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, 1 μm, Molecular Probes, Carlsbad, USA). Cells (10,000/well) were plated the day before the experiment, and CM-H2DCFDA was added with treatments in DMEM supplemented with 0.5 % fetal bovine serum (FBS) without bicarbonate. Cells were maintained at 37 °C without CO2 during the experiment, and the fluorescence (λex=485 nm, λem=538 nm) was recorded using a Fluoroskan Ascent FL plate reader (Thermo Fisher Scientific, Waltham, USA) at different time points.
Measurement of mitochondrial Ca2+ retention capacity
The CRC assay was used to assess PTP opening by following trains of Ca2+ pulses which were measured fluorimetrically at 25 °C in the presence of the Ca2+ indicator Calcium Green-5N (1 μm, λex=505 nm, λem=535 nm, Molecular Probes). We performed CRC experiments either on isolated mitochondria or on whole cells placed in an isotonic buffer (130 μm KCl, 1 μm Pi-Tris (Pi: inorganic phosphate), 10 mm Tris/3-(N-morpholino)propansulfonic acid (MOPS), 10 mm Tris/ethylene glycol tetraacetic acid (EGTA), 5 mm glutamate/2.5 mm malate, pH 7.4). Whole-cell CRC was carried out after 3 h treatment with compound 1, followed by the plasma membrane permeabilization with the nonionic detergent digitonin, which is highly selective for cholesterol-enriched membranes and does not damage the mitochondrial membranes. Cells were washed with phosphate-buffered saline (PBS, pH 7.4) and then permeabilized with 150 μm digitonin (15 min, 4 °C) in a buffer composed of 130 μm KCl, 1 μm Pi-Tris, 10 mm Tris/MOPS, and 1 mm Tris/EGTA (pH 7.4). Digitonin was then washed away by spinning cells in the same buffer (15,000 rpm, 30 min) and changing the EGTA/Tris concentration to 0.1 mm, and the number of cells was carefully assessed before starting each experiment. Mitochondria (0.5 mg mL−1) or permeabilized cells (7×106 cells per experiment) were then placed in the presence of the Ca2+ indicator Calcium-Green-5N, which does not permeate mitochondria, and were exposed to Ca2+ spikes (10 μm and 5 μm, respectively). Drops in fluorescence were used to assess mitochondrial Ca2+ uptake. PTP opening was detected as an increase in fluorescence. Pore inhibitors, such as cyclosporine A, are expected to enhance the threshold of Ca2+ concentration required to trigger the permeability transition, that is, the number of spikes before a sudden and marked fluorescence increase occurs.
Acknowledgments
The authors would like to thank the Cariparo Foundation, Italy, Banca Popolare di Marostica, and Associazione per la Ricerca di Terapie Mirate in Oncologia (A.R.TE.M.O). The authors also thank Dr. Renato Schiesari and Dr. Loris Calore (Department of Chemical Sciences, University of Padova, Italy), Fabiola Pasqualato and Isabella Bortolato (Department of Cardiac, Thoracic, and Vascular Sciences, University of Padova, Italy), and Elena Trevisan (Department of Biomedical Sciences, University of Padova, Italy) for their technical assistance and inexhaustible support. A. R. is supported by “Progetti di Ateneo dell′Università di Padova”, [12.06.41] grant number CPDA123598.
References
- 1.Rosenberg B, Van Camp L, Krigas T. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 2.Mjos KD, Orvig C. Chem. Rev. 2014;114:4540–4563. doi: 10.1021/cr400460s. [DOI] [PubMed] [Google Scholar]
- 3.Lippert B. Coord. Chem. Rev. 1999;182:263–295. [Google Scholar]
- 4.Alderden RA, Hall MD, Hambley TW. J. Chem. Educ. 2006;83:728–734. [Google Scholar]
- 5.Casini A, Kelter G, Gabbiani C, Cinellu MA, Minghetti G, Fregona D, Fiebig HH, Messori L. J. Biol. Inorg. Chem. 2009;14:1139–1149. doi: 10.1007/s00775-009-0558-9. [DOI] [PubMed] [Google Scholar]
- 6.Biersack B, Ahmad A, Sarkar FH, Schobert R. Curr. Med. Chem. 2012;19:3949–3956. doi: 10.2174/092986712802002482. [DOI] [PubMed] [Google Scholar]
- 7.Colotti G, Ilari A, Boffi A, Morea V. Mini-Rev. Med. Chem. 2013;13:211–221. [PubMed] [Google Scholar]
- 8.Jungwirth U, Kowol CR, Keppler BK, Hartinger CG, Berger W, Heffeter P. Antioxid. Redox Signaling. 2011;15:1085–1127. doi: 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casini A, Messori L. Curr. Top. Med. Chem. 2011;11:2647–2660. doi: 10.2174/156802611798040732. [DOI] [PubMed] [Google Scholar]
- 10.Barry NPE, Sadler PJ. Chem. Commun. 2013;49:5106–5131. doi: 10.1039/c3cc41143e. [DOI] [PubMed] [Google Scholar]
- 11.de Almeida A, Oliveira BL, Correia JDG, Soveral G, Casini A. Coord. Chem. Rev. 2013;257:2689–2704. [Google Scholar]
- 12.Márta Nagy E, Ronconi L, Nardon C, Fregona D. Mini-Rev. Med. Chem. 2012;12:1216–1229. doi: 10.2174/138955712802762004. [DOI] [PubMed] [Google Scholar]
- 13.Nagy EM, Pettenuzzo A, Boscutti G, Marchiò L, Dalla Via L, Fregona D. Chem. Eur. J. 2012;18:14464–14472. doi: 10.1002/chem.201202171. [DOI] [PubMed] [Google Scholar]
- 14.Nagy EM, Nardon C, Giovagnini L, Marchiò L, Trevisan A, Fregona D. Dalton Trans. 2011;40:11885–11895. doi: 10.1039/c1dt11504a. [DOI] [PubMed] [Google Scholar]
- 15.Kelland L. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 16.Köberle B, Tomicic MT, Usanova S, Kaina B. Biochim. et Biophys. Acta - Rev. on Cancer. 2010;1806:172–182. doi: 10.1016/j.bbcan.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Reedijk J. Plat. Met. Rev. 2008;52:2–11. [Google Scholar]
- 18. www.cancer.gov/cancertopics/druginfo/fda.
- 19.Ronconi L, Fregona D. Dalton Trans. 2009:10670–10680. doi: 10.1039/b913597a. [DOI] [PubMed] [Google Scholar]
- 20.Nardon C, Boscutti G, Fregona D. Anticancer Res. 2014;34:487–492. [PubMed] [Google Scholar]
- 21.Ronconi L, Nardon C, Boscutti G, Fregona D. In: Advances in Anti-Cancer Agents in Medicinal Chemistry, Vol. 2. Prudhomme M, editor. Bussum: Bentham Science Publishers; 2013. pp. 130–172. (Ed.:, chap. 3, pp. . [Google Scholar]
- 22.Nardon C, Schmitt SM, Yang H, Zuo J, Fregona D, Dou QP. PLoS ONE. 2014;9:84248. doi: 10.1371/journal.pone.0084248. , e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP. Cancer Res. 2006;66:10478–10486. doi: 10.1158/0008-5472.CAN-06-3017. [DOI] [PubMed] [Google Scholar]
- 24.Negom Kouodom M, Ronconi L, Celegato M, Nardon C, Marchiò L, Dou QP, Aldinucci D, Formaggio F, Fregona D. J. Med. Chem. 2012;55:2212–2226. doi: 10.1021/jm201480u. [DOI] [PubMed] [Google Scholar]
- 25.Marzano C, Ronconi L, Chiara F, Giron MC, Faustinelli I, Cristofori P, Trevisan A, Fregona D. Int. J. Cancer. 2011;129:487–496. doi: 10.1002/ijc.25684. [DOI] [PubMed] [Google Scholar]
- 26.Cattaruzza L, Fregona D, Mongiat M, Ronconi L, Fassina A, Colombatti A, Aldinucci D. Int. J. Cancer. 2011;128:206–215. doi: 10.1002/ijc.25311. [DOI] [PubMed] [Google Scholar]
- 27.Giovagnini L, Sitran S, Montopoli M, Caparrotta L, Corsini M, Rosani C, Zanello P, Dou QP, Fregona D. Inorg. Chem. 2008;47:6336–6343. doi: 10.1021/ic800404e. [DOI] [PubMed] [Google Scholar]
- 28.Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP. Toxicol. Appl. Pharmacol. 2008;231:24–33. doi: 10.1016/j.taap.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Criado JJ, Lopez-Arias JA, Macias B, Fernandez-Lago LR, Salas JM. Inorg. Chim. Acta. 1992;193:229–235. [Google Scholar]
- 30.Criado JJ, Fernandez I, Macias B, Salas JM, Medarde M. Inorg. Chim. Acta. 1990;174:67–75. [Google Scholar]
- 31.Criado JJ, Carrasco A, Macias B, Salas JM, Medarde M, Castillo M. Inorg. Chim. Acta. 1989;160:37–42. [Google Scholar]
- 32.Macías B, Criado JJ, Vaquero MV, Villa MV. Thermochim. Acta. 1993;223:213–221. [Google Scholar]
- 33.Castillo M, Criado JJ, Macias B, Vaquero MV. Inorg. Chim. Acta. 1986;124:127–132. [Google Scholar]
- 34.Brown DA, Glass WK, Burke MA. Spectrochim. Acta Part A. 1976;32:137. [Google Scholar]
- 35.Ito H, Fujita J, Saito K. Bull. Chem. Soc. Jpn. 1967;40:2584–2591. [Google Scholar]
- 36.Gianelli L, Amendola V, Fabbrizzi L, Pallavicini P, Mellerio GG. Rapid Commun. Mass Spectrom. 2001;15:2347–2353. [Google Scholar]
- 37.Bishop P, Marsh P, Brisdon AK, Brisdon BJ, Mahon MF. J. Chem. Soc. Dalton Trans. 1998:675–682. [Google Scholar]
- 38.Forghieri F, Preti C, Tassi L, Tosi G. Polyhedron. 1988;7:1231–1237. [Google Scholar]
- 39.Casellato U, Fracasso G, Graziani R, Sindellari L, González AS, Nicolini M. Inorg. Chim. Acta. 1990;167:21–24. [Google Scholar]
- 40.Radanovic DJ, Matović ZD, Miletić VD, Battaglia LP, Ianelli S, Efimenko IA, Ponticelli G. Transition Met. Chem. 1996;21:169–175. [Google Scholar]
- 41.Desseyn HO, Fabretti AC, Forghieri F, Preti C. Spectrochim. Acta Part A. 1985;41:1105–1108. [Google Scholar]
- 42.Ronconi L, Giovagnini L, Marzano C, Bettìo F, Graziani R, Pilloni G, Fregona D. Inorg. Chem. 2005;44:1867–1881. doi: 10.1021/ic048260v. [DOI] [PubMed] [Google Scholar]
- 43.Beurskens PT, Blaauw HJA, Cras JA, Steggerda JJ. Inorg. Chem. 1968;7:805–810. [Google Scholar]
- 44.Coates GE, Parkin C. J. Chem. Soc. 1963:421–429. [Google Scholar]
- 45.Lee AWM, Chan WH, Ho MF. Anal. Chim. Acta. 1991;246:443–445. [Google Scholar]
- 46.Franchini GC, Giusti A, Preti C, Tassi L, Zannini P. Polyhedron. 1985;4:1553–1558. [Google Scholar]
- 47.Hadjikostas CC, Katsoulos GA, Shakhatreh SK. Inorg. Chim. Acta. 1987;133:129–132. [Google Scholar]
- 48.Gangopadhayay AK, Chakravorty A. J. Chem. Phys. 1961;35:2206–2209. [Google Scholar]
- 49.Ronconi L, Marzano C, Zanello P, Corsini M, Miolo G, Maccà C, Trevisan A, Fregona D. J. Med. Chem. 2006;49:1648–1657. doi: 10.1021/jm0509288. [DOI] [PubMed] [Google Scholar]
- 50.Messori L, Abbate F, Marcon G, Orioli P, Fontani M, Mini E, Mazzei T, Carotti S, OConnell T, Zanello P. J. Med. Chem. 2000;43:3541–3548. doi: 10.1021/jm990492u. [DOI] [PubMed] [Google Scholar]
- 51.Cotton FA, Wilkinson G. Advanced Inorganic Chemistry, A Comprehensive Text, 3rd ed. New York: Interscience; 1972. p. 1145. , p. . [Google Scholar]
- 52.Chiara F, Gambalunga A, Sciacovelli M, Nicolli A, Ronconi L, Fregona D, Bernardi P, Rasola A, Trevisan A. Cell Death Dis. 2012;3:444. doi: 10.1038/cddis.2012.184. , e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rasola A, Bernardi P. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 54.Rasola A, Bernardi P. Cell Calcium. 2011;50:222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 55.Trovó-Marqui AB, Tajara EH. Clin. Genet. 2006;70:1–13. doi: 10.1111/j.1399-0004.2006.00639.x. [DOI] [PubMed] [Google Scholar]
- 56.Schubbert S, Shannon K, Bollag G. Nat. Rev. Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 57.Brems H, Beert E, de Ravel T, Legius E. Lancet Oncol. 2009;10:508–515. doi: 10.1016/S1470-2045(09)70033-6. [DOI] [PubMed] [Google Scholar]
- 58.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. PLoS ONE. 2008;3:1852. doi: 10.1371/journal.pone.0001852. , e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saggioro D, Rigobello MP, Paloschi L, Folda A, Moggach SA, Parsons S, Ronconi L, Fregona D, Bindoli A. Chem. Biol. 2007;14:1128–1139. doi: 10.1016/j.chembiol.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 60.Frezza C, Gottlieb E. Semin. Cancer Biol. 2009;19:4–11. doi: 10.1016/j.semcancer.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 61.Levine AJ, Puzio-Kuter AM. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 62.Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 63.Murphy MP. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cairns RA, Harris IS, Mak TW. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]